VEGF and Pleiotrophin Modulate the Immune Profile of Breast Cancer


Abstract
:1. Introduction
2. Bone Marrow
3. Macrophages

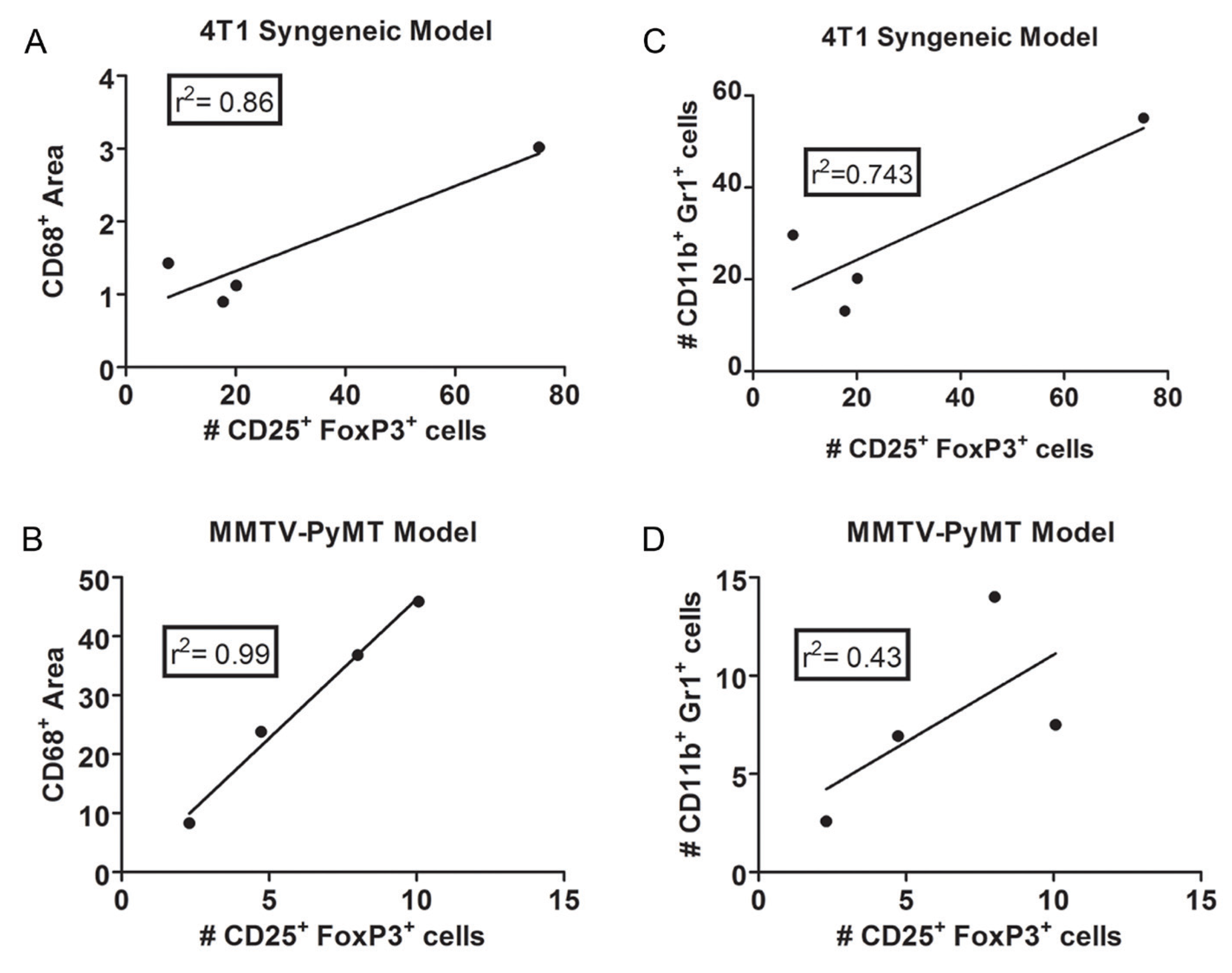{kind=link}
| Cell Type | VEGF Effects | Pre-Clinical Anti-VEGF | Clinical Anti-VEGF | Pleiotrophin Effects |
|---|---|---|---|---|
| Hematopoietic stem cells (HSCs) | Regulates pluripotency, survival, and mobilization from the bone marrow [25,26] | Anti-VEGFR2 inhibits reconstitution following sublethal irradiation [30,31] Anti-VEGFR1 prevents mobilization and recruitment to pre-metastatic niche in a Lewis lung carcinoma model [33] | Sunitinib and sorafenib result in myelosuppression as monotherapies [27,28,29] | Unknown |
| Macrophages | Macrophage chemotaxis [7] | Reduces macrophage infiltration in multiple breast cancer and other cancer models [48,49,50,51,52] | Unknown | Induces macrophage VEGFR2 expression and promotes an angiogenic phenotype [48,54,55,56] |
| Myeloid derived suppressor cells (MDSCs) | Promotes differentiation into neutrophils, macrophages, and dendritic cells [59] | VEGFR2 specific inhibition decreases MDSC in MDA-MB-231 xenograft and MMTV-PyMT transgenic models [50] Inhibition of both VEGFR1 and VEGFR2 increases MDSC number in the tumor in MDA-MB-231 xenograft and MMTV-PyMT transgenic models [50] | Sunitinib decreases MDSCs in in patients with renal cell carcinoma (RCC) [91] Bevacizumab decreases MDSCs in patients with a variety of cancers [77] | Unknown |
