Epigenetics and Colorectal Cancer Pathogenesis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Colorectal Cancer
2. Genetic, Microenvironment and Epigenetic Regulation of CRC Pathogenesis
3. Genetic Regulation of CRC Development
4. Colon Anatomy and Microenvironment
5. CRC Epigenetics
6. DNA Hypermethylation

7. Characteristics of DNA Hypermethylation in CRC
8. DNA Hypomethylation and CRC Development
9. Histone Modifications and CRC Development
10. microRNAs as Epigenetic Regulators in CRC
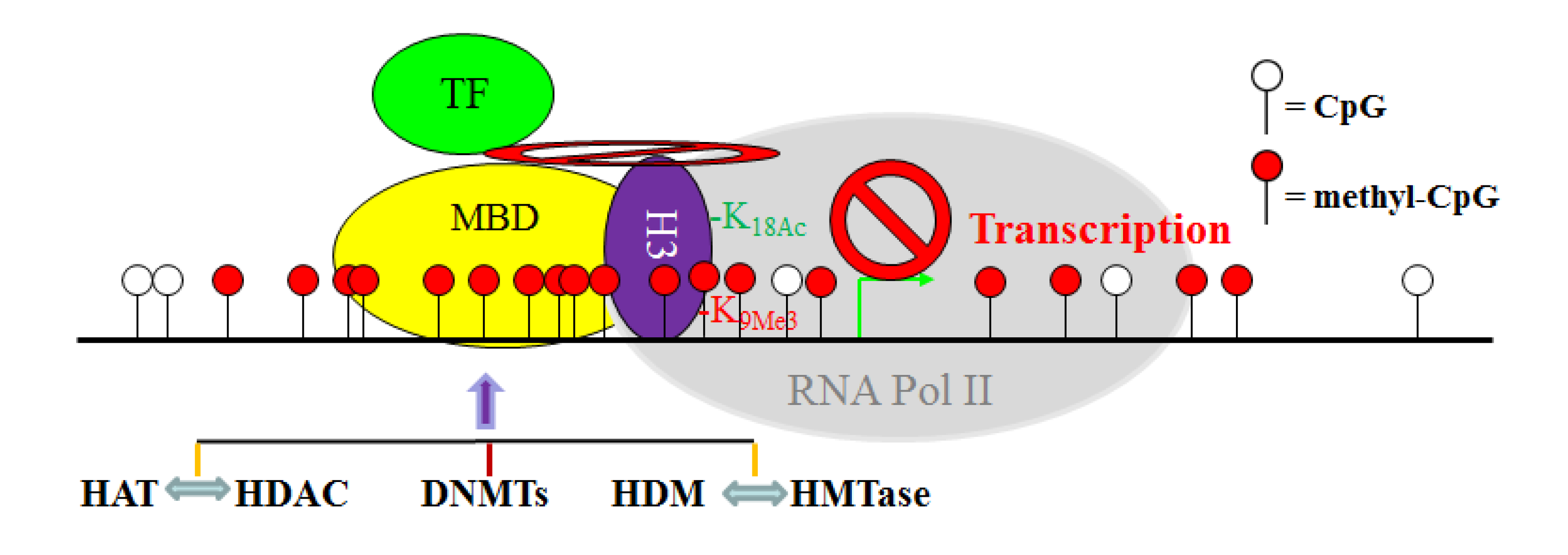
11. The Crosstalk Network among DNMT, HAT, HDAC, HMTase and HDM: A Complex Network

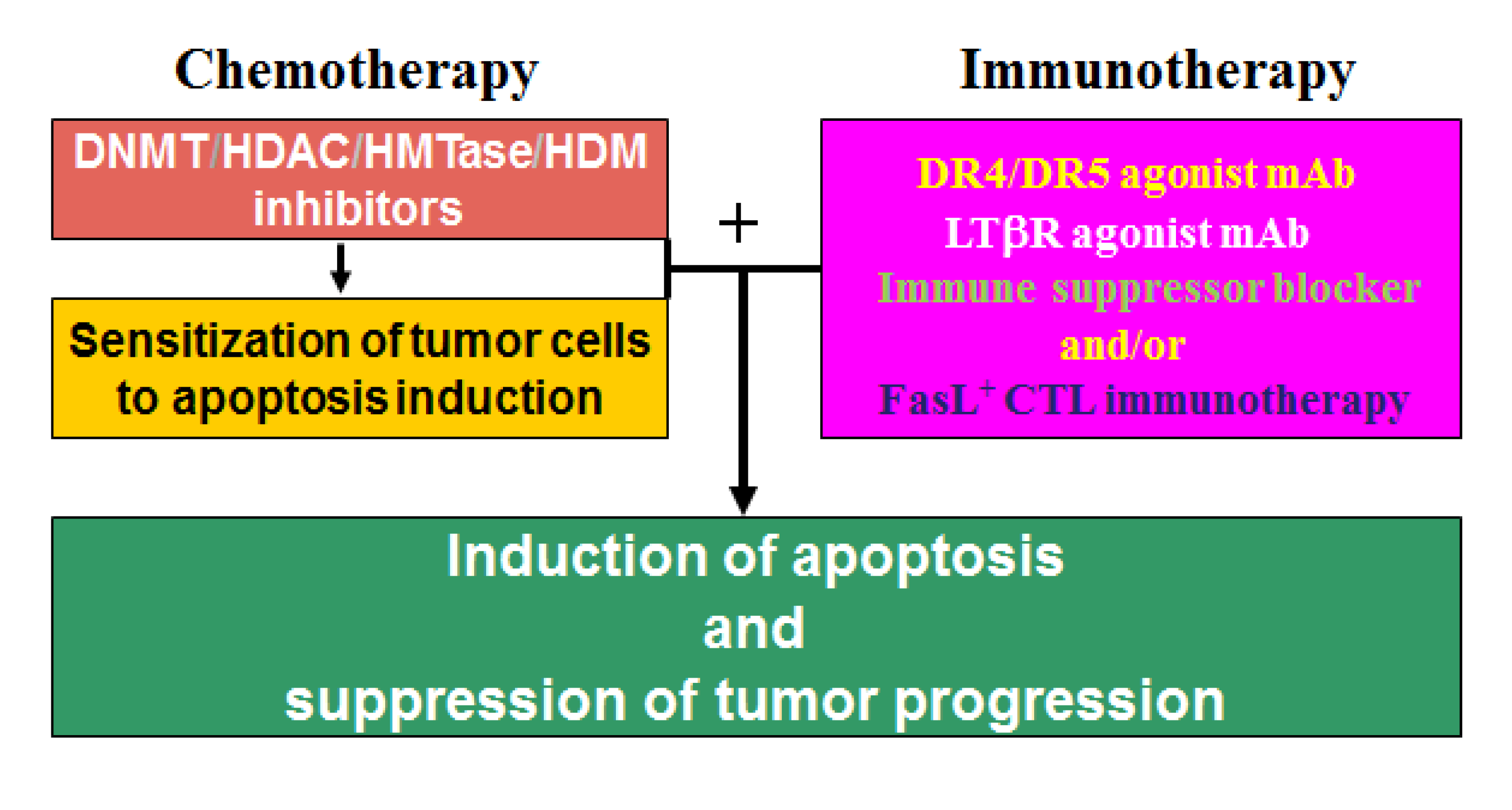
12. Epigenetic Therapy

13. Epigenetic Regulation of Chemosistance in CRC
14. Conclusions
Acknowledgements
References
- Kopetz, S.; Chang, G.J.; Overman, M.J.; Eng, C.; Sargent, D.J.; Larson, D.W.; Grothey, A.; Vauthey, J.N.; Nagorney, D.M.; McWilliams, R.R. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J. Clin. Oncol. 2009, 27, 3677–3683. [Google Scholar] [CrossRef]
- Rustgi, A.K. The genetics of hereditary colon cancer. Genes Dev. 2007, 21, 2525–2538. [Google Scholar] [CrossRef]
- Yang, K.; Kurihara, N.; Fan, K.; Newmark, H.; Rigas, B.; Bancroft, L.; Corner, G.; Livote, E.; Lesser, M.; Edelmann, W.; et al. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res. 2008, 68, 7803–7810. [Google Scholar] [CrossRef]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar]
- Chen, L.W.; Egan, L.; Li, Z.W.; Greten, F.R.; Kagnoff, M.F.; Karin, M. The two faces of IKK and NF-kappaB inhibition: Prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 2003, 9, 575–581. [Google Scholar] [CrossRef]
- Hanada, T.; Kobayashi, T.; Chinen, T.; Saeki, K.; Takaki, H.; Koga, K.; Minoda, Y.; Sanada, T.; Yoshioka, T.; Mimata, H.; et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J. Exp. Med. 2006, 203, 1391–1397. [Google Scholar] [CrossRef]
- Feagins, L.A.; Souza, R.F.; Spechler, S.J. Carcinogenesis in IBD: Potential targets for the prevention of colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 297–305. [Google Scholar] [CrossRef]
- Loeb, L.A. Cancer cells exhibit a mutator phenotype. Adv. Cancer Res. 1998, 72, 25–56. [Google Scholar]
- Klein, G. Foulds’ dangerous idea revisited: The multistep development of tumors 40 years later. Adv. Cancer Res. 1998, 72, 1–23. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Goldstein, N.S. Serrated pathway and APC (conventional)-type colorectal polyps: Molecular-morphologic correlations, genetic pathways, and implications for classification. Am. J. Clin. Pathol. 2006, 125, 146–153. [Google Scholar]
- DePinho, R.A. The age of cancer. Nature 2000, 408, 248–254. [Google Scholar]
- Noffsinger, A.E. Serrated polyps and colorectal cancer: New pathway to malignancy. Annu. Rev. Pathol. 2009, 4, 343–364. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Smigal, C.; Thun, M.J. Cancer statistics, 2006. CA Cancer J. Clin. 2006, 56, 106–130. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci.USA 1999, 96, 8681–8686. [Google Scholar]
- Cummins, J.M.; He, Y.; Leary, R.J.; Pagliarini, R.; Diaz, L.A., Jr.; Sjoblom, T.; Barad, O.; Bentwich, Z.; Szafranska, A.E.; Labourier, E.; et al. The colorectal microRNAome. Proc. Natl. Acad. Sci. USA 2006, 103, 3687–3692. [Google Scholar]
