Epithelial-Mesenchymal Transition in Pancreatic Carcinoma

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Epithelial-Mesenchymal Transition in Tumor Progression

1.2. Pancreatic Adenocarcinoma
2. EMT Signaling Pathways
2.1. Principles of EMT Signaling
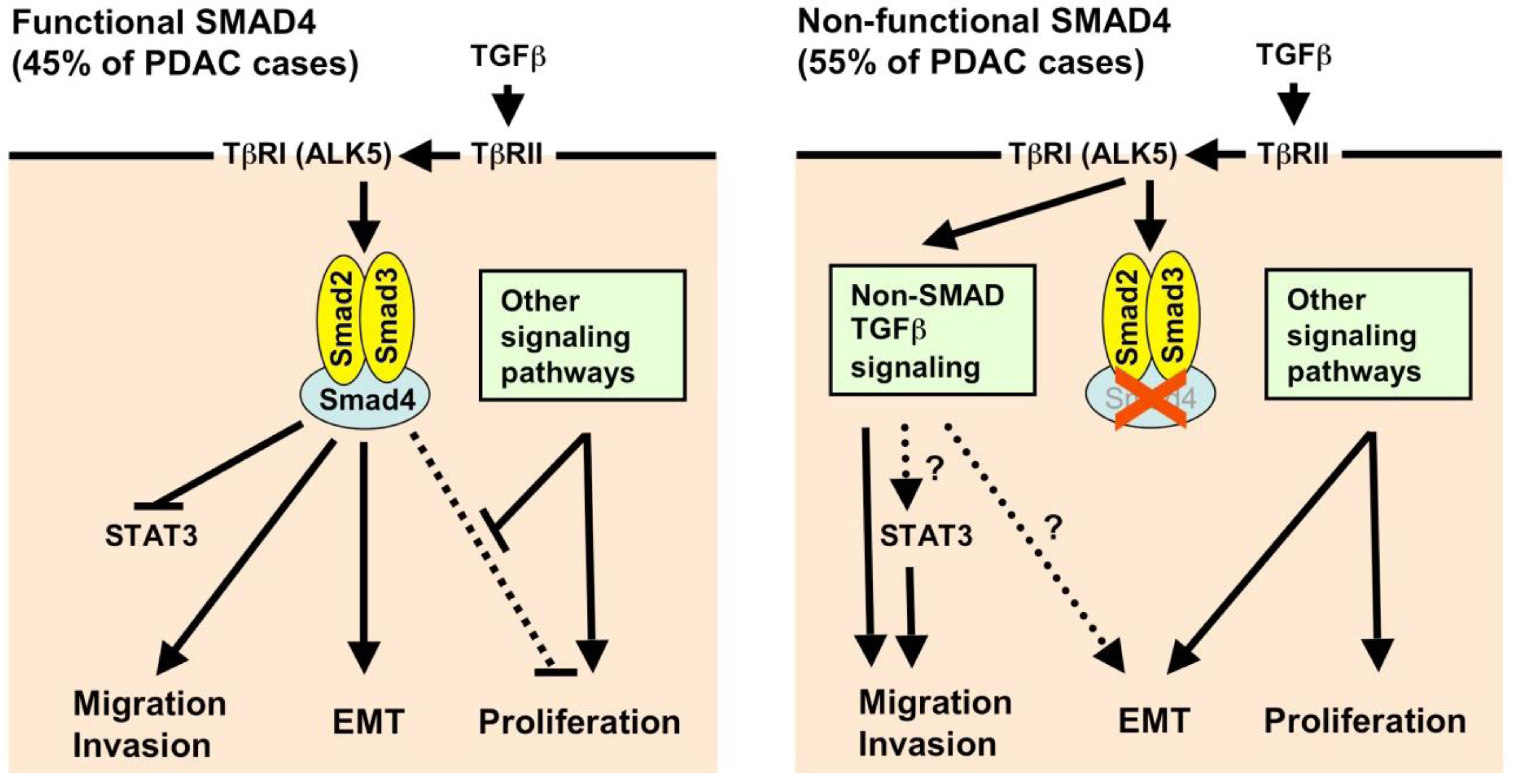
2.2. Transforming Growth Factor-β (TGF-β) and SMAD Signaling
2.3. Other Soluble Factors and Their Signaling Pathways

2.4. RAS Signaling
2.5. Transcriptional Repressors: Snail, ZEB, and bHLH Families
2.6. Other Signaling Pathways
2.7. Cadherins
2.8. The Extracellular Matrix and Proteases
2.9. MicroRNAs and Epigenetic Mechanisms
3. Evidence for EMT in Human Tissue Samples of Pancreatic Carcinoma
4. EMT, Hypoxia, Inflammation, Invasion, and Metastasis
5. EMT and Cancer Stem Cells
6. EMT and Drug Resistance
7. Conclusions and Outlook
References
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef]
- Hugo, H.; Ackland, M.L.; Blick, T.; Lawrence, M.G.; Clements, J.A.; Williams, E.D.; Thompson, E.W. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef]
- Ikushima, H.; Miyazono, K. TGFbeta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef]
- Heldin, C.H.; Landstrom, M.; Moustakas, A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Giehl, K.; Seidel, B.; Gierschik, P.; Adler, G.; Menke, A. TGFbeta1 represses proliferation of pancreatic carcinoma cells which correlates with Smad4-independent inhibition of ERK activation. Oncogene 2000, 19, 4531–4541. [Google Scholar] [CrossRef]
- Ellenrieder, V.; Hendler, S.F.; Boeck, W.; Seufferlein, T.; Menke, A.; Ruhland, C.; Adler, G.; Gress, T.M. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001, 61, 4222–4228. [Google Scholar]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Geng, M.M.; Ellenrieder, V.; Wallrapp, C.; Muller-Pillasch, F.; Sommer, G.; Adler, G.; Gress, T.M. Use of representational difference analysis to study the effect of TGFB on the expression profile of a pancreatic cancer cell line. Genes Chromosomes Cancer 1999, 26, 70–79. [Google Scholar] [CrossRef]
- Ungefroren, H.; Schniewind, B.; Groth, S.; Chen, W.B.; Muerkoster, S.S.; Kalthoff, H.; Fandrich, F. Antitumor activity of ALK1 in pancreatic carcinoma cells. Int. J. Cancer 2007, 120, 1641–1651. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Eshleman, J.R.; Goggins, M.; Jaffee, E.M.; Iacobuzio-Donahue, C.A.; Maitra, A.; Cameron, J.L.; Olino, K.; Schulick, R.; Winter, J.; Herman, J.M.; Laheru, D.; Klein, A.P.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E.; Hruban, R.H. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar]
- Friess, H.; Yamanaka, Y.; Buchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar]
- Bellone, G.; Smirne, C.; Mauri, F.A.; Tonel, E.; Carbone, A.; Buffolino, A.; Dughera, L.; Robecchi, A.; Pirisi, M.; Emanuelli, G. Cytokine expression profile in human pancreatic carcinoma cells and in surgical specimens: Implications for survival. Cancer Immunol. Immunother. 2006, 55, 684–698. [Google Scholar] [CrossRef]