| Neutrophils | Neutrophil chemotaxis [65] | VEGFR2 specific inhibition increases neutrophil infiltration into tumors in multiple breast cancer models [49,50] Inhibition of both VEGFR1 and VEGFR2 decrease neutrophil infiltration into tumors in multiple breast cancer models [49,50] | Unknown | Neutrophil chemotaxis [68] |
| Dendritic cells (DCs) | VEGF:VEGFR1 activation inhibits the differentiation of HSCs down the DC lineage [71,72] VEGF:VEGFR2 activation inhibits DC antigen presenting cell functions [73,74] | Specific inhibition of VEGF: VEGFR2 activation increases the number of mature dendritic cells in the MDA-MB-231 xenograft and 4T1 syngeneic breast cancer models [49,50] | Sorafenib reverses defects in DC maturation in patients with RCC [78] Bevacizumab reverses defects in DC maturation in colorectal cancer patients [78] | Unknown |
| Regulatory T-cells (Tregs) | VEGF expression can be correlated to high FoxP3 expression in breast carcinoma [83] | VEGFR2 specific inhibition decreases Tregs MMTV-PyMT transgenic model [50] Inhibition of both VEGFR1 and VEGFR2 increases Tregs in the MMTV-PyMT transgenic model [50] | Sunitinib decreased Tregs in patients with RCC [91] | Unknown |
4. Myeloid Derived Suppressor Cells
5. Neutrophils
6. Dendritic Cells
7. Regulatory T-cells

8. Anti-VEGF Therapy in Breast Cancer
9. Conclusions
Acknowledgements
Financial Support
References
- American Cancer Society, Cancer Facts and Figures 2009; American Cancer Society: Atlanta, GA, USA, 2009.
