Simple Summary
Sestrins (SESN1-3) are a family of stress-responsive proteins that serve as direct targets of several transcription factors, including tumour suppressor protein p53. Structural studies have identified three core molecular functions of Sestrins: scavenging of reactive oxygen species (ROS), inhibition of mTORC1 signalling, and leucine binding. As leucine-dependent mTORC1 inhibitors and antioxidants, Sestrins regulate metabolism and redox balance, both of which are frequently dysregulated in cancer. Dysregulation of Sestrin expression, most commonly downregulation, is widely observed in cancers and is often associated with increased tumour growth and poor prognosis. However, in certain contexts, Sestrin overexpression may promote tumour survival, and this review explores the molecular mechanisms underlying these seemingly contradictory effects on carcinogenesis, evaluates the evidence for Sestrin involvement across various cancer types, and discusses the potential of Sestrins as diagnostic markers and therapeutic targets.
Abstract
Sestrins (SESN1-3) are a family of stress-responsive proteins that regulate cellular metabolism and redox balance, both of which are frequently disrupted in cancer. As direct targets of stress-responsive transcription factors, including tumour suppressor p53, Sestrins function as leucine-dependent inhibitors of mTORC1 and potent antioxidants. Their downregulation is widely observed across multiple cancers and is associated with increased tumour growth and poor prognosis. Despite their consistent tumour-suppressive effects through mTORC1 inhibition and promotion of p53-dependent apoptosis, Sestrins exhibit a limited role in tumour initiation, which appears to be context-dependent. Their antioxidant activity reduces oxidative damage, thereby protecting against genomic instability and other cancer-promoting events. However, in certain contexts, Sestrins may promote tumour survival and progression by stimulating pro-survival pathways, such as AKT signalling through mTORC2 activation. This review examines the molecular mechanisms underlying these dual functions, with a particular focus on mTOR signalling and oxidative stress. We also discuss Sestrin expression patterns and functional outcomes in various cancer types, including lung, liver, colon, skin, prostate, and follicular lymphomas, highlighting their potential as diagnostic markers and therapeutic targets.
1. Introduction
Sestrins are a family of stress-responsive proteins discovered in the 1990s. There are three Sestrin genes (SESN1-3), which encode functionally similar proteins but are regulated by different transcription factors and environmental stimuli. Sestrins are ubiquitously expressed among most human tissues and the early Sestrins can be traced back to invertebrate animals, such as Caenorhabditis elegans [1]. With the arrival of vertebrates, the single Sestrin gene appears to have duplicated into the three Sestrin genes we know today, presumably to provide redundancy and broaden the range of responses, as each Sestrin gene is regulated by a distinct set of transcription factors [2].
Sestrins are built from two somewhat symmetrical domains, which serve distinct, yet concordant molecular functions (Figure 1). The three established molecular functions of a Sestrin protein are direct reactive oxygen species (ROS) scavenging, sensing of cellular leucine, and inhibition of the mammalian target of rapamycin complex 1 (mTORC1). Sestrins directly neutralise hydrophobic ROS molecules [3,4], but may also support antioxidative cell signalling. They also directly bind leucine via their leucine-binding pocket and thus change their conformation [5]. The conformational change prevents their inhibition of mTORC1, as they are unable to associate with their partner—the GATOR2 protein complex, through which they inhibit mTORC1 [6]. Their inhibition of the mTORC1 complex pauses catabolic metabolism and protein synthesis, redirecting the cell from growth to recycling and repair, thereby halting proliferation and biomass accumulation while promoting cellular restoration through the endogenous recycling process—autophagy [7].
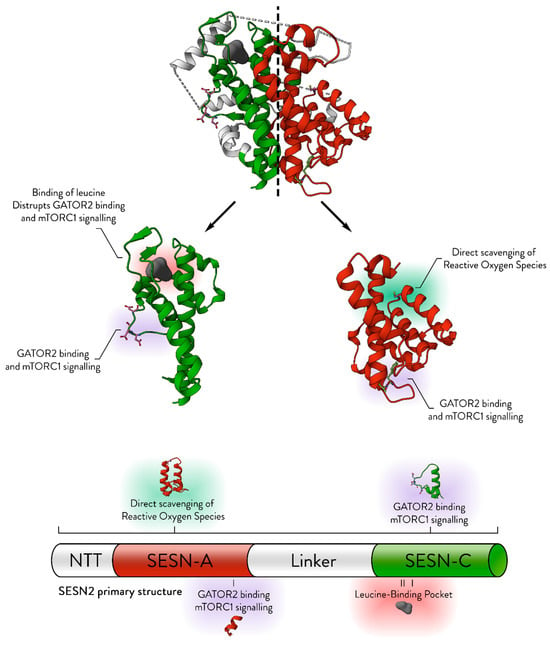
Figure 1.
The structural basis for the established molecular roles of Sestrin. The structure of SESN2 (PDB ID: 5DJ4) is displayed, with its symmetrical nature emphasised by a central dashed line. The SESN-A and SESN-C domains, without the linker domain, are shown separately for detailed examination. Key structural features essential for the molecular functions of Sestrins are annotated. NTT refers to the N-terminal tail. The colour scheme highlights different domains: red for SESN-A, green for SESN-C, and white for the linker region. Modified from [2].
mTORC1 is tightly regulated by two sets of small GTPases: RHEB and RagA-D, through separate signalling pathways. RHEB, anchored to the lysosomal membrane, becomes active in response to ATP, cytokines, glucose, growth factors, and endocrine signals. These signals regulate RHEB through the tuberous sclerosis complex (TSC), which consists of TSC1, TSC2, and TDC1D7. TSC functions as a GTPase-activating protein (GAP) for RHEB, converting active GTP-bound RHEB to its inactive GDP-bound state [8,9]. Growth factor signalling activates the AKT kinase, which phosphorylates TSC2, reducing TSC activity and relieving its inhibition of RHEB and mTORC1 [10,11,12]. In contrast, the AMP-activated protein kinase (AMPK) complex, a central sensor of the AMP/ATP ratio, inhibits mTORC1 indirectly by activating TSC2 and phosphorylating the mTORC1 subunit RAPTOR [13,14,15]. Thus, AKT, and AMPK act in opposition. Early studies indicated that SESN2 may inhibit mTORC1 via AMPK. SESN2 reduces RHEB activation in a TSC2-dependent manner and interacts with both TSC and AMPK, indicating its association with these complexes. A mutant form of SESN2 lacking the N-terminal fails to bind AMPK [16]. SESN2 may facilitate AMPK activation by acting as a platform for LKB1-mediated phosphorylation of the AMPK catalytic subunit [17]. However, the precise SESN–AMPK mechanism remains unclear.
In addition to intercellular signals, mTORC1 is also regulated by an intracellular amino acid-sensing axis. The Rag GTPases function as heterodimers, with RagA/B bound to GTP and RagC/D bound to GDP in their active state [18]. The Ragulator complex anchors Rags to the lysosome [19]. The protruding RAPTOR subunit of mTORC1 binds to the active Rags complex, docking mTORC1 at the lysosome, which is essential for its activation by lysosome-anchored RHEB [20]. The activity of Rags is controlled by the GATOR complexes (GATOR1 and GATOR2). In the absence of leucine, Sestrins bind GATOR2, lifting its inhibition of GATOR1. GATOR1 functions as a GTPase-activating protein (GAP) for RagA/B, inactivating it and preventing mTORC1 activation [6,21,22,23,24].
In addition to mTORC1, the mTOR kinase also forms a second distinct complex called mTORC2. While both complexes house the same mTOR protein in mammals, the accessory subunits of mTORC2 are distinct from those of mTORC1. Instead of RAPTOR, which binds mTORC1 to Rags at the lysosomal membrane, mTORC2 contains RICTOR and thus is not recruited to the lysosome. The precise subcellular localisation of mTORC2 remains unclear, although evidence suggests that its mSIN1 subunit may direct the complex to the plasma membrane [25], and it has additionally been shown to localise at the endoplasmic reticulum [26]. mTORC2 has a distinct set of substrates. Through PKC, this complex influences cytoskeletal organisation [27,28], thus affecting the metastatic potential and survival of some cancers [29]. Notably, mTORC2 also phosphorylates AKT, a protein whose activity is frequently promoted by Sestrins in cancers [30,31]. While initial findings demonstrated that silencing RICTOR dramatically diminished AKT activation in response to SESN2 induction in the MCF7 breast carcinoma cell line [32], the precise role of mTORC2 in Sestrin-mediated AKT regulation continues to be explored [33,34].