- Maier, H.J.; Schmidt-Strassburger, U.; Huber, M.A.; Wiedemann, E.M.; Beug, H.; Wirth, T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010, 295, 214–228. [Google Scholar] [CrossRef]
- Fensterer, H.; Giehl, K.; Buchholz, M.; Ellenrieder, V.; Buck, A.; Kestler, H.A.; Adler, G.; Gierschik, P.; Gress, T.M. Expression profiling of the influence of RAS mutants on the TGFB1-induced phenotype of the pancreatic cancer cell line PANC-1. Genes Chromosomes Cancer 2004, 39, 224–235. [Google Scholar] [CrossRef]
- Zhao, S.; Venkatasubbarao, K.; Lazor, J.W.; Sperry, J.; Jin, C.; Cao, L.; Freeman, J.W. Inhibition of STAT3 Tyr705 phosphorylation by Smad4 suppresses transforming growth factor beta-mediated invasion and metastasis in pancreatic cancer cells. Cancer Res. 2008, 68, 4221–4228. [Google Scholar] [CrossRef]
- Unno, J.; Satoh, K.; Hirota, M.; Kanno, A.; Hamada, S.; Ito, H.; Masamune, A.; Tsukamoto, N.; Motoi, F.; Egawa, S.; Unno, M.; Horii, A.; Shimosegawa, T. LIV-1 enhances the aggressive phenotype through the induction of epithelial to mesenchymal transition in human pancreatic carcinoma cells. Int. J. Oncol. 2009, 35, 813–821. [Google Scholar]
- Jungert, K.; Buck, A.; von Wichert, G.; Adler, G.; Konig, A.; Buchholz, M.; Gress, T.M.; Ellenrieder, V. Sp1 is required for transforming growth factor-beta-induced mesenchymal transition and migration in pancreatic cancer cells. Cancer Res. 2007, 67, 1563–1570. [Google Scholar] [CrossRef]
- Schniewind, B.; Groth, S.; Sebens Muerkoster, S.; Sipos, B.; Schafer, H.; Kalthoff, H.; Fandrich, F.; Ungefroren, H. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene 2007, 26, 4850–4862. [Google Scholar] [CrossRef]
- Giehl, K.; Imamichi, Y.; Menke, A. Smad4-independent TGF-beta signaling in tumor cell migration. Cells Tissues Organs 2007, 185, 123–130. [Google Scholar] [CrossRef]
- Vogelmann, R.; Nguyen-Tat, M.D.; Giehl, K.; Adler, G.; Wedlich, D.; Menke, A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J. Cell Sci. 2005, 118, 4901–4912. [Google Scholar] [CrossRef]
- Imamichi, Y.; Waidmann, O.; Hein, R.; Eleftheriou, P.; Giehl, K.; Menke, A. TGF beta-induced focal complex formation in epithelial cells is mediated by activated ERK and JNK MAP kinases and is independent of Smad4. Biol. Chem. 2005, 386, 225–236. [Google Scholar]
- Levy, L.; Hill, C.S. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; DePinho, R.A. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Wang, P.; Chen, Z.; Meng, Z.Q.; Fan, J.; Luo, J.M.; Liang, W.; Lin, J.H.; Zhou, Z.H.; Chen, H.; Wang, K.; Shen, Y.H.; Xu, Z.D.; Liu, L.M. Dual role of Ski in pancreatic cancer cells: Tumor-promoting versus metastasis-suppressive function. Carcinogenesis 2009, 30, 1497–1506. [Google Scholar] [CrossRef]
- Hamada, S.; Satoh, K.; Hirota, M.; Kimura, K.; Kanno, A.; Masamune, A.; Shimosegawa, T. Bone morphogenetic protein 4 induces epithelial-mesenchymal transition through MSX2 induction on pancreatic cancer cell line. J. Cell. Physiol. 2007, 213, 768–774. [Google Scholar] [CrossRef]
- Gordon, K.J.; Kirkbride, K.C.; How, T.; Blobe, G.C. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis 2009, 30, 238–248. [Google Scholar]