- Van Engeland, M.; Derks, S.; Smits, K.M.; Meijer, G.A.; Herman, J.G. Colorectal cancer epigenetics: Complex simplicity. J. Clin. Oncol. 2011, 29, 1382–1391. [Google Scholar]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Jass, J.R. Molecular heterogeneity of colorectal cancer: Implications for cancer control. Surg. Oncol. 2007, 16, S7–S9. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Vazquez, A.; Bond, E.E.; Levine, A.J.; Bond, G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 979–987. [Google Scholar]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Eppert, K.; Scherer, S.W.; Ozcelik, H.; Pirone, R.; Hoodless, P.; Kim, H.; Tsui, L.C.; Bapat, B.; Gallinger, S.; Andrulis, I.L.; et al. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 1996, 86, 543–552. [Google Scholar] [CrossRef]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998, 92, 645–656. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Albertsen, H.; Herrick, J.; Levin, T.R.; Sweeney, C.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology 2005, 129, 837–845. [Google Scholar] [CrossRef]
- Nosho, K.; Irahara, N.; Shima, K.; Kure, S.; Kirkner, G.J.; Schernhammer, E.S.; Hazra, A.; Hunter, D.J.; Quackenbush, J.; Spiegelman, D.; et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One 2008, 3, e3698. [Google Scholar] [CrossRef]
- Soreide, K.; Janssen, E.A.; Soiland, H.; Korner, H.; Baak, J.P. Microsatellite instability in colorectal cancer. Br. J. Surg. 2006, 93, 395–406. [Google Scholar] [CrossRef]
- Hermsen, M.; Postma, C.; Baak, J.; Weiss, M.; Rapallo, A.; Sciutto, A.; Roemen, G.; Arends, J.W.; Williams, R.; Giaretti, W.; et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology 2002, 123, 1109–1119. [Google Scholar] [CrossRef]
- Douglas, E.J.; Fiegler, H.; Rowan, A.; Halford, S.; Bicknell, D.C.; Bodmer, W.; Tomlinson, I.P.; Carter, N.P. Array comparative genomic hybridization analysis of colorectal cancer cell lines and primary carcinomas. Cancer Res. 2004, 64, 4817–4825. [Google Scholar]
- Martin, E.S.; Tonon, G.; Sinha, R.; Xiao, Y.; Feng, B.; Kimmelman, A.C.; Protopopov, A.; Ivanova, E.; Brennan, C.; Montgomery, K.; et al. Common and distinct genomic events in sporadic colorectal cancer and diverse cancer types. Cancer Res. 2007, 67, 10736–10743. [Google Scholar]
- Yamauchi, M.; Lochhead, P.; Morikawa, T.; Huttenhower, C.; Chan, A.T.; Giovannucci, E.; Fuchs, C.; Ogino, S. Colorectal cancer: A tale of two sides or a continuum? Gut 2012, 61, 794–797. [Google Scholar]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar]
- Pritchard, C.C.; Grady, W.M. Colorectal cancer molecular biology moves into clinical practice. Gut 2011, 60, 116–129. [Google Scholar]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar]
- Tanaka, N.; Huttenhower, C.; Nosho, K.; Baba, Y.; Shima, K.; Quackenbush, J.; Haigis, K.M.; Giovannucci, E.; Fuchs, C.S.; Ogino, S. Novel application of structural equation modeling to correlation structure analysis of CpG island methylation in colorectal cancer. Am. J. Pathol. 2010, 177, 2731–2740. [Google Scholar] [CrossRef]
- Zlobec, I.; Bihl, M.; Foerster, A.; Rufle, A.; Lugli, A. Comprehensive analysis of CpG island methylator phenotype (CIMP)-high, -low, and -negative colorectal cancers based on protein marker expression and molecular features. J. Pathol. 2011, 225, 336–343. [Google Scholar] [CrossRef]