While Sestrins contain a catalytic cysteine which is essential for neutralisation of hydrophobic ROS, such as cumene hydroperoxide [3,4], they also may support other antioxidative mechanisms. Sestrins were shown to support the activation of the antioxidant transcription factor Nrf2 [35,36]. Some experiments indicated that SESN2 may interact with Keap1, the inhibitor of Nrf2, and induce Keap1’s selective degradation, thereby activating Nrf2 [35,37]. Other studies supported the notion that Sestrins protect mitochondria from ROS-induced damage. Mitophagy, a selective form of autophagy that recycles damaged mitochondria to maintain cellular function, has been proposed to involve Sestrins. One study showed that Sestrins promote mitophagy of impaired mitochondria in macrophages [38], and another work has demonstrated that Sestrins colocalise with mitochondria [39]. Also, SESN2 has been shown to directly support mitophagy, through association with ULK1, which promotes Beclin-1 phosphorylation, thereby initiating mitophagy via the PINK1–Parkin pathway [40].
While Sestrins are expressed at varying basal levels in most tissues [41], they are drastically upregulated in response to a multitude of stress insults (Figure 2). Most important for regulation of carcinogenesis are the p53-inducible SESN1 and SESN2 genes, which are direct targets of p53 [1,42] and are both expressed at low levels in p53-deficient cells [1,43]. The TP53 gene is frequently mutated in cancer, with over 29,000 identified mutations and an overall prevalence of 29% across all cancers [44]. While p53 mutations vary by cancer type, they are nearly ubiquitous in malignancies, with rates varying between 10% in haematological malignancies to over 90% in high-grade serous ovarian carcinoma [45]. As oxidative damage can inadvertently damage DNA [46], SESN1 and SESN2 have been found to be essential for the protective effect against oxidative damage mediated by p53 [43]. Hypoxia, under which tumours frequently find themselves, steadily upregulates SESN2 [1]. In some cases, SESN2 expression has been shown to correlate with HIF-1α activity; however, a direct regulatory relationship remains unconfirmed [47,48]. ER stress upregulates SESN2 through transcription factors ATF6 [49], XBP1 [50], and ATF4, for which a SESN2 promoter region has been identified [51]. Oxidative stress can upregulate SESN2 via the Nrf2 antioxidative transcription factor [52], and the SESN3 gene is a confirmed direct target of the antioxidative transcription factors FOXO1&3 [53,54].
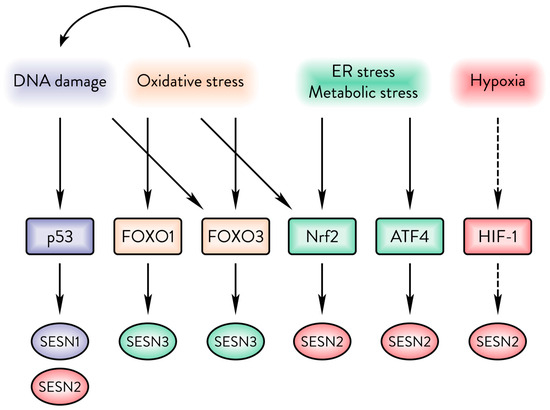
Figure 2.
The Sestrin family responds to a variety of cellular stresses. DNA damage upregulates SESN1&2. Oxidative damage upregulates SESN3 but can also cause upregulation of SESN1&2 via p53. ER and energy stresses can upregulate SESN2 via Nrf2 or ATF4. Hypoxia upregulates SESN2, but it is unclear whether that occurs in a HIF-1α-dependent manner.
Recent statistics have shown that approximately 20% of people in the world will develop cancer in their lifetime [55], underscoring the urgent need for reliable diagnostic markers and therapeutic targets. The multifaceted nature of Sestrin proteins and the network of signals governing their upregulation complicate their physiological and pathological roles. In normal cells, their functions reinforce each other, as their antioxidative activity protects cells from damage while mTORC1 inhibition halts catabolic processes, thereby shifting the cell into a protective recovery state. In cancer cells, Sestrins hamper aberrant growth, promote DNA-surveillance mechanisms by facilitating apoptotic cell death, and limit oxidative stress to protect genomic integrity. However, when Sestrin activators such as p53 are nonfunctional, their tumour-suppressive capacity is compromised. Reduced Sestrin expression, which is frequently observed across many tumours, impairs their ability to inhibit mTORC1 effectively. In other cases, disruptions in their signalling partners such as GATOR1 may prevent effective mTORC1 inhibition. As a result, when Sestrins fail to inhibit mTORC1, they may be repurposed by the tumour to support its growth and survival, with their ability to protect against oxidative stress and metabolic challenges inadvertently promoting tumour progression.
Overall, we observe that Sestrins are downregulated across multiple cancers. According to the database GENT2, the three SESN genes are downregulated across all cancers [56]. Poor expression of Sestrins is linked to poor prognosis in several cancers such as lung and colorectal cancers [30,57,58]. Yet, conversely, we encounter cases in which raised Sestrin expression can carry a poor prognosis [31,59]. For example, in melanoma, Sestrin can support the survival of cancerous cells exposed to ultraviolet B (UVB) radiation, and also protect metastatic cells from oxidative damage, thus supporting growth and survival of the cancer [31,60].
This review will explore the available evidence and attempt to clarify the seemingly contradictory roles of Sestrins in cancer, highlighting the molecular mechanisms that underlie their dual function as both tumour suppressors and facilitators of malignancy.
2. Sestrins in Cancer
Carcinogenesis is distinguished by critical hallmarks such as continuous cell growth, resistance to apoptosis, evasion of tumour suppressors, genomic instability, and persistent inflammation, among others [61]. Sestrins act as crucial mediators of the p53 pathway, influencing these key hallmarks of carcinogenesis [62,63]. However, their influence on cancer can be ambiguous depending on the context. Sestrins inhibit mTORC1 while also stimulating the AKT pathway [63], both of which are frequently activated in cancers and can have opposing effects on tumorigenesis, either supporting or suppressing it. These parallel but interdependent pathways indicate that Sestrins could inhibit tumour growth by repressing mTORC1 or promote survival and proliferation through AKT activation [64]. Additionally, their role in autophagy activation can either deter or facilitate carcinogenesis, depending on the carcinogenesis stage, and type [65]. Consequently, the impact of Sestrins on cell viability may differ based on the stress type, resulting in varying effects within tumours [66,67].
2.1. Sestrins in Follicular Lymphomas
The 6q21 locus, which includes SESN1, frequently undergoes deletion in human follicular lymphomas, and SESN1 expression is notably reduced in lymphomas that express the mutant EZH2X641Y protein [68]. EZH2, a crucial component of the Polycomb Repressive Complex 2 and a histone-lysine N-methyltransferase, catalyses the methylation of histone H3 on lysine 27, effectively repressing gene expression—a process vital for lymphocyte maturation [69]. The EZH2X641Y mutation in lymphoma cells leads to mTORC1 activation via suppression of SESN1 transcription. This activation of mTORC1 is implicated in the progression of cancer, as evidenced by the observation that mTORC1 inhibition suppresses stem cell proliferation and lymphoma advancement [70]. Furthermore, the knockout of SESN1 in the SU-DHL-10 lymphoma cell line abolished the apoptosis induced by GSK126, an inhibitor of EZH2’s methyltransferase activity [68].
2.2. Sestrins in Lung Cancer
SESN1 and SESN2 frequently exhibit reduced expression in lung tumours, with decreased SESN2 levels marking a poor prognosis for lung cancer patients [30,58]. The loss of SESN1 and SESN2 in A549 lung adenocarcinoma cells facilitates tumour growth and enhances resistance to apoptosis [30,43,66]. Interestingly, Sesn2 knockout in mice suppresses early-stage lung tumour growth, which may be linked to diminished Akt kinase activity [30]. However, in the A549 cell line obtained from advanced lung adenocarcinoma, SESN2 did not contribute to the activation of AKT and sensitised cancer cells to cell death and pro-apoptotic cytokines [30,43,66]. A recent study in A549 cells suggests that SESN1 and SESN2 regulate aberrant STAT3 activation in lung cancer, limiting malignant proliferation and enhancing sensitivity to apoptosis [71].
2.3. Sestrins in Liver Cancer
Unpublished findings from Budanov and Karin suggested that Sesn2 knockout in mice leads to reduced liver tumour growth in diethylnitrosamine (DEN)-induced liver carcinogenesis models. In contrast, a later study indicated that increased expression of Sesn3, another member of the Sestrin family, correlates with improved survival rates in animal models of liver cancer [72]. Experiments involving mice injected with the carcinogen DEN and subsequently fed a choline-deficient high-fat diet showed that Sesn3 knockout animals developed a greater number of tumours than control subjects, and these tumours were also larger and more metastatic, underscoring the tumour-suppressive role of the Sesn3 gene [72].