- Gordon, K.J.; Dong, M.; Chislock, E.M.; Fields, T.A.; Blobe, G.C. Loss of type III transforming growth factor beta receptor expression increases motility and invasiveness associated with epithelial to mesenchymal transition during pancreatic cancer progression. Carcinogenesis 2008, 29, 252–262. [Google Scholar] [CrossRef]
- Satoh, K.; Hamada, S.; Kimura, K.; Kanno, A.; Hirota, M.; Umino, J.; Fujibuchi, W.; Masamune, A.; Tanaka, N.; Miura, K.; Egawa, S.; Motoi, F.; Unno, M.; Vonderhaar, B.K.; Shimosegawa, T. Up-regulation of MSX2 enhances the malignant phenotype and is associated with twist 1 expression in human pancreatic cancer cells. Am. J. Pathol. 2008, 172, 926–939. [Google Scholar] [CrossRef]
- Brinkmann, V.; Foroutan, H.; Sachs, M.; Weidner, K.M.; Birchmeier, W. Hepatocyte growth factor/scatter factor induces a variety of tissue-specific morphogenic programs in epithelial cells. J. Cell Biol. 1995, 131, 1573–1586. [Google Scholar] [CrossRef]
- Camp, E.R.; Yang, A.; Gray, M.J.; Fan, F.; Hamilton, S.R.; Evans, D.B.; Hooper, A.T.; Pereira, D.S.; Hicklin, D.J.; Ellis, L.M. Tyrosine kinase receptor RON in human pancreatic cancer: Expression, function, and validation as a target. Cancer 2007, 109, 1030–1039. [Google Scholar] [CrossRef]
- Koorstra, J.B.; Karikari, C.A.; Feldmann, G.; Bisht, S.; Rojas, P.L.; Offerhaus, G.J.; Alvarez, H.; Maitra, A. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol. Ther. 2009, 8, 618–626. [Google Scholar] [CrossRef]
- Yang, A.D.; Camp, E.R.; Fan, F.; Shen, L.; Gray, M.J.; Liu, W.; Somcio, R.; Bauer, T.W.; Wu, Y.; Hicklin, D.J.; Ellis, L.M. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006, 66, 46–51. [Google Scholar] [CrossRef]
- Baran, B.; Bechyne, I.; Siedlar, M.; Szpak, K.; Mytar, B.; Sroka, J.; Laczna, E.; Madeja, Z.; Zembala, M.; Czyz, J. Blood monocytes stimulate migration of human pancreatic carcinoma cells in vitro: The role of tumour necrosis factor - alpha. Eur. J. Cell Biol. 2009, 88, 743–752. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Buchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464. [Google Scholar] [CrossRef]
- Baril, P.; Gangeswaran, R.; Mahon, P.C.; Caulee, K.; Kocher, H.M.; Harada, T.; Zhu, M.; Kalthoff, H.; Crnogorac-Jurcevic, T.; Lemoine, N.R. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: Role of the beta4 integrin and the PI3k pathway. Oncogene 2007, 26, 2082–2094. [Google Scholar] [CrossRef]
- Kanno, A.; Satoh, K.; Masamune, A.; Hirota, M.; Kimura, K.; Umino, J.; Hamada, S.; Satoh, A.; Egawa, S.; Motoi, F.; Unno, M.; Shimosegawa, T. Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells. Int. J. Cancer 2008, 122, 2707–2718. [Google Scholar] [CrossRef]
- Ruan, K.; Bao, S.; Ouyang, G. The multifaceted role of periostin in tumorigenesis. Cell. Mol. Life Sci. 2009, 66, 2219–2230. [Google Scholar] [CrossRef]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grunert, S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef]