- Tysnes, B.B.; Bjerkvig, R. Cancer initiation and progression: involvement of stem cells and the microenvironment. Biochim. Biophys. Acta 2007, 1775, 283–297. [Google Scholar]
- DeNardo, D.G.; Coussens, L.M. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007, 9, 212. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef]
- Pradeep, C.R.; Sunila, E.S.; Kuttan, G. Expression of vascular endothelial growth factor (VEGF) and VEGF receptors in tumor angiogenesis and malignancies. Integr. Cancer Ther. 2005, 4, 315–321. [Google Scholar] [CrossRef]
- Sawano, A.; Iwai, S.; Sakurai, Y.; Ito, M.; Shitara, K.; Nakahata, T.; Shibuya, M. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte–macrophages in humans. Blood 2001, 97, 785–791. [Google Scholar] [CrossRef]
- Lee, T.H.; Seng, S.; Sekine, M.; Hinton, C.; Fu, Y.; Avraham, H.K.; Avraham, S. Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed VEGFR1/FLT1. PLoS Med. 2007, 4, e186. [Google Scholar] [CrossRef]
- Yang, A.D.; Camp, E.R.; Fan, F.; Shen, L.; Gray, M.J.; Liu, W.; Somcio, R.; Bauer, T.W.; Wu, Y.; Hicklin, D.J.; Ellis, L.M. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006, 66, 46–51. [Google Scholar] [CrossRef]
- Ellis, L.M.; Hicklin, D.J. Pathways mediating resistance to vascular endothelial growth factor-targeted therapy. Clin. Cancer Res. 2008, 14, 6371–6375. [Google Scholar] [CrossRef]
- Grepin, R.; Pages, G. Molecular mechanisms of resistance to tumour anti-angiogenic strategies. J. Oncol. 2010, 2010, 835680. [Google Scholar]
- Berardi, A.C.; Wang, A.; Abraham, J.; Scadden, D.T. Basic fibroblast growth factor mediates its effects on committed myeloid progenitors by direct action and has no effect on hematopoietic stem cells. Blood 1995, 86, 2123–2129. [Google Scholar]
- Byrd, V.M.; Ballard, D.W.; Miller, G.G.; Thomas, J.W. Fibroblast growth factor-1 (FGF-1) enhances IL-2 production and nuclear translocation of NF-kappaB in FGF receptor-bearing Jurkat T cells. J. Immunol. 1999, 162, 5853–5859. [Google Scholar]
- Kitayama, J.; Nagawa, H.; Yasuhara, H.; Tsuno, N.; Kimura, W.; Shibata, Y.; Muto, T. Suppressive effect of basic fibroblast growth factor on transendothelial emigration of CD4(+) T-lymphocyte. Cancer Res. 1994, 54, 4729–4733. [Google Scholar]
- Nakayama, T.; Mutsuga, N.; Tosato, G. Effect of fibroblast growth factor 2 on stromal cell-derived factor 1 production by bone marrow stromal cells and hematopoiesis. J. Natl. Cancer Inst. 2007, 99, 223–235. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Vacca, A.; Roncali, L.; Presta, M. Endogenous and exogenous fibroblast growth factor-2 modulate wound healing in the chick embryo chorioallantoic membrane. Angiogenesis 1999, 3, 89–95. [Google Scholar] [CrossRef]
- Takagi, S.; Takahashi, K.; Katsura, Y.; Matsuoka, T.; Ohsaka, A. Basic fibroblast growth factor modulates the surface expression of effector cell molecules and primes respiratory burst activity in human neutrophils. Acta Haematol. 2000, 103, 78–83. [Google Scholar] [CrossRef]
- Zhao, X.M.; Citrin, B.S.; Miller, G.G.; Frist, W.H.; Merrill, W.H.; Fischell, T.A.; Atkinson, J.B.; Yeoh, T.K. Association of acidic fibroblast growth factor and untreated low grade rejection with cardiac allograft vasculopathy. Transplantation 1995, 59, 1005–1010. [Google Scholar] [CrossRef]
- Lewis, C.E.; De Palma, M.; Naldini, L. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. 2007, 67, 8429–8432. [Google Scholar] [CrossRef]
- Lewis, C.E.; Hughes, R. Inflammation and breast cancer. Microenvironmental factors regulating macrophage function in breast tumours: hypoxia and angiopoietin-2. Breast Cancer Res. 2007, 9, 209. [Google Scholar] [CrossRef]
- Lin, Y.L.; Liang, Y.C.; Chiang, B.L. Placental growth factor down-regulates type 1 T helper immune response by modulating the function of dendritic cells. J. Leukoc. Biol. 2007, 82, 1473–1480. [Google Scholar] [CrossRef]
- Selvaraj, S.K.; Giri, R.K.; Perelman, N.; Johnson, C.; Malik, P.; Kalra, V.K. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood 2003, 102, 1515–1524. [Google Scholar] [CrossRef]
- Burger, J.A.; Kipps, T.J. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef]
- Schulz, C.; von Andrian, U.H.; Massberg, S. Hematopoietic stem and progenitor cells: their mobilization and homing to bone marrow and peripheral tissue. Immunol. Res. 2009, 44, 160–168. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Chang, Y.; Deuel, T.F. Pleiotrophin, a multifunctional tumor promoter through induction of tumor angiogenesis, remodeling of the tumor microenvironment, and activation of stromal fibroblas. Cell Cycle 2007, 6, 2877–2883. [Google Scholar] [CrossRef]
- Lynn, K.D.; Roland, C.L.; Brekken, R.A. Unpulished Data. 2010.