2.4. Sestrins in Prostate Cancer
While SESN3 exhibits antiproliferative properties, it has been identified as upregulated in hormone-refractory prostate cancers [73]. This increase in SESN3 expression occurs in prostate carcinoma cells transitioning into hormone-refractory neuroendocrine prostate cancer (NEPC). Such changes may be associated with the reduced expression of micro-RNA-708, which directly interacts with SESN3 mRNA, thereby diminishing its expression [74]. NEPC is a highly aggressive form marked by loss of androgen signalling, gain of neuroendocrine phenotype, and resistance to hormone therapies [75]. TP53 loss is a common feature of NEPC, found in about 67% of cases [76], and likely leads to a reduction in SESN1 and SESN2 expression. In this context, the p53-independent SESN3 may be upregulated to manage oxidative stress. Since this occurs during the transition to NEPC, mTORC1 inhibition by Sestrins may no longer be a concern, making SESN3’s antioxidant role selectively advantageous.
2.5. Sestrins in Skin Cancer
In contrast to Sestrins’ recognised tumour-suppressive function across multiple tissues, SESN2 frequently shows elevated expression in various skin cancers, including melanomas and squamous cell carcinomas, acting as an oncogene to promote skin carcinogenesis [31]. This effect may be attributed to SESN2’s protective role against ultraviolet and other stressors that threaten cancer cell viability, primarily through the activation of AKT [31,60]. In sebaceous carcinoma, SESN2 was found to be significantly downregulated as a result of p53 mutations, and low SESN2 expression correlated with poor tumour differentiation, which is an indicator of tumour aggressiveness and poor prognosis [77].
2.6. Sestrins in Colon Cancer
Expression analysis of SESN2 in colorectal cancers revealed its downregulation, with its reduced levels linked to advanced tumour stages marked by vascular invasion, lymph node metastasis, and liver metastasis. Furthermore, lower SESN2 expression correlates with a poor prognosis and a reduced patient life expectancy [57]. Since this downregulation was observed in advanced stages of cancer, it is likely attributable to p53 inactivation, which is frequently observed in later stages of tumour progression. Another study showed that Sesn2 knockout in a mouse model where colorectal cancer was induced through dietary administration of dextran sodium sulphate (DSS) led to accelerated tumour growth. This growth was associated with increased mTORC1 activity and inflammation in the colon [78].
2.7. Sestrins in Endometrial Cancer
A study of SESN2 in primary endometrial cancer tissue revealed that SESN2 was upregulated in cancer tissues. Interestingly, mTORC1 activity remained elevated in tumour samples despite increased SESN2 expression. Subsequent experiments on endometrial cancer cell lines (HEC-1-A and Ishikawa) produced contradictory results: silencing SESN2 in these cell lines enhanced mTORC1 activity, increased proliferation, and raised intracellular ROS levels. Furthermore, SESN2 silencing promoted the growth of larger and heavier xenografts from these cell lines. Although SESN2 functions as expected in endometrial cell lines, SESN–mTORC1 signalling may be disrupted in the tumour microenvironment, as SESN2 levels do not correlate with reduced mTORC1 activity in endometrial tumours [79]. Another study has shown that SESN3 is highly methylated in endometrial cancers, suggesting that its epigenetic repression may facilitate oncogenic processes in this context [80].
2.8. Sestrins in Brain Cancer
In the context of neuroblastoma, a study showed that lysine-specific demethylase 1 (LSD1) influences the histone modifications of the SESN2 promoter, thereby repressing its expression. Inhibition of LSD1 was shown to activate autophagy and repress mTORC1, and SESN2 was required for this effect. Furthermore, the study found an inverse correlation between LSD1 and SESN2 expression in differentiated neuroblastomas, and reported that high LSD1 and low SESN2 levels were each significantly associated with poorer survival [81]. In glioblastoma, silencing SESN2 in U87 tumour cells sensitised them to ionising radiation (IR), reducing proliferation, and significantly increasing ROS levels. SESN2 silencing also elevated platelet-derived growth factor receptor beta (PDGFRβ) expression. Mechanistically, SESN2 was shown to promote proteasomal degradation of PDGFRβ, as its ubiquitination and degradation were impaired upon SESN2 knockdown. Co-silencing SESN2 and PDGFRβ partially normalised ROS levels and proliferation, although not to baseline [82]. These findings are supported by studies in mouse lung fibroblasts, where SESN2 knockout increased PDGFRβ expression and this increase was reversed by SESN2 reconstitution. Notably, reconstitution with the catalytically inactive SESN2 mutant (C125G) that is incapable of reducing ROS did not affect PDGFRβ expression, indicating SESN’s ROS regulation as the potential mechanism [83]. Further work demonstrated that SESN2 knockout impaired PDGFRβ degradation via reduced proteasomal activity, distinct from the mechanism observed in glioblastoma. Moreover, SESN2 may modulate PDGFRβ expression indirectly through Nrf2 activation and antioxidant gene induction [36]. Overall, the interaction between SESN2 and PDGFRβ in glioblastoma remains incompletely understood, and it is unclear whether PDGFRβ modulation by SESN2 significantly influences glioblastoma progression or reflects a broader regulatory mechanism.
2.9. Sestrins and Cancer Stem Cells
Tumours are recognised for their heterogeneous cell composition, where a single cell can be markedly genetically different from the rest of the tumour. Various cell types collaborate to support the tumour with adaptive mechanisms for survival and propagation within the host. Recent research highlights that cancer growth and spread are often propelled by cancer stem cells. They are noted for their unlimited proliferative capacity, stress resistance, and ability to differentiate into various cancer cell subtypes. These cells also exhibit heightened resistance to cancer treatments [84]. Putative cancer stem cells, or so-called tumour-initiating cells with stem-like properties, have been identified in a range of cancers, including leukaemia [85], lung carcinoma [86,87], hepatocarcinoma [88,89], colon carcinoma [90], melanoma [91], squamous carcinoma [92], prostate carcinoma, and other types of cancer [84]. While the involvement of cancer stem cells in follicular lymphoma remains debated, tumour-initiating cells possessing stem-like qualities have been reported for these cancer types [93].
The role of Sestrins in the regulation of cancer stem cells remains unclear. However, a study examining the activity of Sesn3 in liver cancer investigated its impact on cancer stemness. Tumours lacking Sesn3 exhibited increased activation of Akt and Stat3, along with elevated levels of stem cell markers such as Acta2, Cd44, and Cd133 [72]. Moreover, the same study showed that SESN3 might regulate stemness by sequestering the transcription factor Gli2 in the cytoplasm through direct protein–protein interactions. However, as the SESN3–Gli2 interaction was shown via an overexpression study in 293T cells, this hypothesis requires validation using endogenous protein from liver cells [72]. Nevertheless, the support of stemness through the regulation of Gli2, AKT, and STAT3 may represent a crucial pathway through which SESN3 may counteract liver carcinogenesis. Overexpression of SESN2 in colorectal cell lines (HCT-116, SW620) lowered both mRNA and protein levels of stemness-related Sox2, Oct4, Cxcr4, and CD44. The same study showed that mouse xenografts of SESN2 overexpression had lower protein levels of Sox2 [94].
A summary of Sestrin expression changes, genetic manipulations, and biological effects across the tumour types discussed above is provided in Table 1.

Table 1.
Expression and the biological role of Sestrins across various tumour types. The table summarises the expression patterns of Sestrin proteins in different cancers, the expression differences vs. normal tissues or modes of genetic manipulation, the impact on tumour progression (e.g., suppression or support), and the associated molecular mechanisms. Abbreviations: FL, follicular lymphoma; ↓, downregulation; ↑, upregulation; NSCLC, non-small cell lung cancer; TCGA, The Cancer Genome Atlas; HSPC, hormone-sensitive prostate cancer; HRPC, hormone-refractory prostate cancers; CRC, colorectal cancer; EMT, Epithelial-Mesenchymal Transition; N/A, not available.
2.10. Sestrins’ Role in Early Stages of Carcinogenesis
Despite their importance in suppressing cancer, Sestrins seem to have a limited impact on tumour initiation. A study on lung cancer in a mouse model showed that knockout of either Sesn1, Sesn2, or both does not impact tumour formation [30]. In a study of the impact of Sestrins in colon cancer, mice knockouts of Sesn2 did not develop more tumours [78]. However, the same study revealed a crucial finding: Sesn2 expression in the colon epithelium is essential, as its absence compromises the colon lining, leading to increased chronic inflammation and subsequent tumour formation [78]. Additionally, while Sestrins may not directly influence tumour initiation they support the integrity of DNA. Sestrins were shown to be the frontline in p53-mediated protection of DNA from oxidative damage [43], and another study showed that inhibition of Sestrins directly contributed to chromosome breaks due to the build-up of ROS in Ras-transformed fibroblasts [95]. Therefore, current evidence suggests that Sestrins do not have a direct molecular role in tumour initiation. However, they appear to play a protective role against cancer-promoting events. Thus, while Sestrins are important for maintaining tissue integrity, their role seems more indirect, as they mitigate cancer-promoting events rather than directly influencing tumour initiation.