- Agbunag, C.; Bar-Sagi, D. Oncogenic K-ras drives cell cycle progression and phenotypic conversion of primary pancreatic duct epithelial cells. Cancer Res. 2004, 64, 5659–5663. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; Kawaguchi, Y.; Johann, D.; Liotta, L.A.; Crawford, H.C.; Putt, M.E.; Jacks, T.; Wright, C.V.; Hruban, R.H.; Lowy, A.M.; Tuveson, D.A. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Brembeck, F.H.; Schreiber, F.S.; Deramaudt, T.B.; Craig, L.; Rhoades, B.; Swain, G.; Grippo, P.; Stoffers, D.A.; Silberg, D.G.; Rustgi, A.K. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003, 63, 2005–2009. [Google Scholar]
- Luttges, J.; Schlehe, B.; Menke, M.A.; Vogel, I.; Henne-Bruns, D.; Kloppel, G. The K-ras mutation pattern in pancreatic ductal adenocarcinoma usually is identical to that in associated normal, hyperplastic, and metaplastic ductal epithelium. Cancer 1999, 85, 1703–1710. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with "K-Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Wellner, U.; Brabletz, T.; Keck, T. ZEB1 in Pancreatic Cancer. Cancers 2010, 2, 1617–1628. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; Brunton, V.G.; Morton, J.; Sansom, O.; Schuler, J.; Stemmler, M.P.; Herzberger, C.; Hopt, U.; Keck, T.; Brabletz, S.; Brabletz, T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Imamichi, Y.; Konig, A.; Gress, T.; Menke, A. Collagen type I-induced Smad-interacting protein 1 expression downregulates E-cadherin in pancreatic cancer. Oncogene 2007, 26, 2381–2385. [Google Scholar] [CrossRef]
- Schaeffer, D.F.; Assi, K.; Chan, K.; Buczkowski, A.K.; Chung, S.W.; Scudamore, C.H.; Weiss, A.; Salh, B.; Owen, D.A. Tumor expression of integrin-linked kinase (ILK) correlates with the expression of the E-cadherin repressor snail: An immunohistochemical study in ductal pancreatic adenocarcinoma. Virchows Arch. 2010, 456, 261–268. [Google Scholar] [CrossRef]
- Nishioka, R.; Itoh, S.; Gui, T.; Gai, Z.; Oikawa, K.; Kawai, M.; Tani, M.; Yamaue, H.; Muragaki, Y. SNAIL induces epithelial-to-mesenchymal transition in a human pancreatic cancer cell line (BxPC3) and promotes distant metastasis and invasiveness in vivo. Exp. Mol. Pathol. 2010, 89, 149–157. [Google Scholar] [CrossRef]
- Yin, T.; Wang, C.; Liu, T.; Zhao, G.; Zha, Y.; Yang, M. Expression of snail in pancreatic cancer promotes metastasis and chemoresistance. J. Surg. Res. 2007, 141, 196–203. [Google Scholar] [CrossRef]
- Cates, J.M.; Byrd, R.H.; Fohn, L.E.; Tatsas, A.D.; Washington, M.K.; Black, C.C. Epithelial-mesenchymal transition markers in pancreatic ductal adenocarcinoma. Pancreas 2009, 38, e1–6. [Google Scholar] [CrossRef]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.J.; Hotz, H.G. Epithelial to mesenchymal transition: Expression of the regulators snail, slug, and twist in pancreatic cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef]
- Ohuchida, K.; Mizumoto, K.; Ohhashi, S.; Yamaguchi, H.; Konomi, H.; Nagai, E.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M. Twist, a novel oncogene, is upregulated in pancreatic cancer: Clinical implication of Twist expression in pancreatic juice. Int. J. Cancer 2007, 120, 1634–1640. [Google Scholar] [CrossRef]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Invest. 2004, 114, 569–581. [Google Scholar]