- Rauvala, H. An 18-kd heparin-binding protein of developing brain that is distinct from fibroblast growth factors. EMBO J. 1989, 8, 2933–2941. [Google Scholar]
- Silos-Santiago, I.; Yeh, H.J.; Gurrieri, M.A.; Guillerman, R.P.; Li, Y.S.; Wolf, J.; Snider, W.; Deuel, T.F. Localization of pleiotrophin and its mRNA in subpopulations of neurons and their corresponding axonal tracts suggests important roles in neural-glial interactions during development and in maturity. J. Neurobiol. 1996, 31, 283–296. [Google Scholar] [CrossRef]
- Vanderwinden, J.M.; Mailleux, P.; Schiffmann, S.N.; Vanderhaeghen, J.J. Cellular distribution of the new growth factor pleiotrophin (HB-GAM) mRNA in developing and adult rat tissues. Anat. Embryol. (Berl.) 1992, 186, 387–406. [Google Scholar]
- Yeh, H.J.; He, Y.Y.; Xu, J.; Hsu, C.Y.; Deuel, T.F. Upregulation of pleiotrophin gene expression in developing microvasculature, macrophages, and astrocytes after acute ischemic brain injury. J. Neurosci. 1998, 18, 3699–3707. [Google Scholar]
- Courty, J.; Dauchel, M.C.; Caruelle, D.; Perderiset, M.; Barritault, D. Mitogenic properties of a new endothelial cell growth factor related to pleiotrophin. Biochem. Biophys. Res. Commun. 1991, 180, 145–151. [Google Scholar] [CrossRef]
- Fang, W.; Hartmann, N.; Chow, D.T.; Riegel, A.T.; Wellstein, A. Pleiotrophin stimulates fibroblasts and endothelial and epithelial cells and is expressed in human cancer. J. Biol. Chem. 1992, 267, 25889–25897. [Google Scholar]
- Laaroubi, K.; Delbe, J.; Vacherot, F.; Desgranges, P.; Tardieu, M.; Jaye, M.; Barritault, D.; Courty, J. Mitogenic and in vitro angiogenic activity of human recombinant heparin affin regulatory peptide. Growth Factors 1994, 10, 89–98. [Google Scholar] [CrossRef]
- Milner, P.G.; Li, Y.S.; Hoffman, R.M.; Kodner, C.M.; Siegel, N.R.; Deuel, T.F. A novel 17 kD heparin-binding growth factor (HBGF-8) in bovine uterus: purification and N-terminal amino acid sequence. Biochem. Biophys. Res. Commun. 1989, 165, 1096–1103. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Alcantara, S.; Dimitrov, T.; Vega, J.A.; Deuel, T.F. Pleiotrophin disrupts calcium-dependent homophilic cell-cell adhesion and initiates an epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2006, 103, 17795–17800. [Google Scholar] [CrossRef]
- Riegel, A.T.; Wellstein, A. The potential role of the heparin-binding growth factor pleiotrophin in breast cancer. Breast Cancer Res. Treat. 1994, 31, 309–314. [Google Scholar] [CrossRef]
- Choudhuri, R.; Zhang, H.T.; Donnini, S.; Ziche, M.; Bicknell, R. An angiogenic role for the neurokines midkine and pleiotrophin in tumorigenesis. Cancer Res. 1997, 57, 1814–1819. [Google Scholar]
- Zhang, N.; Zhong, R.; Wang, Z.Y.; Deuel, T.F. Human breast cancer growth inhibited in vivo by a dominant negative pleiotrophin mutant. J. Biol. Chem. 1997, 272, 16733–16736. [Google Scholar]
- Chang, Y.; Zuka, M.; Perez-Pinera, P.; Astudillo, A.; Mortimer, J.; Berenson, J.R.; Deuel, T.F. Secretion of pleiotrophin stimulates breast cancer progression through remodeling of the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2007, 104, 10888–10893. [Google Scholar]