- Ogino, S.; Shima, K.; Meyerhardt, J.A.; McCleary, N.J.; Ng, K.; Hollis, D.; Saltz, L.B.; Mayer, R.J.; Schaefer, P.; Whittom, R.; et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: Results from intergroup trial CALGB 89803. Clin. Cancer Res. 2012, 18, 890–900. [Google Scholar] [CrossRef]
- LaPointe, L.C.; Dunne, R.; Brown, G.S.; Worthley, D.L.; Molloy, P.L.; Wattchow, D.; Young, G.P. Map of differential transcript expression in the normal human large intestine. Physiol. Genomics 2008, 33, 50–64. [Google Scholar] [CrossRef]
- Campbell, P.T.; Jacobs, E.T.; Ulrich, C.M.; Figueiredo, J.C.; Poynter, J.N.; McLaughlin, J.R.; Haile, R.W.; Jacobs, E.J.; Newcomb, P.A.; Potter, J.D.; et al. Case-control study of overweight, obesity, and colorectal cancer risk, overall and by tumor microsatellite instability status. J. Natl. Cancer Inst. 2010, 102, 391–400. [Google Scholar] [CrossRef]
- Slattery, M.L.; Curtin, K.; Anderson, K.; Ma, K.N.; Ballard, L.; Edwards, S.; Schaffer, D.; Potter, J.; Leppert, M.; Samowitz, W.S. Associations between cigarette smoking, lifestyle factors, and microsatellite instability in colon tumors. J. Natl. Cancer Inst. 2000, 92, 1831–1836. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Karlsson, M.; Marits, P.; Dahl, K.; Dagoo, T.; Enerback, S.; Thorn, M.; Winqvist, O. Pilot study of sentinel-node-based adoptive immunotherapy in advanced colorectal cancer. Ann. Surg. Oncol. 2010, 17, 1747–1757. [Google Scholar] [CrossRef]
- Hu, X.; Zimmerman, A.M.; Bardhan, K.; Yang, D.; Waller, J.L.; Liles, G.B.; Lee, J.R.; Pollock, R.; Lev, D.; Ware, C.F.; et al. Lymphotoxin beta receptor mediates caspase-dependent tumor cell apoptosis in vitro and tumor suppression in vivo despite induction of NF-kappaB activation. Carcinogenesis 2013. [Google Scholar] [PubMed]
- Liu, F.; Bardhan, K.; Yang, D.; Thangaraju, M.; Ganapathy, V.; Waller, J.L.; Liles, G.B.; Lee, J.R.; Liu, K. NF-kappaB directly regulates Fas transcription to modulate Fas-mediated apoptosis and tumor suppression. J. Biol. Chem. 2012, 287, 25530–25540. [Google Scholar]
- Zimmerman, M.A.; Rahman, N.T.; Yang, D.; Lahat, G.; Lazar, A.J.; Pollock, R.E.; Lev, D.; Liu, K. Unphosphorylated STAT1 promotes sarcoma development through repressing expression of Fas and bad and conferring apoptotic resistance. Cancer Res. 2012, 72, 4724–4732. [Google Scholar]
- Zimmerman, M.A.; Singh, N.; Martin, P.M.; Thangaraju, M.; Ganapathy, V.; Waller, J.L.; Shi, H.; Robertson, K.D.; Munn, D.H.; Liu, K. Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1405–1415. [Google Scholar]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar]
- Pages, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Chiba, T.; Ohtani, H.; Mizoi, T.; Naito, Y.; Sato, E.; Nagura, H.; Ohuchi, A.; Ohuchi, K.; Shiiba, K.; Kurokawa, Y.; et al. Intraepithelial CD8+ T-cell-count becomes a prognostic factor after a longer follow-up period in human colorectal carcinoma: Possible association with suppression of micrometastasis. Br. J. Cancer 2004, 91, 1711–1717. [Google Scholar]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Zlobec, I.; Terracciano, L.M.; Lugli, A. Local recurrence in mismatch repair-proficient colon cancer predicted by an infiltrative tumor border and lack of CD8+ tumor-infiltrating lymphocytes. Clin. Cancer Res. 2008, 14, 3792–3797. [Google Scholar] [CrossRef]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar]