- Huber, M.A.; Beug, H.; Wirth, T. Epithelial-mesenchymal transition: NF-kappaB takes center stage. Cell Cycle 2004, 3, 1477–1480. [Google Scholar] [CrossRef]
- Fujioka, S.; Sclabas, G.M.; Schmidt, C.; Frederick, W.A.; Dong, Q.G.; Abbruzzese, J.L.; Evans, D.B.; Baker, C.; Chiao, P.J. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin. Cancer Res. 2003, 9, 346–354. [Google Scholar]
- Huber, M.A.; Maier, H.J.; Alacakaptan, M.; Wiedemann, E.; Braunger, J.; Boehmelt, G.; Madwed, J.B.; Young, E.R.R.; Marshall, D.R.; Pehamberger, H.; Wirth, T.; Kraut, N.; Beug, H. BI 5700, a selective chemical inhibitor of IkappaB kinase 2, specifically suppresses epithelial-mesenchymal transition and metastasis in mouse models of tumor progression. Genes Cancer 2010, 1, 101–114. [Google Scholar] [CrossRef]
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2. Oncogene 2007, 26, 711–724. [Google Scholar] [CrossRef]
- Hidalgo, M.; Maitra, A. The hedgehog pathway and pancreatic cancer. N. Engl. J. Med. 2009, 361, 2094–2096. [Google Scholar] [CrossRef]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; Gabrielson, K.L.; Matsui, W.; Maitra, A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- Dhar, G.; Mehta, S.; Banerjee, S.; Gardner, A.; McCarty, B.M.; Mathur, S.C.; Campbell, D.R.; Kambhampati, S.; Banerjee, S.K. Loss of WISP-2/CCN5 signaling in human pancreatic cancer: A potential mechanism for epithelial-mesenchymal-transition. Cancer Lett. 2007, 254, 63–70. [Google Scholar] [CrossRef]
- Ripka, S.; Konig, A.; Buchholz, M.; Wagner, M.; Sipos, B.; Kloppel, G.; Downward, J.; Gress, T.; Michl, P. WNT5A--target of CUTL1 and potent modulator of tumor cell migration and invasion in pancreatic cancer. Carcinogenesis 2007, 28, 1178–1187. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef]
- Song, Y.; Washington, M.K.; Crawford, H.C. Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res. 2010, 70, 2115–2125. [Google Scholar] [CrossRef]
- Berx, G.; van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef]
- Seidel, B.; Braeg, S.; Adler, G.; Wedlich, D.; Menke, A. E- and N-cadherin differ with respect to their associated p120ctn isoforms and their ability to suppress invasive growth in pancreatic cancer cells. Oncogene 2004, 23, 5532–5542. [Google Scholar] [CrossRef]
- von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; Schnieke, A.; Schmid, R.M.; Schneider, G.; Saur, D. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361-371, e361-365. [Google Scholar] [CrossRef]
- Nakajima, S.; Doi, R.; Toyoda, E.; Tsuji, S.; Wada, M.; Koizumi, M.; Tulachan, S.S.; Ito, D.; Kami, K.; Mori, T.; Kawaguchi, Y.; Fujimoto, K.; Hosotani, R.; Imamura, M. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res. 2004, 10, 4125–4133. [Google Scholar] [CrossRef]