- Gerber, H.P.; Malik, A.K.; Solar, G.P.; Sherman, D.; Liang, X.H.; Meng, G.; Hong, K.; Marsters, J.C.; Ferrara, N. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 2002, 417, 954–958. [Google Scholar] [CrossRef]
- Robertson, S.; Kennedy, M.; Keller, G. Hematopoietic commitment during embryogenesis. Ann. N. Y. Acad. Sci. 1999, 872, 9–16. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; Desai, J.; Fletcher, C.D.; George, S.; Bello, C.L.; Huang, X.; Baum, C.M.; Casali, P.G. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Motzer, R.J.; Michaelson, M.D.; Rosenberg, J.; Bukowski, R.M.; Curti, B.D.; George, D.J.; Hudes, G.R.; Redman, B.G.; Margolin, K.A.; Wilding, G. Sunitinib efficacy against advanced renal cell carcinoma. J. Urol. 2007, 178, 1883–1887. [Google Scholar] [CrossRef]
- Nexavar (sorafenib) package insert, 17.5 FDA-approved patient labeling. Bayer Healthcare Pharmaceuticals Inc.: West Haven, CT, USA, 2009.
- Hooper, A.T.; Butler, J.M.; Nolan, D.J.; Kranz, A.; Iida, K.; Kobayashi, M.; Kopp, H.G.; Shido, K.; Petit, I.; Yanger, K.; James, D.; Witte, L.; Zhu, Z.; Wu, Y.; Pytowski, B.; Rosenwaks, Z.; Mittal, V.; Sato, T.N.; Rafii, S. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 2009, 4, 263–274. [Google Scholar] [CrossRef]
- Kopp, H.G.; Hooper, A.T.; Avecilla, S.T.; Rafii, S. Functional heterogeneity of the bone marrow vascular niche. Ann. N. Y. Acad. Sci. 2009, 1176, 47–54. [Google Scholar] [CrossRef]
- Grunewald, M.; Avraham, I.; Dor, Y.; Bachar-Lustig, E.; Itin, A.; Jung, S.; Chimenti, S.; Landsman, L.; Abramovitch, R.; Keshet, E. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 2006, 124, 175–189. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; Zhu, Z.; Hicklin, D.; Wu, Y.; Port, J.L.; Altorki, N.; Port, E.R.; Ruggero, D.; Shmelkov, S.V.; Jensen, K.K.; Rafii, S.; Lyden, D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Tare, R.S.; Oreffo, R.O.; Clarke, N.M.; Roach, H.I. Pleiotrophin/Osteoblast-stimulating factor 1: dissecting its diverse functions in bone formation. J. Bone Miner. Res. 2002, 17, 2009–2020. [Google Scholar] [CrossRef]
- Vazin, T.; Becker, K.G.; Chen, J.; Spivak, C.E.; Lupica, C.R.; Zhang, Y.; Worden, L.; Freed, W.J. A novel combination of factors, termed SPIE, which promotes dopaminergic neuron differentiation from human embryonic stem cells. PLoS One 2009, 4, e6606. [Google Scholar]
- Roger, J.; Brajeul, V.; Thomasseau, S.; Hienola, A.; Sahel, J.A.; Guillonneau, X.; Goureau, O. Involvement of Pleiotrophin in CNTF-mediated differentiation of the late retinal progenitor cells. Dev. Biol. 2006, 298, 527–539. [Google Scholar] [CrossRef]
- Caruelle, D.; Mazouzi, Z.; Husmann, I.; Delbe, J.; Duchesnay, A.; Gautron, J.; Martelly, I.; Courty, J. Upregulation of HARP during in vitro myogenesis and rat soleus muscle regeneration. J. Muscle Res. Cell Motil. 2004, 25, 45–53. [Google Scholar] [CrossRef]
- Heiss, C.; Wong, M.L.; Block, V.I.; Lao, D.; Real, W.M.; Yeghiazarians, Y.; Lee, R.J.; Springer, M.L. Pleiotrophin induces nitric oxide dependent migration of endothelial progenitor cells. J. Cell. Physiol. 2008, 215, 366–373. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer. 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjallman, A.H.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar] [CrossRef]