- George, S.; Primrose, J.; Talbot, R.; Smith, J.; Mullee, M.; Bailey, D.; du Boulay, C.; Jordan, H. Will Rogers revisited: Prospective observational study of survival of 3592 patients with colorectal cancer according to number of nodes examined by pathologists. Br. J. Cancer 2006, 95, 841–847. [Google Scholar] [CrossRef]
- Goldstein, N.S. Lymph node recoveries from 2427 pT3 colorectal resection specimens spanning 45 years: Recommendations for a minimum number of recovered lymph nodes based on predictive probabilities. Am. J. Surg. Pathol. 2002, 26, 179–189. [Google Scholar] [CrossRef]
- Le Voyer, T.E.; Sigurdson, E.R.; Hanlon, A.L.; Mayer, R.J.; Macdonald, J.S.; Catalano, P.J.; Haller, D.G. Colon cancer survival is associated with increasing number of lymph nodes analyzed: A secondary survey of intergroup trial INT-0089. J. Clin. Oncol. 2003, 21, 2912–2919. [Google Scholar] [CrossRef]
- Baxter, N.N.; Virnig, D.J.; Rothenberger, D.A.; Morris, A.M.; Jessurun, J.; Virnig, B.A. Lymph node evaluation in colorectal cancer patients: A population-based study. J. Natl. Cancer Inst. 2005, 97, 219–225. [Google Scholar] [CrossRef]
- Bui, L.; Rempel, E.; Reeson, D.; Simunovic, M. Lymph node counts, rates of positive lymph nodes, and patient survival for colon cancer surgery in Ontario, Canada: A population-based study. J. Surg. Oncol. 2006, 93, 439–445. [Google Scholar] [CrossRef]
- Chang, G.J.; Rodriguez-Bigas, M.A.; Skibber, J.M.; Moyer, V.A. Lymph node evaluation and survival after curative resection of colon cancer: Systematic review. J. Natl. Cancer Inst. 2007, 99, 433–441. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Irahara, N.; Meyerhardt, J.A.; Baba, Y.; Shima, K.; Glickman, J.N.; Ferrone, C.R.; Mino-Kenudson, M.; Tanaka, N.; et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin. Cancer Res. 2009, 15, 6412–6420. [Google Scholar] [CrossRef]
- Nosho, K.; Baba, Y.; Tanaka, N.; Shima, K.; Hayashi, M.; Meyerhardt, J.A.; Giovannucci, E.; Dranoff, G.; Fuchs, C.S.; Ogino, S. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: Cohort study and literature review. J. Pathol. 2010, 222, 350–366. [Google Scholar] [CrossRef]
- Michael-Robinson, J.M.; Biemer-Huttmann, A.; Purdie, D.M.; Walsh, M.D.; Simms, L.A.; Biden, K.G.; Young, J.P.; Leggett, B.A.; Jass, J.R.; Radford-Smith, G.L. Tumour infiltrating lymphocytes and apoptosis are independent features in colorectal cancer stratified according to microsatellite instability status. Gut 2001, 48, 360–366. [Google Scholar] [CrossRef]
- Guidoboni, M.; Gafa, R.; Viel, A.; Doglioni, C.; Russo, A.; Santini, A.; Del Tin, L.; Macri, E.; Lanza, G.; Boiocchi, M.; et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am. J. Pathol. 2001, 159, 297–304. [Google Scholar] [CrossRef]
- Menon, A.G.; Janssen-van Rhijn, C.M.; Morreau, H.; Putter, H.; Tollenaar, R.A.; van de Velde, C.J.; Fleuren, G.J.; Kuppen, P.J. Immune system and prognosis in colorectal cancer: A detailed immunohistochemical analysis. Lab. Invest. 2004, 84, 493–501. [Google Scholar]
- Ogino, S.; Odze, R.D.; Kawasaki, T.; Brahmandam, M.; Kirkner, G.J.; Laird, P.W.; Loda, M.; Fuchs, C.S. Correlation of pathologic features with CpG island methylator phenotype (CIMP) by quantitative DNA methylation analysis in colorectal carcinoma. Am. J. Surg. Pathol. 2006, 30, 1175–1183. [Google Scholar]