- Schreiber, S.C.; Giehl, K.; Kastilan, C.; Hasel, C.; Muhlenhoff, M.; Adler, G.; Wedlich, D.; Menke, A. Polysialylated NCAM represses E-cadherin-mediated cell-cell adhesion in pancreatic tumor cells. Gastroenterology 2008, 134, 1555–1566. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Habisch, H.; Zhou, S.; Siech, M.; Bachem, M.G. Interaction of Stellate Cells with Pancreatic Carcinoma Cells. Cancers 2010, 2, 1661–1682. [Google Scholar] [CrossRef]
- Menke, A.; Philippi, C.; Vogelmann, R.; Seidel, B.; Lutz, M.P.; Adler, G.; Wedlich, D. Down-regulation of E-cadherin gene expression by collagen type I and type III in pancreatic cancer cell lines. Cancer Res. 2001, 61, 3508–3517. [Google Scholar]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef]
- Shintani, Y.; Hollingsworth, M.A.; Wheelock, M.J.; Johnson, K.R. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 2006, 66, 11745–11753. [Google Scholar] [CrossRef]
- Imamichi, Y.; Menke, A. Signaling pathways involved in collagen-induced disruption of the E-cadherin complex during epithelial-mesenchymal transition. Cells Tissues Organs 2007, 185, 180–190. [Google Scholar] [CrossRef]
- Shintani, Y.; Fukumoto, Y.; Chaika, N.; Svoboda, R.; Wheelock, M.J.; Johnson, K.R. Collagen I-mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J. Cell Biol. 2008, 180, 1277–1289. [Google Scholar] [CrossRef]
- Orlichenko, L.S.; Radisky, D.C. Matrix metalloproteinases stimulate epithelial-mesenchymal transition during tumor development. Clin. Exp. Metastasis 2008, 25, 593–600. [Google Scholar] [CrossRef]
- Cheng, H.; Fukushima, T.; Takahashi, N.; Tanaka, H.; Kataoka, H. Hepatocyte growth factor activator inhibitor type 1 regulates epithelial to mesenchymal transition through membrane-bound serine proteinases. Cancer Res. 2009, 69, 1828–1835. [Google Scholar] [CrossRef]
- Wang, F.; Sloss, C.; Zhang, X.; Lee, S.W.; Cusack, J.C. Membrane-bound heparin-binding epidermal growth factor like growth factor regulates E-cadherin expression in pancreatic carcinoma cells. Cancer Res. 2007, 67, 8486–8493. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Lai, M. The microRNA network and tumor metastasis. Oncogene 29, 937–948. [CrossRef]
- Kent, O.A.; Mullendore, M.; Wentzel, E.A.; Lopez-Romero, P.; Tan, A.C.; Alvarez, H.; West, K.; Ochs, M.F.; Hidalgo, M.; Arking, D.E.; Maitra, A.; Mendell, J.T. A resource for analysis of microRNA expression and function in pancreatic ductal adenocarcinoma cells. Cancer Biol. Ther. 2009, 8, 2013–2024. [Google Scholar] [CrossRef]
- Li, A.; Omura, N.; Hong, S.M.; Vincent, A.; Walter, K.; Griffith, M.; Borges, M.; Goggins, M. Pancreatic Cancers Epigenetically Silence SIP1 and Hypomethylate and Overexpress miR-200a/200b in Association with Elevated Circulating miR-200a and miR-200b Levels. Cancer Res. 2010, 70, 5226–5237. [Google Scholar] [CrossRef]
- Li, Y.; VandenBoom, T.G., 2nd; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef]
- Abe, N.; Watanabe, T.; Suzuki, Y.; Matsumoto, N.; Masaki, T.; Mori, T.; Sugiyama, M.; Chiappetta, G.; Fusco, A.; Atomi, Y. An increased high-mobility group A2 expression level is associated with malignant phenotype in pancreatic exocrine tissue. Br. J. Cancer 2003, 89, 2104–2109. [Google Scholar] [CrossRef]