- Lin, E.Y.; Li, J.F.; Bricard, G.; Wang, W.; Deng, Y.; Sellers, R.; Porcelli, S.A.; Pollard, J.W. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol. Oncol. 2007, 1, 288–302. [Google Scholar] [CrossRef]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar]
- Leek, R. D.; Harris, A. L. Tumor-associated macrophages in breast cancer. J. Mammary Gland Biol. Neoplasia 2002, 7, 177–189. [Google Scholar] [CrossRef]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One 2009, 4, e6562. [Google Scholar]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar] [CrossRef]
- Roland, C.L.; Dineen, S.P.; Lynn, K.D.; Sullivan, L.A.; Dellinger, M.T.; Sadegh, L.; Sullivan, J.P.; Shames, D.S.; Brekken, R.A. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol. Cancer Ther. 2009, 8, 1761–1771. [Google Scholar] [CrossRef]
- Roland, C.L.; Lynn, K.D.; Toombs, J.E.; Dineen, S.P.; Udugamasooriya, D.G.; Brekken, R.A. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS One 2009, 4, e7669. [Google Scholar]
- Salnikov, A.V.; Heldin, N.E.; Stuhr, L.B.; Wiig, H.; Gerber, H.; Reed, R.K.; Rubin, K. Inhibition of carcinoma cell-derived VEGF reduces inflammatory characteristics in xenograft carcinoma. Int. J. Cancer 2006, 119, 2795–2802. [Google Scholar] [CrossRef]
- Whitehurst, B.; Flister, M.J.; Bagaitkar, J.; Volk, L.; Bivens, C.M.; Pickett, B.; Castro-Rivera, E.; Brekken, R.A.; Gerard, R.D.; Ran, S. Anti-VEGF-A therapy reduces lymphatic vessel density and expression of VEGFR-3 in an orthotopic breast tumor model. Int. J. Cancer 2007, 121, 2181–2191. [Google Scholar] [CrossRef]
- Vroling, L.; Yuana, Y.; Schuurhuis, G.J.; van Hinsbergh, V.W.; Gundy, C.; de Haas, R.; van Cruijsen, H.; Boven, E.; Hoekman, K.; Broxterman, H.J. VEGFR2 expressing circulating (progenitor) cell populations in volunteers and cancer patients. Thromb. Haemost. 2007, 98, 440–450. [Google Scholar]
- Chen, H.; Campbell, R.A.; Chang, Y.; Li, M.; Wang, C.S.; Li, J.; Sanchez, E.; Share, M.; Steinberg, J.; Berenson, A.; Shalitin, D.; Zeng, Z.; Gui, D.; Perez-Pinera, P.; Berenson, R.J.; Said, J.; Bonavida, B.; Deuel, T.F.; Berenson, J.R. Pleiotrophin produced by multiple myeloma induces transdifferentiation of monocytes into vascular endothelial cells: a novel mechanism of tumor-induced vasculogenesis. Blood 2009, 113, 1992–2002. [Google Scholar] [CrossRef]
- Sharifi, B.G.; Zeng, Z.; Wang, L.; Song, L.; Chen, H.; Qin, M.; Sierra-Honigmann, M.R.; Wachsmann-Hogiu, S.; Shah, P.K. Pleiotrophin induces transdifferentiation of monocytes into functional endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1273–1280. [Google Scholar] [CrossRef]
- Collino, F.; Revelli, A.; Massobrio, M.; Katsaros, D.; Schmitt-Ney, M.; Camussi, G.; Bussolati, B. Epithelial-mesenchymal transition of ovarian tumor cells induces an angiogenic monocyte cell population. Exp. Cell Res. 2009, 315, 2982–2994. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gabrilovich, D.I. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008, 68, 2561–2563. [Google Scholar] [CrossRef]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef]