- Dahlin, A.M.; Henriksson, M.L.; van Guelpen, B.; Stenling, R.; Oberg, A.; Rutegard, J.; Palmqvist, R. Colorectal cancer prognosis depends on T-cell infiltration and molecular characteristics of the tumor. Mod. Pathol. 2011, 24, 671–682. [Google Scholar]
- Ogino, S.; Galon, J.; Fuchs, C.S.; Dranoff, G. Cancer immunology—Analysis of host and tumor factors for personalized medicine. Nat. Rev. Clin. Oncol. 2011, 8, 711–719. [Google Scholar]
- Galon, J.; Pages, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer classification using the Immunoscore: A worldwide task force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Goel, A.; Xicola, R.M.; Nguyen, T.P.; Doyle, B.J.; Sohn, V.R.; Bandipalliam, P.; Rozek, L.S.; Reyes, J.; Cordero, C.; Balaguer, F.; et al. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology 2010, 138, 1854–1862. [Google Scholar] [CrossRef]
- Migheli, F.; Migliore, L. Epigenetics of colorectal cancer. Clin. Genet. 2012, 81, 312–318. [Google Scholar] [CrossRef]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Akhtar-Zaidi, B.; Cowper-Sal-lari, R.; Corradin, O.; Saiakhova, A.; Bartels, C.F.; Balasubramanian, D.; Myeroff, L.; Lutterbaugh, J.; Jarrar, A.; Kalady, M.F.; Willis, J.; et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science 2012, 336, 736–739. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Kondo, Y.; Issa, J.P. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 29–39. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Singer-Sam, J.; Riggs, A.D. X chromosome inactivation and DNA methylation. EXS 1993, 64, 358–384. [Google Scholar]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef]
- Esteller, M.; Herman, J.G. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J. Pathol. 2002, 196, 1–7. [Google Scholar] [CrossRef]
- Wu, C.; Bekaii-Saab, T. CpG Island Methylation, Microsatellite Instability, and BRAF Mutations and Their Clinical Application in the Treatment of Colon Cancer. Chemother. Res. Pract. 2012, 2012, 359041. [Google Scholar]
- Suzuki, K.; Suzuki, I.; Leodolter, A.; Alonso, S.; Horiuchi, S.; Yamashita, K.; Perucho, M. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell 2006, 9, 199–207. [Google Scholar]
- Chen, R.Z.; Pettersson, U.; Beard, C.; Jackson-Grusby, L.; Jaenisch, R. DNA hypomethylation leads to elevated mutation rates. Nature 1998, 395, 89–93. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. DNA methylation and genetic instability in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 1997, 94, 2545–2550. [Google Scholar] [CrossRef]
- Ha, K.; Lee, G.E.; Palii, S.S.; Brown, K.D.; Takeda, Y.; Liu, K.; Bhalla, K.N.; Robertson, K.D. Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery. Hum. Mol. Genet. 2011, 20, 126–140. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar]
- Milicic, A.; Harrison, L.A.; Goodlad, R.A.; Hardy, R.G.; Nicholson, A.M.; Presz, M.; Sieber, O.; Santander, S.; Pringle, J.H.; Mandir, N.; et al. Methylation subtypes and large-scale epigenetic alterations in gastric cancer. Sci. Transl. Med. 2012, 4, 156ra140. [Google Scholar]
- Yamada, Y.; Jackson-Grusby, L.; Linhart, H.; Meissner, A.; Eden, A.; Lin, H.; Jaenisch, R. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 13580–13585. [Google Scholar]