- Watanabe, S.; Ueda, Y.; Akaboshi, S.; Hino, Y.; Sekita, Y.; Nakao, M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am. J. Pathol. 2009, 174, 854–868. [Google Scholar] [CrossRef]
- Tarin, D.; Thompson, E.W.; Newgreen, D.F. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005, 65, 5996–6000. [Google Scholar] [CrossRef]
- Thompson, E.W.; Newgreen, D.F.; Tarin, D. Carcinoma invasion and metastasis: A role for epithelial-mesenchymal transition? Cancer Res. 2005, 65, 5991–5995, discussion 5995. [Google Scholar] [CrossRef]
- Waerner, T.; Alacakaptan, M.; Tamir, I.; Oberauer, R.; Gal, A.; Brabletz, T.; Schreiber, M.; Jechlinger, M.; Beug, H. ILEI: A cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 2006, 10, 227–239. [Google Scholar] [CrossRef]
- Moody, S.E.; Perez, D.; Pan, T.C.; Sarkisian, C.J.; Portocarrero, C.P.; Sterner, C.J.; Notorfrancesco, K.L.; Cardiff, R.D.; Chodosh, L.A. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005, 8, 197–209. [Google Scholar] [CrossRef]
- Blanco, M.J.; Moreno-Bueno, G.; Sarrio, D.; Locascio, A.; Cano, A.; Palacios, J.; Nieto, M.A. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002, 21, 3241–3246. [Google Scholar] [CrossRef]
- Elloul, S.; Elstrand, M.B.; Nesland, J.M.; Trope, C.G.; Kvalheim, G.; Goldberg, I.; Reich, R.; Davidson, B. Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer 2005, 103, 1631–1643. [Google Scholar] [CrossRef]
- Franci, C.; Takkunen, M.; Dave, N.; Alameda, F.; Gomez, S.; Rodriguez, R.; Escriva, M.; Montserrat-Sentis, B.; Baro, T.; Garrido, M.; Bonilla, F.; Virtanen, I.; Garcia de Herreros, A. Expression of Snail protein in tumor-stroma interface. Oncogene 2006, 25, 5134–5144. [Google Scholar]
- Natalwala, A.; Spychal, R.; Tselepis, C. Epithelial-mesenchymal transition mediated tumourigenesis in the gastrointestinal tract. World J. Gastroenterol. 2008, 14, 3792–3797. [Google Scholar] [CrossRef]
- Javle, M.M.; Gibbs, J.F.; Iwata, K.K.; Pak, Y.; Rutledge, P.; Yu, J.; Black, J.D.; Tan, D.; Khoury, T. Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Ann. Surg. Oncol. 2007, 14, 3527–3533. [Google Scholar] [CrossRef]
- Shin, S.J.; Kim, K.O.; Kim, M.K.; Lee, K.H.; Hyun, M.S.; Kim, K.J.; Choi, J.H.; Song, H.S. Expression of E-cadherin and uPA and their association with the prognosis of pancreatic cancer. Jpn. J. Clin. Oncol. 2005, 35, 342–348. [Google Scholar] [CrossRef]
- Tomita, K.; van Bokhoven, A.; van Leenders, G.J.; Ruijter, E.T.; Jansen, C.F.; Bussemakers, M.J.; Schalken, J.A. Cadherin switching in human prostate cancer progression. Cancer Res. 2000, 60, 3650–3654. [Google Scholar]
- Cannito, S.; Novo, E.; Compagnone, A.; Valfre di Bonzo, L.; Busletta, C.; Zamara, E.; Paternostro, C.; Povero, D.; Bandino, A.; Bozzo, F.; Cravanzola, C.; Bravoco, V.; Colombatto, S.; Parola, M. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 2008, 29, 2267–2278. [Google Scholar] [CrossRef]
- Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 101, 293–299.