2.11. Sestrins Inhibit Tumour Growth
2.11.1. Sestrin—Tumour Suppressor
Although the absence of Sestrins typically does not affect tumour initiation, evidence shows that Sestrin expression has a pronounced influence on tumour volume, weight, and cell proliferation. The earliest studies on Sestrins showed that inhibition of SESN1 accelerated the proliferation of fibroblast cells [16]. Subsequent studies explored the role of Sestrins in specific cancer models. Detailed studies of inflammation-associated colon cancer showed that mice knockout of Sesn2 did not influence tumour initiation but dramatically increased the size of the tumours [78]. Several xenograft experiments on colorectal cancer cells showed that SESN2 overexpression dramatically decreased the size and weight of tumours [94,96]. Knockout of SESN1&2 increased the proliferation of A549 lung cancer cells, with the rate of proliferation increasing progressively with each Sestrin knocked out [30]. A subsequent study revealed that this proliferation is facilitated by altered STAT3 signalling in the absence of Sestrin [71]. Xenografts of endometrial cancer also have shown to grow larger and heavier with inhibition of SESN2 [79].
A likely molecular mechanism underlying Sestrins’ repression of aberrant growth is their inhibition of mTORC1, as this complex is frequently overactivated in cancer and essential for cell growth and biomass accumulation [97,98]. Loss of SESN2 increased mTORC1 activation and correlated with bigger tumours in the context of CRC [78]. Loss of p53, a common event in cancer progression, promotes mTORC1 activation [99]. Inhibition of overactivated mTORC1 can display tumour-suppressing effects in vivo and is therefore a common target for clinical trials [100]. In addition to inhibiting mTORC1, a recent study shows that Sestrins may also be inhibitors of STAT3 phosphorylation, thereby constraining aberrant cell proliferation [71]. Multiple studies demonstrate that Sestrins influence the cell cycle. Overexpression of SESN1/2 increased the distribution of cells in the G1 phase (growth phase) in the 293T cell line [16]. This effect could be related to expression of the cell cycle protein Cyclin D1, which Sestrins appear to suppress in particular contexts [16,71]. Sesn overexpression in the gut stem cells of the D. melanogaster also arrested cells in the G1 phase, which the authors suggested was due to mTORC1 inhibition [101]. Therefore, a substantial body of evidence supports the role of Sestrins as suppressors of tumour growth and proliferation.
Given the role of Sestrins in suppressing tumour growth and proliferation, cancers may develop mechanisms to bypass Sestrin-mediated inhibition of mTORC1 (Figure 3). One likely strategy is to decrease the activity of Sestrin-activating transcription factors, such as p53, thereby reducing the availability of Sestrins that inhibit cell growth. Another potential mechanism involves the dysfunction of GATOR1, the protein complex through which Sestrins regulate mTORC1. GATOR1 was initially identified as a tumour suppressor [22]. Moreover, the genes encoding subunits of GATOR1 have been found to be mutated and downregulated in certain cancers [102,103,104]. Although the frequency and specific effects of GATOR1 dysfunction in cancer remain to be fully explored, it is possible that an inactive GATOR1 could impair Sestrins’ ability to suppress tumour growth. Conversely, inactivation of GATOR2 in cancer could mimic Sestrin activation by lifting the inhibitory effect on GATOR1. A study demonstrated that knockout of the WDR59 subunit of GATOR2 significantly reduced the size of mammary tumours, while overexpression of SESN3 produced a similar effect [105]. Therefore, dysfunction of the SESN–GATOR–Rags signalling axis may serve as a mechanism to promote tumour growth. Post-translational modifications can influence the inhibition of mTORC1 by Sestrins. A recent study shows that K63-chain ubiquitination of SESN2 regulates its interaction with GATOR2. In colorectal cancer, this ubiquitination may be disrupted by dysregulation of the E3 ligase RNF167 and the deubiquitinase STAMBPL1, thereby impairing SESN2-mediated inhibition of mTORC1 [106]. Further research into Sestrin post-translational modifications could provide additional insights into how tumours evade Sestrin-mediated mTORC1 inhibition.
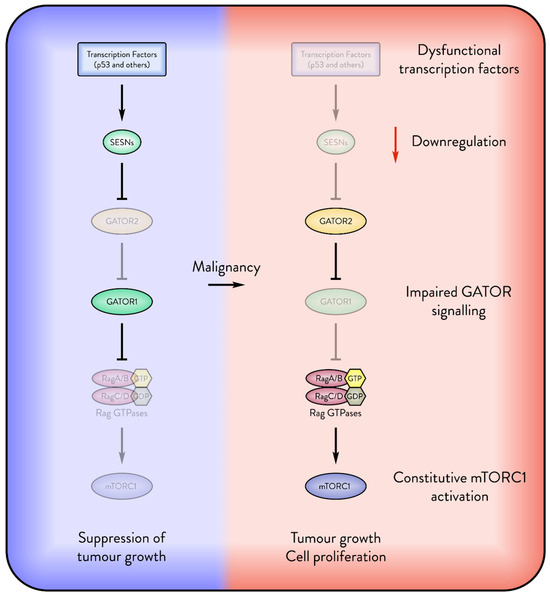
Figure 3.
Cancers attempt to suppress the Sestrin inhibition of mTORC1. The figure displays several possible mechanisms by which malignant cells override the inhibition of mTORC1 by Sestrins (SESNs). The absence of transcription factors such as p53 downregulates the expression of Sestrins. Dysfunctional GATOR signalling may override Sestrin inhibition of mTORC1, enabling the cancer to freely express Sestrin for its pro-survival functions.
2.11.2. Sestrin—Tumour Protector
Although most evidence shows that Sestrins suppress tumours, some specific cases have shown that Sestrins can instead support tumour growth. Experiments on melanoma xenografts in mice showed that cancer cells with silenced SESN2 produced smaller tumours, which the authors attributed to increased survival of inoculant cells due to SESN2’s protection from ROS and possibly the activation of pro-survival AKT [31]. Further studies of melanoma showed that Sestrins support the survival of metastatic cancer cells. The authors showed that intracellular ROS was significantly increased in suspended cells with silenced SESN2, and ROS correlated with the number of metastases induced via intravenous injection of cancer cells [59].
The antioxidative function of Sestrins may enable tumour survival by protecting aberrant cells, particularly when mTORC1 signalling is compromised. For example, a study of primary endometrial tumours showed that SESN2 expression was correlated with lower patient survival, and mTORC1 activity levels were higher in these tumours in the presence of Sestrins, suggesting dysfunctional Sestrin–mTORC1 signalling [79]. As previously discussed, Sestrins can support Nrf2 activation and a fraction of the Sestrin proteins was found to translocate into the nucleus and possibly associate with the transcription factor Nrf2, where it had a supporting effect on its activity [36]. Overactivation of Nrf2 in cancer leads to increased expression of metabolic enzymes, which could support metabolism in favour of cell proliferation [107].
Another example of Sestrin supporting tumour growth is in lung cancer under glutamine deprivation. SESN2 was significantly upregulated in multiple glutamine-deprived lung cancer cell lines. Furthermore, SESN2 knockouts of these lines were significantly more sensitive to glutamine deprivation-induced cell death and formed markedly smaller tumours under inhibition of the glutamine transporter [33]. This study, along with another, attributed this effect to Sestrins’ support of AKT activity [33,34], potentially via a possible association with mTORC2 via the GATOR2 complex [34], suggesting that while Sestrin–mTORC1 signalling is well defined, their cooperation with mTORC2 could be an important area for further cancer research.
Therefore, common patterns in cases where Sestrins support cancer include impaired mTORC1 signalling, activation of mTORC2 and AKT, and protection against metabolic and oxidative stress (Figure 4). Future studies on these mechanisms could clarify why Sestrins sometimes promote tumour survival and growth instead of suppressing tumour progression.
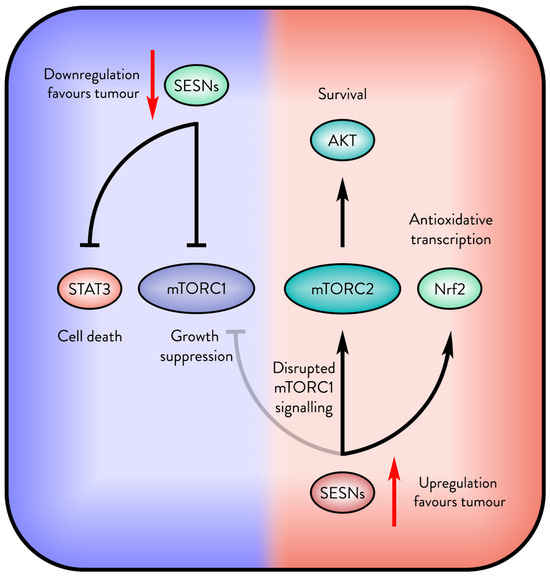
Figure 4.