- Silver, A.; Sengupta, N.; Propper, D.; Wilson, P.; Hagemann, T.; Patel, A.; Parker, A.; Ghosh, A.; Feakins, R.; Dorudi, S.; et al. A distinct DNA methylation profile associated with microsatellite and chromosomal stable sporadic colorectal cancers. Int. J. Cancer 2012, 130, 1082–1092. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Ohnishi, M.; Fuchs, C.S. 18q loss of heterozygosity in microsatellite stable colorectal cancer is correlated with CpG island methylator phenotype-negative (CIMP-0) and inversely with CIMP-low and CIMP-high. BMC Cancer 2007, 7, 72. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Nosho, K.; Ohnishi, M.; Suemoto, Y.; Kirkner, G.J.; Fuchs, C.S. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int. J. Cancer 2008, 122, 2767–2773. [Google Scholar]
- Issa, J.P.; Vertino, P.M.; Wu, J.; Sazawal, S.; Celano, P.; Nelkin, B.D.; Hamilton, S.R.; Baylin, S.B. Increased cytosine DNA-methyltransferase activity during colon cancer progression. J. Natl. Cancer Inst. 1993, 85, 1235–1240. [Google Scholar] [CrossRef]
- Nosho, K.; Shima, K.; Irahara, N.; Kure, S.; Baba, Y.; Kirkner, G.J.; Chen, L.; Gokhale, S.; Hazra, A.; Spiegelman, D.; et al. DNMT3B expression might contribute to CpG island methylator phenotype in colorectal cancer. Clin. Cancer Res. 2009, 15, 3663–3671. [Google Scholar] [CrossRef]
- Schmidt, W.M.; Sedivy, R.; Forstner, B.; Steger, G.G.; Zochbauer-Muller, S.; Mader, R.M. Progressive up-regulation of genes encoding DNA methyltransferases in the colorectal adenoma-carcinoma sequence. Mol. Carcinog. 2007, 46, 766–772. [Google Scholar] [CrossRef]
- Ibrahim, A.E.; Arends, M.J.; Silva, A.L.; Wyllie, A.H.; Greger, L.; Ito, Y.; Vowler, S.L.; Huang, T.H.; Tavare, S.; Murrell, A.; et al. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut 2011, 60, 499–508. [Google Scholar] [CrossRef]
- Jones, P.A. The DNA methylation paradox. Trends Genet. 1999, 15, 34–37. [Google Scholar] [CrossRef]
- Wajed, S.A.; Laird, P.W.; DeMeester, T.R. DNA methylation: An alternative pathway to cancer. Ann. Surg. 2001, 234, 10–20. [Google Scholar] [CrossRef]
- McGough, J.M.; Yang, D.; Huang, S.; Georgi, D.; Hewitt, S.M.; Rocken, C.; Tanzer, M.; Ebert, M.P.; Liu, K. DNA methylation represses IFN-gamma-induced and signal transducer and activator of transcription 1-mediated IFN regulatory factor 8 activation in colon carcinoma cells. Mol. Cancer Res. 2008, 6, 1841–1851. [Google Scholar]
- Yang, D.; Thangaraju, M.; Greeneltch, K.; Browning, D.D.; Schoenlein, P.V.; Tamura, T.; Ozato, K.; Ganapathy, V.; Abrams, S.I.; Liu, K. Repression of IFN regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer Res. 2007, 67, 3301–3309. [Google Scholar]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar]
- Hung, M.S.; Shen, C.K. Eukaryotic methyl-CpG-binding domain proteins and chromatin modification. Eukaryot. Cell 2003, 2, 841–846. [Google Scholar] [CrossRef]
- Parry, L.; Clarke, A.R. The Roles of the Methyl-CpG Binding Proteins in Cancer. Genes Cancer 2011, 2, 618–630. [Google Scholar]
- Jubb, A.M.; Bell, S.M.; Quirke, P. Methylation and colorectal cancer. J. Pathol. 2001, 195, 111–134. [Google Scholar] [CrossRef]
- Pancione, M.; Sabatino, L.; Fucci, A.; Carafa, V.; Nebbioso, A.; Forte, N.; Febbraro, A.; Parente, D.; Ambrosino, C.; Normanno, N.; et al. Epigenetic silencing of peroxisome proliferator-activated receptor gamma is a biomarker for colorectal cancer progression and adverse patients’ outcome. PLoS One 2010, 5, e14229. [Google Scholar]