- Shojaei, F.; Wu, X.; Zhong, C.; Yu, L.; Liang, X.H.; Yao, J.; Blanchard, D.; Bais, C.; Peale, F.V.; van Bruggen, N.; Ho, C.; Ross, J.; Tan, M.; Carano, R.A.; Meng, Y.G.; Ferrara, N. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 2007, 450, 825–831. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; Hanahan, D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef]
- Scapini, P.; Morini, M.; Tecchio, C.; Minghelli, S.; Di Carlo, E.; Tanghetti, E.; Albini, A.; Lowell, C.; Berton, G.; Noonan, D.M.; Cassatella, M.A. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J. Immunol. 2004, 172, 5034–5040. [Google Scholar]
- Ancelin, M.; Chollet-Martin, S.; Herve, M.A.; Legrand, C.; El Benna, J.; Perrot-Applanat, M. Vascular endothelial growth factor VEGF189 induces human neutrophil chemotaxis in extravascular tissue via an autocrine amplification mechanism. Lab. Invest. 2004, 84, 502–512. [Google Scholar] [CrossRef]
- Pahler, J.C.; Tazzyman, S.; Erez, N.; Chen, Y.Y.; Murdoch, C.; Nozawa, H.; Lewis, C.E.; Hanahan, D. Plasticity in tumor-promoting inflammation: impairment of macrophage recruitment evokes a compensatory neutrophil response. Neoplasia 2008, 10, 329–340. [Google Scholar]
- Heng, D.Y.; Xie, W.; Regan, M.M.; Warren, M.A.; Golshayan, A.R.; Sahi, C.; Eigl, B.J.; Ruether, J.D.; Cheng, T.; North, S.; Venner, P.; Knox, J.J.; Chi, K.N.; Kollmannsberger, C.; McDermott, D.F.; Oh, W.K.; Atkins, M.B.; Bukowski, R.M.; Rini, B.I.; Choueiri, T.K. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J. Clin. Oncol. 2009, 27, 5794–5799. [Google Scholar]
- Ochiai, K.; Muramatsu, H.; Yamamoto, S.; Ando, H.; Muramatsu, T. The role of midkine and pleiotrophin in liver regeneration. Liver Int. 2004, 24, 484–491. [Google Scholar] [CrossRef]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibitis the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, X.; Dikov, M.M.; Novitskiy, S.V.; Mosse, C.A.; Yang, L.; Carbone, D.P. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood 2007, 110, 624–631. [Google Scholar] [CrossRef]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D. P.; Gabrilovich, D. I. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar]
- Mimura, K.; Kono, K.; Takahashi, A.; Kawaguchi, Y.; Fujii, H. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol. Immunother. 2007, 56, 761–770. [Google Scholar] [CrossRef]
- Almand, B.; Resser, J.R.; Lindman, B.; Nadaf, S.; Clark, J.I.; Kwon, E.D.; Carbone, D.P.; Gabrilovich, D.I. Clinical significance of defective dendritic cell differentiation in cancer. Clin. Cancer Res. 2000, 6, 1755–1766. [Google Scholar]
- Osada, T.; Chong, G.; Tansik, R.; Hong, T.; Spector, N.; Kumar, R.; Hurwitz, H.I.; Dev, I.; Nixon, A.B.; Lyerly, H.K.; Clay, T.; Morse, M.A. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol. Immunother. 2008, 57, 1115–1124. [Google Scholar] [CrossRef]
- Fricke, I.; Mirza, N.; Dupont, J.; Lockhart, C.; Jackson, A.; Lee, J.H.; Sosman, J.A.; Gabrilovich, D.I. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin. Cancer Res. 2007, 13, 4840–4848. [Google Scholar] [CrossRef]
- Hipp, M.M.; Hilf, N.; Walter, S.; Werth, D.; Brauer, K.M.; Radsak, M.P.; Weinschenk, T.; Singh-Jasuja, H.; Brossart, P. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood 2008, 111, 5610–5620. [Google Scholar] [CrossRef]