The opposite outcomes of Sestrin in cancer. Sestrins (SESNs) support tumour suppression, but when mTORC1 signalling is disrupted, Sestrins may be upregulated by the tumour for their antioxidative and survival signalling.
3. Sestrins as Therapeutic Targets in Cancer
Sestrin activity is tightly regulated by leucine sensing, and pharmacological modulation of the Sestrin–GATOR2 interaction has emerged as a potential therapeutic strategy. Small molecules such as leucine analogues have been developed to mimic or antagonise leucine’s effect on Sestrins.
Navitor Pharmaceuticals’ lead compound, NV-5138, targets the leucine-binding pocket of SESN2 and binds with comparable affinity to leucine. Structural studies revealed that SESN2 bound to NV-5138 adopts a conformation essentially identical to that of the leucine-bound protein. Unlike leucine, NV-5138 is neither incorporated into protein synthesis nor metabolised by the branched-chain amino transaminase, which is the possible reason for its high bioavailability in the brain. Thus, NV-5138 is being investigated for activation of mTORC1 in neurological disorders associated with reduced mTORC1 activity, including depression [108]. NV-5138 demonstrated antidepressant effects in rodent models in an mTORC1-dependent manner [109]. However, the utility of leucine analogues in cancer remains uncertain. Given that mTORC1 is frequently hyperactivated in malignancies, and that Sestrin-mediated mTORC1 inhibition is its core mode of tumour suppression, antagonising Sestrins may inadvertently promote tumorigenesis. Furthermore, leucine mimetics are unlikely to impact Sestrins’ antioxidative function, which may be exploited by tumours to mitigate oxidative stress.
Navitor’s patent (AU2021215177B2) describes several compounds classified as “leucine antagonists” for the SESN–GATOR2 interaction, defined as molecules that increase SESN2–GATOR2 binding by more than 40% [110]. Given that impaired SESN–GATOR2 interaction is associated with enhanced mTORC1 activity and worsened cancer progression, molecules that strengthen this interaction may have therapeutic potential in oncology. However, NV-5138 remains the company’s lead candidate and is currently in Phase 2 clinical trials. It is unclear whether the development of leucine antagonists for oncological applications is planned; nonetheless, evaluating their potential anti-cancer properties represents an intriguing research avenue.
Similarly, a cosmetic industry patent (US20170275694A1) seeks to identify compounds that upregulate Sestrin expression to combat skin ageing and pigmentation [111], although no compounds have been reported in connection to this patent.
Several natural and pharmacological agents have been identified as activators of Sestrin expression. Resveratrol, found in red grapes and wine, activates SESN2 at concentrations of 10–30 µM [112]. However, its poor oral bioavailability (~0.3% as free unconjugated product) limits plasma concentrations to approximately 300 nM [113]. Quercetin, abundant in fruits and vegetables, induces SESN2 expression in HCT116 cells at 50–100 µM [114,115] but similarly suffers from low bioavailability (typically ~0.5–2 µM plasma concentration) [116].
Pharmacological agents such as nelfinavir, an ER stress inducer, and bortezomib, a proteasome inhibitor, have been shown to activate SESN2 via the ATF4 transcription factor [117]. However, therapeutic concentrations of nelfinavir (~15 µg/mL) exceed typical plasma levels achieved in patients (3–4 µg/mL) [118,119]. Bortezomib requires intravenous or subcutaneous administration, limiting its application solely as a SESN2 activator.
Metformin, a widely used anti-diabetic agent, activates SESN2 through ATF4 induction and suppression of the SESN2 negative regulator miR-21-5p [120,121]. With high oral bioavailability and tolerable side effects, metformin remains an attractive candidate, although its efficacy as an anti-cancer adjuvant remains inconclusive [122], and its utility for SESN2 activation in oncological contexts requires further investigation.
Conversely, in contexts where Sestrin activity supports tumour survival, strategies to downregulate Sestrins may be beneficial. For example, cabazitaxel, a microtubule-binding agent used in cancer therapy, was shown to moderately reduce SESN3 expression, thereby increasing intracellular ROS levels [123].
In addition to direct pharmacological modulation, tumour leucine metabolism may indirectly influence Sestrin activity. Cancers can display elevated levels of leucine [124], and leucine transporters such as LAT1 (SLC7A5) are frequently found overexpressed across various cancers [125]. Elevated leucine levels inhibit Sestrin-mediated mTORC1 suppression, potentially contributing to tumour progression. While Sestrin upregulation under stress conditions may counterbalance leucine-mediated inhibition, the functional impact of high leucine availability in cancers with low Sestrin expression remains unclear. Further studies targeting leucine transporters or modulating tumour amino acid metabolism could reveal additional strategies to restore Sestrin tumour-suppressive functions [126].
Overall, modulation of Sestrin activity through small molecules or metabolic interventions presents multiple avenues for enhancing cancer therapies (Figure 5). While direct Sestrin activation may reinforce tumour-suppressive mechanisms via mTORC1 inhibition, strategies aiming to inhibit Sestrins must carefully consider the context, due to the risk of inadvertently supporting tumour progression. Further research is required to define the specific contexts in which pharmacological or metabolic modulation of Sestrins may yield therapeutic benefit.
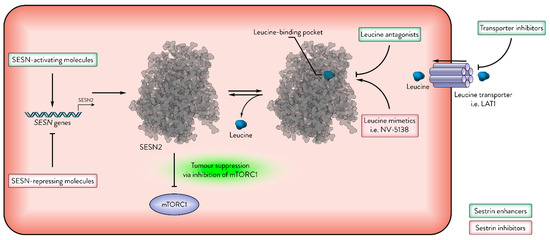
Figure 5.
Sestrins as therapeutic targets in cancer. The activity of Sestrins (e.g., SESN2) can be modulated by leucine and other molecules. This figure illustrates several mechanisms of enhancing or inhibiting Sestrin activity. Green indicates Sestrin-enhancing molecules; red indicates Sestrin-inhibiting molecules.
4. Future Directions
Despite substantial progress, the role of Sestrins in cancer remains complex and, at times, contradictory. To resolve these discrepancies and effectively leverage Sestrins in oncological contexts, several key areas require further investigation.
First, it is critical to elucidate the molecular pathways underlying Sestrin function in cancer. This includes confirming the regulation of STAT3 by Sestrins across various tumour contexts, defining the transcriptional interplay between Sestrins and Nrf2 in oncogenic settings, and clarifying the mechanisms by which Sestrins interact with mTORC2 and activate AKT signalling in cancer. Given the established regulation of Sestrins by leucine, future studies should determine whether additional Sestrin-mediated signalling pathways are similarly modulated by leucine availability.
Second, the relationship between Sestrins and metabolic stress requires deeper exploration. Pathways such as glutamine metabolism, amino acid metabolism, and ROS management are highly relevant, especially considering parallels between Sestrin function in metabolic diseases such as diabetes and in cancer. Given the complexity of cancer metabolism, this area represents a substantial and underexplored field for Sestrin research. The development of Sestrin–GATOR modulators, currently under investigation for neurological disorders, presents an exciting therapeutic avenue. Rational design of leucine analogues and leucine antagonists, based on structural insights into the Sestrin leucine-binding pocket, should be extended to cancer studies. In parallel, targeting deregulated leucine metabolism in tumours could enhance the therapeutic efficacy of Sestrin modulation.
Third, a comprehensive understanding of Sestrin post-translational modifications may reveal novel opportunities for therapeutic intervention, particularly in tumour contexts where such modifications may impair Sestrin function.
Finally, beyond their direct involvement in carcinogenesis, Sestrins contribute to cancer prevention by maintaining tissue integrity, as evidenced by their role in protecting the colon epithelium from inflammatory damage. Given the strong association between ageing and cancer incidence, the anti-ageing functions of Sestrins highlight their potential role in cancer prevention [41].
5. Conclusions
Sestrins are ubiquitously expressed proteins that are markedly upregulated in response to stress, including DNA damage and metabolic stress, both of which are frequently observed in transformed malignant cells. In response to a diverse array of transcription factors, Sestrins are on the frontline in the fight against cancer, but their implication in tumour initiation is limited. Evidence suggests that although Sestrins do not play a direct role in tumour initiation, they counteract several cancer-promoting events and are often impaired by oncogenic disruptions, such as the loss of p53 function. Studies consistently show that a reduction in Sestrin levels is highly correlated with tumour growth, impaired apoptosis, and poor prognosis across multiple tumour types. However, in certain contexts or metabolic conditions, increased Sestrin expression may instead support tumour progression.