- Bader, S.; Walker, M.; McQueen, H.A.; Sellar, R.; Oei, E.; Wopereis, S.; Zhu, Y.; Peter, A.; Bird, A.P.; Harrison, D.J. MBD1, MBD2 and CGBP genes at chromosome 18q21 are infrequently mutated in human colon and lung cancers. Oncogene 2003, 22, 3506–3510. [Google Scholar] [CrossRef]
- Howard, J.H.; Frolov, A.; Tzeng, C.W.; Stewart, A.; Midzak, A.; Majmundar, A.; Godwin, A.; Heslin, M.; Bellacosa, A.; Arnoletti, J.P. Epigenetic downregulation of the DNA repair gene MED1/MBD4 in colorectal and ovarian cancer. Cancer Biol. Ther. 2009, 8, 94–100. [Google Scholar]
- Riccio, A.; Aaltonen, L.A.; Godwin, A.K.; Loukola, A.; Percesepe, A.; Salovaara, R.; Masciullo, V.; Genuardi, M.; Paravatou-Petsotas, M.; Bassi, D.E.; et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat. Genet. 1999, 23, 266–268. [Google Scholar]
- Bader, S.; Walker, M.; Hendrich, B.; Bird, A.; Bird, C.; Hooper, M.; Wyllie, A. Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene 1999, 18, 8044–8047. [Google Scholar]
- Bader, S.; Walker, M.; Harrison, D. Most microsatellite unstable sporadic colorectal carcinomas carry MBD4 mutations. Br. J. Cancer 2000, 83, 1646–1649. [Google Scholar] [CrossRef]
- Bader, S.A.; Walker, M.; Harrison, D.J. A human cancer-associated truncation of MBD4 causes dominant negative impairment of DNA repair in colon cancer cells. Br. J. Cancer 2007, 96, 660–666. [Google Scholar]
- Millar, C.B.; Guy, J.; Sansom, O.J.; Selfridge, J.; MacDougall, E.; Hendrich, B.; Keightley, P.D.; Bishop, S.M.; Clarke, A.R.; Bird, A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 2002, 297, 403–405. [Google Scholar]
- Jin, B.; Yao, B.; Li, J.L.; Fields, C.R.; Delmas, A.L.; Liu, C.; Robertson, K.D. DNMT1 and DNMT3B modulate distinct polycomb-mediated histone modifications in colon cancer. Cancer Res. 2009, 69, 7412–7421. [Google Scholar]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Cai, Y.; McGarvey, K.M.; Easwaran, H.; van Neste, L.; Ohm, J.E.; O’Hagan, H.M.; Baylin, S.B. Polycomb CBX7 promotes initiation of heritable repression of genes frequently silenced with cancer-specific DNA hypermethylation. Cancer Res. 2009, 69, 6322–6330. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.S.; Cui, Z.Y.; Lee, H.S.; Ahn, J.S.; Park, C.K.; Park, K.; Ahn, M.J. Clinicopathological implications of EpCAM expression in adenocarcinoma of the lung. Anticancer Res. 2009, 29, 1817–1822. [Google Scholar]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar]
- Fussbroich, B.; Wagener, N.; Macher-Goeppinger, S.; Benner, A.; Falth, M.; Sultmann, H.; Holzer, A.; Hoppe-Seyler, K.; Hoppe-Seyler, F. EZH2 depletion blocks the proliferation of colon cancer cells. PLoS One 2011, 6, e21651. [Google Scholar]
- Wheeler, J.M.; Bodmer, W.F.; Mortensen, N.J. DNA mismatch repair genes and colorectal cancer. Gut 2000, 47, 148–153. [Google Scholar] [CrossRef]
- Chan, T.L.; Yuen, S.T.; Kong, C.K.; Chan, Y.W.; Chan, A.S.; Ng, W.F.; Tsui, W.Y.; Lo, M.W.; Tam, W.Y.; Li, V.S.; et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat. Genet. 2006, 38, 1178–1183. [Google Scholar]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar]
- Xu, Y.; Hu, B.; Choi, A.J.; Gopalan, B.; Lee, B.H.; Kalady, M.F.; Church, J.M.; Ting, A.H. Unique DNA methylome profiles in CpG island methylator phenotype colon cancers. Genome Res. 2012, 22, 283–291. [Google Scholar]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef]