- van Cruijsen, H.; Hoekman, K.; Stam, A.G.; van den Eertwegh, A.J.; Kuenen, B.C.; Scheper, R.J.; Giaccone, G.; de Gruijl, T.D. Defective differentiation of myeloid and plasmacytoid dendritic cells in advanced cancer patients is not normalized by tyrosine kinase inhibition of the vascular endothelial growth factor receptor. Clin. Dev. Immunol. 2007, 2007, 17315. [Google Scholar]
- Dieckmann, D.; Plottner, H.; Berchtold, S.; Berger, T.; Schuler, G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J. Exp. Med. 2001, 193, 1303–1310. [Google Scholar] [CrossRef]
- Gupta, S.; Joshi, K.; Wig, J.D.; Arora, S.K. Intratumoral FOXP3 expression in infiltrating breast carcinoma: Its association with clinicopathologic parameters and angiogenesis. Acta Oncol. 2007, 46, 792–797. [Google Scholar] [CrossRef]
- Donovan, D.; Harmey, J.H.; Toomey, D.; Osborne, D.H.; Redmond, H.P.; Bouchier-Hayes, D.J. TGF beta-1 regulation of VEGF production by breast cancer cells. Ann. Surg. Oncol. 1997, 4, 621–627. [Google Scholar] [CrossRef]
- Qin, F.X. Dynamic behavior and function of Foxp3+ regulatory T cells in tumor bearing host. Cell Mol. Immunol. 2009, 6, 3–13. [Google Scholar] [CrossRef]
- Perruche, S.; Zhang, P.; Liu, Y.; Saas, P.; Bluestone, J.A.; Chen, W. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat. Med. 2008, 14, 528–535. [Google Scholar] [CrossRef]
- Cortes-Funes, H. The role of antiangiogenesis therapy: bevacizumab and beyond. Clin. Transl. Oncol. 2009, 11, 349–355. [Google Scholar] [CrossRef]
- Miller, K.D.; Chap, L.I.; Holmes, F.A.; Cobleigh, M.A.; Marcom, P.K.; Fehrenbacher, L.; Dickler, M.; Overmoyer, B.A.; Reimann, J.D.; Sing, A.P.; Langmuir, V.; Rugo, H.S. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J. Clin. Oncol. 2005, 23, 792–799. [Google Scholar] [CrossRef]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef]
- Schneider, B.P.; Wang, M.; Radovich, M.; Sledge, G.W.; Badve, S.; Thor, A.; Flockhart, D.A.; Hancock, B.; Davidson, N.; Gralow, J.; Dickler, M.; Perez, E.A.; Cobleigh, M.; Shenkier, T.; Edgerton, S.; Miller, K.D. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J. Clin. Oncol. 2008, 26, 4672–4678. [Google Scholar] [CrossRef]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; Dreicer, R.; Bukowski, R.; Finke, J.H. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef]
- Connolly, D.T.; Heuvelman, D.M.; Nelson, R.; Olander, J.V.; Eppley, B.L.; Delfino, J.J.; Siegel, N.R.; Leimgruber, R.M.; Feder, J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Invest. 1989, 84, 1470–1478. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lynn, K.D.; Roland, C.L.; Brekken, R.A. VEGF and Pleiotrophin Modulate the Immune Profile of Breast Cancer. Cancers 2010, 2, 970-988. https://doi.org/10.3390/cancers2020970
Lynn KD, Roland CL, Brekken RA. VEGF and Pleiotrophin Modulate the Immune Profile of Breast Cancer. Cancers. 2010; 2(2):970-988. https://doi.org/10.3390/cancers2020970
Chicago/Turabian StyleLynn, Kristi D., Christina L. Roland, and Rolf A. Brekken. 2010. "VEGF and Pleiotrophin Modulate the Immune Profile of Breast Cancer" Cancers 2, no. 2: 970-988. https://doi.org/10.3390/cancers2020970
APA StyleLynn, K. D., Roland, C. L., & Brekken, R. A. (2010). VEGF and Pleiotrophin Modulate the Immune Profile of Breast Cancer. Cancers, 2(2), 970-988. https://doi.org/10.3390/cancers2020970