Although the involvement of Sestrins in mTORC1 signalling is well established, their relationship with mTORC2 and survival factors like STAT3 remains poorly understood and warrants further study. Exploring these pathways further may provide insight into why Sestrins, despite their usual tumour-suppressive role, can sometimes facilitate cancer progression. Moreover, clarifying the molecular mechanisms of Sestrins could enhance their potential as prognostic and therapeutic markers, informing more precise treatment strategies.
Potentially targeting metabolic pathways, such as glutamine metabolism, is beneficial for tumours that exploit high Sestrin levels to protect themselves from metabolic stress, while supporting Sestrin expression in low-Sestrin tumours could enhance the effectiveness of conventional DNA-targeting therapeutic drugs, such as cisplatin or etoposide. In tumours with lower Sestrin expression, including those with p53 deficiency, Sestrins could be activated via alternative transcription factors.
The role of Sestrins’ leucine binding properties is currently poorly studied in cancers. Elevated leucine levels, common in many cancers, may inhibit Sestrin-mediated mTORC1 suppression, thereby promoting tumour growth. Therefore, future therapies should carefully integrate Sestrin expression levels, leucine metabolism, and broader cancer metabolic profiles to effectively exploit Sestrins as therapeutic targets.
Author Contributions
Conceptualization, A.H.; writing—original draft preparation, A.H.; writing—review and editing, A.H. and A.V.B.; visualization, A.H.; funding acquisition, A.V.B. All authors have read and agreed to the published version of the manuscript.
Funding
The studies were supported by Wellcome Trust Trinity College Dublin—Institutional Strategic Support Fund (204814/Z/16/Z), and Trinity St.James’s Cancer Institute Cancer Research Stimulus Award (TSJCI Crest Award) to A.V.B.
Acknowledgments
We thank Nadushenka Pryadilova for her help with the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Budanov, A.V.; Shoshani, T.; Faerman, A.; Zelin, E.; Kamer, I.; Kalinski, H.; Gorodin, S.; Fishman, A.; Chajut, A.; Einat, P.; et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002, 21, 6017–6031. [Google Scholar] [CrossRef] [PubMed]
- Haidurov, A.; Budanov, A.V. Locked in Structure: Sestrin and GATOR—A Billion-Year Marriage. Cells 2024, 13, 1587. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef]
- Kim, H.; An, S.; Ro, S.-H.; Teixeira, F.; Jin Park, G.; Kim, C.; Cho, C.-S.; Kim, J.-S.; Jakob, U.; Hee Lee, J.; et al. Janus-faced Sestrin2 controls ROS and mTOR signalling through two separate functional domains. Nat. Commun. 2015, 6, 10025. [Google Scholar] [CrossRef]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.-L.; Karin, M.; Budanov, A.V. Sestrins Inhibit mTORC1 Kinase Activation Through the GATOR Complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb Binds and Regulates the mTOR Kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex Toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.T.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin Activation of Rheb, a Mediator of mTOR/S6K/4E-BP Signaling, Is Inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the Tuberous Sclerosis Complex-2 Tumor Suppressor Gene Product Tuberin as a Target of the Phosphoinositide 3-Kinase/Akt Pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of mTORC1 at the Lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Morrison, A.; Chen, L.; Wang, J.; Zhang, M.; Yang, H.; Ma, Y.; Budanov, A.; Lee, J.H.; Karin, M.; Li, J. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 2015, 29, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Rogala, K.B.; Gu, X.; Kedir, J.F.; Abu-Remaileh, M.; Bianchi, L.F.; Bottino, A.M.S.; Dueholm, R.; Niehaus, A.; Overwijn, D.; Fils, A.-C.P.; et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 2019, 366, 468–475. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor Suppressor Complex with GAP Activity for the Rag GTPases That Signal Amino Acid Sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef]
- Shen, K.; Huang, R.K.; Brignole, E.J.; Condon, K.J.; Valenstein, M.L.; Chantranupong, L.; Bomaliyamu, A.; Choe, A.; Hong, C.; Yu, Z.; et al. Architecture of the human GATOR1 and GATOR1–Rag GTPases complexes. Nature 2018, 556, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.X.; Guan, K.L. The SIN1-PH Domain Connects mTORC2 to PI3K. Cancer Discov. 2015, 5, 1127–1129. [Google Scholar] [CrossRef]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef]
- Larsson, C. Protein kinase C and the regulation of the actin cytoskeleton. Cell. Signal. 2006, 18, 276–284. [Google Scholar] [CrossRef]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef]
- Morrison Joly, M.; Williams, M.M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Young, C.D.; Sarbassov, D.D.; Muller, W.J.; Brantley-Sieders, D.; Cook, R.S. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Res. 2017, 19, 74. [Google Scholar] [CrossRef]
- Ding, B.; Haidurov, A.; Chawla, A.; Parmigiani, A.; van de Kamp, G.; Dalina, A.; Yuan, F.; Lee, J.H.; Chumakov, P.M.; Grossman, S.R.; et al. p53-inducible SESTRINs might play opposite roles in the regulation of early and late stages of lung carcinogenesis. Oncotarget 2019, 10, 6997–7009. [Google Scholar] [CrossRef]
- Zhao, B.; Shah, P.; Budanov, A.V.; Qiang, L.; Ming, M.; Aplin, A.; Sims, D.M.; He, Y.Y. Sestrin2 protein positively regulates AKT enzyme signaling and survival in human squamous cell carcinoma and melanoma cells. J. Biol. Chem. 2014, 289, 35806–35814. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Talukdar, S.; Park, E.J.; Park, H.L.; Park, H.-W.; Bandyopadhyay, G.; Li, N.; Aghajan, M.; Jang, I.; et al. Maintenance of Metabolic Homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012, 16, 311–321. [Google Scholar] [CrossRef]
- Byun, J.-K.; Choi, Y.-K.; Kim, J.-H.; Jeong, J.Y.; Jeon, H.-J.; Kim, M.-K.; Hwang, I.; Lee, S.-Y.; Lee, Y.M.; Lee, I.-K.; et al. A Positive Feedback Loop Between Sestrin2 and mTORC2 Is Required for the Survival of Glutamine-Depleted Lung Cancer Cells. Cell Rep. 2017, 20, 586–599. [Google Scholar] [CrossRef]
- Kowalsky, A.H.; Namkoong, S.; Mettetal, E.; Park, H.W.; Kazyken, D.; Fingar, D.C.; Lee, J.H. The GATOR2-mTORC2 axis mediates Sestrin2-induced AKT Ser/Thr kinase activation. J. Biol. Chem. 2020, 295, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins Activate Nrf2 by Promoting p62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Tomasovic, A.; Kurrle, N.; Sürün, D.; Heidler, J.; Husnjak, K.; Poser, I.; Schnütgen, F.; Scheibe, S.; Seimetz, M.; Jaksch, P.; et al. Sestrin 2 Protein Regulates Platelet-derived Growth Factor Receptor β (Pdgfrβ) Expression by Modulating Proteasomal and Nrf2 Transcription Factor Functions. J. Biol. Chem. 2015, 290, 9738–9752. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef]
- Kovaleva, I.E.; Tokarchuk, A.V.; Zheltukhin, A.O.; Dalina, A.A.; Safronov, G.G.; Evstafieva, A.G.; Lyamzaev, K.G.; Chumakov, P.M.; Budanov, A.V. Mitochondrial localization of SESN2. PLoS ONE 2020, 15, e0226862. [Google Scholar] [CrossRef]
- Kumar, A.; Shaha, C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci. Rep. 2018, 8, 615. [Google Scholar] [CrossRef]
- Haidurov, A.; Budanov, A.V. Sestrin family—The stem controlling healthy ageing. Mech. Ageing Dev. 2020, 192, 111379. [Google Scholar] [CrossRef]