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef]
- Masugi, Y.; Yamazaki, K.; Hibi, T.; Aiura, K.; Kitagawa, Y.; Sakamoto, M. Solitary cell infiltration is a novel indicator of poor prognosis and epithelial-mesenchymal transition in pancreatic cancer. Hum. Pathol. 2010, 41, 1061–1068. [Google Scholar] [CrossRef]
- Wicki, A.; Lehembre, F.; Wick, N.; Hantusch, B.; Kerjaschki, D.; Christofori, G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006, 9, 261–272. [Google Scholar] [CrossRef]
- Tsuji, T.; Ibaragi, S.; Hu, G.F. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009, 69, 7135–7139. [Google Scholar] [CrossRef]
- Maschler, S.; Gebeshuber, C.A.; Wiedemann, E.M.; Alacakaptan, M.; Schreiber, M.; Custic, I.; Beug, H. Annexin A1 attenuates EMT and metastatic potential in breast cancer. EMBO Mol. Med. 2010, 2, 401–414. [Google Scholar] [CrossRef]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; Campbell, L.L.; Polyak, K.; Brisken, C.; Yang, J.; Weinberg, R.A. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Radisky, D.C.; LaBarge, M.A. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2008, 2, 511–512. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar]
- Jung, A.; Brabletz, T.; Kirchner, T. The migrating cancer stem cells model--a conceptual explanation of malignant tumour progression. Ernst Schering Found. Symp. Proc. 2006, 109–124. [Google Scholar]
- Sarkar, F.H.; Li, Y.; Wang, Z.; Kong, D. Pancreatic cancer stem cells and EMT in drug resistance and metastasis. Minerva Chir 2009, 64, 489–500. [Google Scholar]
- Cano, C.E.; Motoo, Y.; Iovanna, J.L. Epithelial-to-Mesenchymal Transition in Pancreatic Adenocarcinoma. Sci. World J. 2010, 10, 1947–1957. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; Sorrentino, B.P. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef]
- Kabashima, A.; Higuchi, H.; Takaishi, H.; Matsuzaki, Y.; Suzuki, S.; Izumiya, M.; Iizuka, H.; Sakai, G.; Hozawa, S.; Azuma, T.; Hibi, T. Side population of pancreatic cancer cells predominates in TGF-beta-mediated epithelial to mesenchymal transition and invasion. Int. J. Cancer 2009, 124, 2771–2779. [Google Scholar] [CrossRef]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef]
- Ruckert, F.; Joensson, P.; Saeger, H.D.; Grutzmann, R.; Pilarsky, C. Functional analysis of LOXL2 in pancreatic carcinoma. Int. J. Colorectal Dis. 2010, 25, 303–311. [Google Scholar] [CrossRef]
- Hamada, S.; Satoh, K.; Kimura, K.; Kanno, A.; Masamune, A.; Shimosegawa, T. MSX2 overexpression inhibits gemcitabine-induced caspase-3 activity in pancreatic cancer cells. World J. Gastroenterol. 2005, 11, 6867–6870. [Google Scholar]
- Buck, E.; Eyzaguirre, A.; Barr, S.; Thompson, S.; Sennello, R.; Young, D.; Iwata, K.K.; Gibson, N.W.; Cagnoni, P.; Haley, J.D. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol. Cancer Ther. 2007, 6, 532–541. [Google Scholar] [CrossRef]
- Barr, S.; Thomson, S.; Buck, E.; Russo, S.; Petti, F.; Sujka-Kwok, I.; Eyzaguirre, A.; Rosenfeld-Franklin, M.; Gibson, N.W.; Miglarese, M.; Epstein, D.; Iwata, K.K.; Haley, J.D. Bypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitions. Clin. Exp. Metastasis 2008, 25, 685–693. [Google Scholar] [CrossRef]
- Pino, M.S.; Balsamo, M.; Di Modugno, F.; Mottolese, M.; Alessio, M.; Melucci, E.; Milella, M.; McConkey, D.J.; Philippar, U.; Gertler, F.B.; Natali, P.G.; Nistico, P. Human Mena+11a isoform serves as a marker of epithelial phenotype and sensitivity to epidermal growth factor receptor inhibition in human pancreatic cancer cell lines. Clin. Cancer Res. 2008, 14, 4943–4950. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Beug, H. Breast cancer stem cells: Eradication by differentiation therapy? Cell 2009, 138, 623–625. [Google Scholar] [CrossRef]
- Nelson, W.J. Remodeling epithelial cell organization: Transitions between front-rear and apical-basal polarity. Cold Spring Harb. Perspect. Biol. 2009, 1, a000513. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maier, H.J.; Wirth, T.; Beug, H. Epithelial-Mesenchymal Transition in Pancreatic Carcinoma. Cancers 2010, 2, 2058-2083. https://doi.org/10.3390/cancers2042058
Maier HJ, Wirth T, Beug H. Epithelial-Mesenchymal Transition in Pancreatic Carcinoma. Cancers. 2010; 2(4):2058-2083. https://doi.org/10.3390/cancers2042058
Chicago/Turabian StyleMaier, Harald J., Thomas Wirth, and Hartmut Beug. 2010. "Epithelial-Mesenchymal Transition in Pancreatic Carcinoma" Cancers 2, no. 4: 2058-2083. https://doi.org/10.3390/cancers2042058
APA StyleMaier, H. J., Wirth, T., & Beug, H. (2010). Epithelial-Mesenchymal Transition in Pancreatic Carcinoma. Cancers, 2(4), 2058-2083. https://doi.org/10.3390/cancers2042058