- Velasco-Miguel, S.; Buckbinder, L.; Jean, P.; Gelbert, L.; Talbott, R.; Laidlaw, J.; Seizinger, B.; Kley, N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 1999, 18, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Peuget, S.; Zhou, X.; Selivanova, G. Translating p53-based therapies for cancer into the clinic. Nat. Rev. Cancer 2024, 24, 192–215. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Loeb, L.A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. 2001, 477, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Essler, S.; Dehne, N.; Brüne, B. Role of sestrin2 in peroxide signaling in macrophages. FEBS Lett. 2009, 583, 3531–3535. [Google Scholar] [CrossRef]
- Olson, N.; Hristova, M.; Heintz, N.H.; Lounsbury, K.M.; van der Vliet, A. Activation of hypoxia-inducible factor-1 protects airway epithelium against oxidant-induced barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L993–L1002. [Google Scholar] [CrossRef]
- Jegal, K.H.; Park, S.M.; Cho, S.S.; Byun, S.H.; Ku, S.K.; Kim, S.C.; Ki, S.H.; Cho, I.J. Activating transcription factor 6-dependent sestrin 2 induction ameliorates ER stress-mediated liver injury. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2017, 1864, 1295–1307. [Google Scholar] [CrossRef]
- Saveljeva, S.; Cleary, P.; Mnich, K.; Ayo, A.; Pakos-Zebrucka, K.; Patterson, J.B.; Logue, S.E.; Samali, A. Endoplasmic reticulum stress-mediated induction of SESTRIN 2 potentiates cell survival. Oncotarget 2016, 7, 12254–12266. [Google Scholar] [CrossRef]
- Garaeva, A.A.; Kovaleva, I.E.; Chumakov, P.M.; Evstafieva, A.G. Mitochondrial dysfunction induces SESN2 gene expression through Activating Transcription Factor 4. Cell Cycle 2016, 15, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.Y.; Jin, S.H.; Cho, I.J.; Ki, S.H. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free. Radic. Biol. Med. 2012, 53, 834–841. [Google Scholar] [CrossRef]
- Chen, C.-C.; Jeon, S.-M.; Bhaskar, P.T.; Nogueira, V.; Sundararajan, D.; Tonic, I.; Park, Y.; Hay, N. FoxOs Inhibit mTORC1 and Activate Akt by Inducing the Expression of Sestrin3 and Rictor. Dev. Cell 2010, 18, 592–604. [Google Scholar] [CrossRef]
- Hagenbuchner, J.; Kuznetsov, A.; Hermann, M.; Hausott, B.; Obexer, P.; Ausserlechner, M.J. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J. Cell Sci. 2012, 125, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Yoon, B.-H.; Kim, S.-K.; Kim, S.-Y. GENT2: An updated gene expression database for normal and tumor tissues. BMC Med. Genom. 2019, 12, 101. [Google Scholar] [CrossRef]
- Wei, J.L.; Fu, Z.X.; Fang, M.; Guo, J.B.; Zhao, Q.N.; Lu, W.D.; Zhou, Q.Y. Decreased expression of sestrin 2 predicts unfavorable outcome in colorectal cancer. Oncol. Rep. 2015, 33, 1349–1357. [Google Scholar] [CrossRef]
- Chen, K.B.; Xuan, Y.; Shi, W.J.; Chi, F.; Xing, R.; Zeng, Y.C. Sestrin2 expression is a favorable prognostic factor in patients with non-small cell lung cancer. Am. J. Transl. Res. 2016, 8, 1903–1909. [Google Scholar]
- Zhu, G.; Xu, P.; Guo, S.; Yi, X.; Wang, H.; Yang, Y.; Liu, L.; Shi, Q.; Gao, T.; Li, C. Metastatic Melanoma Cells Rely on Sestrin2 to Acquire Anoikis Resistance via Detoxifying Intracellular ROS. J. Investig. Dermatol. 2020, 140, 666–675.E2. [Google Scholar] [CrossRef]
- Zhao, B.; Shah, P.; Qiang, L.; He, T.C.; Budanov, A.; He, Y.Y. Distinct Role of Sesn2 in Response to UVB-Induced DNA Damage and UVA-Induced Oxidative Stress in Melanocytes. Photochem. Photobiol. 2017, 93, 375–381. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal 2011, 15, 1679–1690. [Google Scholar] [CrossRef]
- Parmigiani, A.; Budanov, A.V. Sensing the Environment Through Sestrins: Implications for Cellular Metabolism. Int. Rev. Cell Mol. Biol. 2016, 327, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Ding, B.; Parmigiani, A.; Yang, C.; Budanov, A.V. Sestrin2 facilitates death receptor-induced apoptosis in lung adenocarcinoma cells through regulation of XIAP degradation. Cell Cycle 2015, 14, 3231–3241. [Google Scholar] [CrossRef]
- Ding, B.; Parmigiani, A.; Divakaruni, A.S.; Archer, K.; Murphy, A.N.; Budanov, A.V. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci. Rep. 2016, 6, 22538. [Google Scholar] [CrossRef]
- Oricchio, E.; Katanayeva, N.; Donaldson, M.C.; Sungalee, S.; Pasion, J.P.; Béguelin, W.; Battistello, E.; Sanghvi, V.R.; Jiang, M.; Jiang, Y.; et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma. Sci. Transl. Med. 2017, 9, eaak9969. [Google Scholar] [CrossRef]
- Nutt, S.L.; Keenan, C.; Chopin, M.; Allan, R.S. EZH2 function in immune cell development. Biol. Chem. 2020, 401, 933–943. [Google Scholar] [CrossRef]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef]
- Haidurov, A.; Zheltukhin, A.O.; Snezhkina, A.V.; Krasnov, G.S.; Kudryavtseva, A.V.; Budanov, A.V. p53-regulated SESN1 and SESN2 regulate cell proliferation and cell death through control of STAT3. Cell Commun. Signal. 2025, 23, 105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kim, H.G.; Dong, E.; Dong, C.; Huang, M.; Liu, Y.; Liangpunsakul, S.; Dong, X.C. Sesn3 deficiency promotes carcinogen-induced hepatocellular carcinoma via regulation of the hedgehog pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2685–2693. [Google Scholar] [CrossRef]
- Tamura, K.; Furihata, M.; Tsunoda, T.; Ashida, S.; Takata, R.; Obara, W.; Yoshioka, H.; Daigo, Y.; Nasu, Y.; Kumon, H.; et al. Molecular features of hormone-refractory prostate cancer cells by genome-wide gene expression profiles. Cancer Res. 2007, 67, 5117–5125. [Google Scholar] [CrossRef]
- Shan, J.; Al-Muftah, M.A.; Al-Kowari, M.K.; Abuaqel, S.W.J.; Al-Rumaihi, K.; Al-Bozom, I.; Li, P.; Chouchane, L. Targeting Wnt/EZH2/microRNA-708 signaling pathway inhibits neuroendocrine differentiation in prostate cancer. Cell Death Discov. 2019, 5, 139. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Ci, X.; Choi, S.Y.C.; Crea, F.; Lin, D.; Wang, Y. Molecular events in neuroendocrine prostate cancer development. Nat. Rev. Urol. 2021, 18, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Jayaraj, P.; Sen, S.; Rangarajan, S.; Ray, N.; Vasu, K.; Singh, V.K.; Phartyal, R.; Yadav, S.; Verma, A. Immunohistochemical evaluation of stress-responsive protein sestrin2 and its correlation with p53 mutational status in eyelid sebaceous gland carcinoma. Br. J. Ophthalmol. 2018, 102, 848. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.H.; Xue, X.; Ramakrishnan, S.K.; Cho, C.S.; Namkoong, S.; Jang, I.; Semple, I.A.; Ho, A.; Park, H.W.; Shah, Y.M.; et al. Tumor suppressive role of sestrin2 during colitis and colon carcinogenesis. Elife 2016, 5, e12204. [Google Scholar] [CrossRef]
- Shin, J.; Bae, J.; Park, S.; Kang, H.G.; Shin, S.M.; Won, G.; Kim, J.S.; Cho, S.G.; Choi, Y.; Oh, S.M.; et al. mTOR-Dependent Role of Sestrin2 in Regulating Tumor Progression of Human Endometrial Cancer. Cancers 2020, 12, 2515. [Google Scholar] [CrossRef]
- Zighelboim, I.; Goodfellow, P.J.; Schmidt, A.P.; Walls, K.C.; Mallon, M.A.; Mutch, D.G.; Yan, P.S.; Huang, T.H.-M.; Powell, M.A. Differential Methylation Hybridization Array of Endometrial Cancers Reveals Two Novel Cancer-Specific Methylation Markers. Clin. Cancer Res. 2007, 13, 2882–2889. [Google Scholar] [CrossRef]
- Ambrosio, S.; Saccà, C.D.; Amente, S.; Paladino, S.; Lania, L.; Majello, B. Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene 2017, 36, 6701–6711. [Google Scholar] [CrossRef]
- Liu, S.Y.; Lee, Y.J.; Lee, T.C. Association of platelet-derived growth factor receptor β accumulation with increased oxidative stress and cellular injury in sestrin 2 silenced human glioblastoma cells. FEBS Lett. 2011, 585, 1853–1858. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heidler, J.; Fysikopoulos, A.; Wempe, F.; Seimetz, M.; Bangsow, T.; Tomasovic, A.; Veit, F.; Scheibe, S.; Pichl, A.; Weisel, F.; et al. Sestrin-2, a repressor of PDGFRβ signalling, promotes cigarette-smoke-induced pulmonary emphysema in mice and is upregulated in individuals with COPD. Dis. Model. Mech. 2013, 6, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, N.; Yusoff, N.M.; Zakaria, Z.; Lim, M.N.; Baharuddin, P.J.; Fakiruddin, K.S.; Yahaya, B. Human non-small cell lung cancer expresses putative cancer stem cell markers and exhibits the transcriptomic profile of multipotent cells. BMC Cancer 2015, 15, 84. [Google Scholar] [CrossRef]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef]
- Wang, N.; Wang, S.; Li, M.Y.; Hu, B.G.; Liu, L.P.; Yang, S.L.; Yang, S.; Gong, Z.; Lai, P.B.S.; Chen, G.G. Cancer stem cells in hepatocellular carcinoma: An overview and promising therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816287. [Google Scholar] [CrossRef]
- Munro, M.J.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2018, 71, 110–116. [Google Scholar] [CrossRef]
- Zabierowski, S.E.; Herlyn, M. Melanoma stem cells: The dark seed of melanoma. J. Clin. Oncol. 2008, 26, 2890–2894. [Google Scholar] [CrossRef] [PubMed]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef]
- Martinez-Climent, J.A.; Fontan, L.; Gascoyne, R.D.; Siebert, R.; Prosper, F. Lymphoma stem cells: Enough evidence to support their existence? Haematologica 2010, 95, 293–302. [Google Scholar] [CrossRef]
- Wei, J.; Zheng, X.; Li, W.; Li, X.; Fu, Z. Sestrin2 reduces cancer stemness via Wnt/β-catenin signaling in colorectal cancer. Cancer Cell Int. 2022, 22, 75. [Google Scholar] [CrossRef]
- Kopnin, P.B.; Agapova, L.S.; Kopnin, B.P.; Chumakov, P.M. Repression of Sestrin Family Genes Contributes to Oncogenic Ras-Induced Reactive Oxygen Species Up-regulation and Genetic Instability. Cancer Res. 2007, 67, 4671–4678. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yin, K.; Falcon, D.M.; Xue, X. The interaction of Hemin and Sestrin2 modulates oxidative stress and colon tumor growth. Toxicol. Appl. Pharmacol. 2019, 374, 77–85. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Lu, J.; Temp, U.; Müller-Hartmann, A.; Esser, J.; Grönke, S.; Partridge, L. Sestrin is a key regulator of stem cell function and lifespan in response to dietary amino acids. Nat. Aging 2021, 1, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Xie, F.; Cao, H.; Wang, C.; Zhu, M.; Liu, X.; Lu, X.; Huang, T.; Shen, Y.; Li, K.; et al. Mutational inactivation of mTORC1 repressor gene DEPDC5 in human gastrointestinal stromal tumors. Proc. Natl. Acad. Sci. USA 2019, 116, 22746–22753. [Google Scholar] [CrossRef] [PubMed]
- Lerman, M.I.; Minna, J.D. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: Identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res. 2000, 60, 6116–6133. [Google Scholar] [PubMed]
- Tang, Y.; Jiang, L.; Tang, W. Decreased expression of NPRL2 in renal cancer cells is associated with unfavourable pathological, proliferation and apoptotic features. Pathol. Oncol. Res. 2014, 20, 829–837. [Google Scholar] [CrossRef]
- Dai, M.; Yan, G.; Wang, N.; Daliah, G.; Edick, A.M.; Poulet, S.; Boudreault, J.; Ali, S.; Burgos, S.A.; Lebrun, J.-J. In vivo genome-wide CRISPR screen reveals breast cancer vulnerabilities and synergistic mTOR/Hippo targeted combination therapy. Nat. Commun. 2021, 12, 3055. [Google Scholar] [CrossRef]
- Wang, D.; Xu, C.; Yang, W.; Chen, J.; Ou, Y.; Guan, Y.; Guan, J.; Liu, Y. E3 ligase RNF167 and deubiquitinase STAMBPL1 modulate mTOR and cancer progression. Mol. Cell 2022, 82, 770–784.E9. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Giaime, E.; Narayan, S.; Hahm, S.; Howell, J.; O’Neill, D.; Vlasuk, G.P.; Saiah, E. Discovery of NV-5138, the first selective Brain mTORC1 activator. Sci. Rep. 2019, 9, 4107. [Google Scholar] [CrossRef]
- Kato, T.; Pothula, S.; Liu, R.J.; Duman, C.H.; Terwilliger, R.; Vlasuk, G.P.; Saiah, E.; Hahm, S.; Duman, R.S. Sestrin modulator NV-5138 produces rapid antidepressant effects via direct mTORC1 activation. J. Clin. Investig. 2019, 129, 2542–2554. [Google Scholar] [CrossRef]
- Fetalvero Kristina, M.; Narayan, S.; O’Neill David, J.; Saiah, E.; Sengupta, S. Modulators of Sestrin-GATOR2 Interaction and Uses Thereof. U.S. Patent 10,752,644, 11 August 2021. [Google Scholar]
- Gendronneau, G.; Berlin, I.; Lejeune, F.; Latreille, J. Sestrin Activators for Preventing and/or Attenuating Skin Ageing and/or Hydrating the Skin and/or for Regulating Skin Pigmentation. U.S. Patent 10,745,755, 25 September 2015. [Google Scholar]
- Jin, S.H.; Yang, J.H.; Shin, B.Y.; Seo, K.; Shin, S.M.; Cho, I.J.; Ki, S.H. Resveratrol inhibits LXRα-dependent hepatic lipogenesis through novel antioxidant Sestrin2 gene induction. Toxicol. Appl. Pharmacol. 2013, 271, 95–105. [Google Scholar] [CrossRef]
- Sergides, C.; Chirilă, M.; Silvestro, L.; Pitta, D.; Pittas, A. Bioavailability and safety study of resveratrol 500 mg tablets in healthy male and female volunteers. Exp. Ther. Med. 2016, 11, 164–170. [Google Scholar] [CrossRef]
- Kim, G.T.; Lee, S.H.; Kim, J.I.; Kim, Y.M. Quercetin regulates the sestrin 2-AMPK-p38 MAPK signaling pathway and induces apoptosis by increasing the generation of intracellular ROS in a p53-independent manner. Int. J. Mol. Med. 2014, 33, 863–869. [Google Scholar] [CrossRef]
- Kim, G.T.; Lee, S.H.; Kim, Y.M. Quercetin Regulates Sestrin 2-AMPK-mTOR Signaling Pathway and Induces Apoptosis via Increased Intracellular ROS in HCT116 Colon Cancer Cells. J. Cancer Prev. 2013, 18, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Aghababaei, F.; Hadidi, M. Recent Advances in Potential Health Benefits of Quercetin. Pharmaceuticals 2023, 16, 1020. [Google Scholar] [CrossRef] [PubMed]
- Brüning, A.; Rahmeh, M.; Friese, K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef]
- Markowitz, M.; Conant, M.; Hurley, A.; Schluger, R.; Duran, M.; Peterkin, J.; Chapman, S.; Patick, A.; Hendricks, A.; Yuen, G.J.; et al. A preliminary evaluation of nelfinavir mesylate, an inhibitor of human immunodeficiency virus (HIV)-1 protease, to treat HIV infection. J. Infect. Dis. 1998, 177, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.A.; Rosenbaum, S.E.; Kerr, B.M.; Pithavala, Y.K.; Yuen, G.; Dudley, M.N. A population pharmacokinetic analysis of nelfinavir mesylate in human immunodeficiency virus-infected patients enrolled in a phase III clinical trial. Antimicrob. Agents Chemother. 2000, 44, 1832–1837. [Google Scholar] [CrossRef]
- Jang, S.-K.; Hong, S.-E.; Lee, D.-H.; Kim, J.-Y.; Kim, J.Y.; Ye, S.-K.; Hong, J.; Park, I.-C.; Jin, H.-O. Inhibition of mTORC1 through ATF4-induced REDD1 and Sestrin2 expression by Metformin. BMC Cancer 2021, 21, 803. [Google Scholar] [CrossRef]
- Pulito, C.; Mori, F.; Sacconi, A.; Goeman, F.; Ferraiuolo, M.; Pasanisi, P.; Campagnoli, C.; Berrino, F.; Fanciulli, M.; Ford, R.J.; et al. Metformin-induced ablation of microRNA 21-5p releases Sestrin-1 and CAB39L antitumoral activities. Cell Discov. 2017, 3, 17022. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Kosaka, T.; Hongo, H.; Miyazaki, Y.; Nishimoto, K.; Miyajima, A.; Oya, M. Reactive oxygen species induction by cabazitaxel through inhibiting Sestrin-3 in castration resistant prostate cancer. Oncotarget 2017, 8, 87675–87683. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.K.; Okekunle, A.P.; Lee, J.E.; Sung, M.K.; Lim, Y.J. Role of Branched-chain Amino Acid Metabolism in Tumor Development and Progression. J. Cancer Prev. 2021, 26, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Häfliger, P.; Charles, R.-P. The L-Type Amino Acid Transporter LAT1—An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y. Amino acid transporter LAT1 (SLC7A5) as a molecular target for cancer diagnosis and therapeutics. Pharmacol. Ther. 2022, 230, 107964. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).