- Ang, P.W.; Loh, M.; Liem, N.; Lim, P.L.; Grieu, F.; Vaithilingam, A.; Platell, C.; Yong, W.P.; Iacopetta, B.; Soong, R. Comprehensive profiling of DNA methylation in colorectal cancer reveals subgroups with distinct clinicopathological and molecular features. BMC Cancer 2010, 10, 227. [Google Scholar] [CrossRef]
- Ward, R.L.; Cheong, K.; Ku, S.L.; Meagher, A.; O’Connor, T.; Hawkins, N.J. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. J. Clin. Oncol. 2003, 21, 3729–3736. [Google Scholar] [CrossRef]
- Van Rijnsoever, M.; Elsaleh, H.; Joseph, D.; McCaul, K.; Iacopetta, B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin. Cancer Res. 2003, 9, 2898–2903. [Google Scholar]
- Ogino, S.; Meyerhardt, J.A.; Kawasaki, T.; Clark, J.W.; Ryan, D.P.; Kulke, M.H.; Enzinger, P.C.; Wolpin, B.M.; Loda, M.; Fuchs, C.S. CpG island methylation, response to combination chemotherapy, and patient survival in advanced microsatellite stable colorectal carcinoma. Virchows Arch. 2007, 450, 529–537. [Google Scholar] [CrossRef]
- Lee, S.; Cho, N.Y.; Choi, M.; Yoo, E.J.; Kim, J.H.; Kang, G.H. Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathol. Int. 2008, 58, 104–113. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Meyerhardt, J.A.; Loda, M.; Giovannucci, E.L.; Fuchs, C.S. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009, 58, 90–96. [Google Scholar] [CrossRef]
- Barault, L.; Charon-Barra, C.; Jooste, V.; de la Vega, M.F.; Martin, L.; Roignot, P.; Rat, P.; Bouvier, A.M.; Laurent-Puig, P.; Faivre, J.; et al. Hypermethylator phenotype in sporadic colon cancer: Study on a population-based series of 582 cases. Cancer Res. 2008, 68, 8541–8546. [Google Scholar] [CrossRef]
- Jover, R.; Nguyen, T.P.; Perez-Carbonell, L.; Zapater, P.; Paya, A.; Alenda, C.; Rojas, E.; Cubiella, J.; Balaguer, F.; Morillas, J.D.; et al. 5-Fluorouracil adjuvant chemotherapy does not increase survival in patients with CpG island methylator phenotype colorectal cancer. Gastroenterology 2011, 140, 1174–1181. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef]
- Thomas, J.H. Genomic imprinting proposed as a surveillance mechanism for chromosome loss. Proc. Natl. Acad. Sci. USA 1995, 92, 480–482. [Google Scholar]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef]
- Zhang, H.; Richards, B.; Wilson, T.; Lloyd, M.; Cranston, A.; Thorburn, A.; Fishel, R.; Meuth, M. Apoptosis induced by overexpression of hMSH2 or hMLH1. Cancer Res. 1999, 59, 3021–3027. [Google Scholar]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.M.; van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Loda, M.; Fuchs, C.S. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: Possible associations with male sex and KRAS mutations. J. Mol. Diagn. 2006, 8, 582–588. [Google Scholar] [CrossRef]
- Issa, J.P.; Ahuja, N.; Toyota, M.; Bronner, M.P.; Brentnall, T.A. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001, 61, 3573–3577. [Google Scholar]
- Sanchez, J.A.; Dejulius, K.L.; Bronner, M.; Church, J.M.; Kalady, M.F. Relative role of methylator and tumor suppressor pathways in ulcerative colitis-associated colon cancer. Inflamm. Bowel Dis. 2011, 17, 1966–1970. [Google Scholar] [CrossRef]
- Olaru, A.V.; Cheng, Y.; Agarwal, R.; Yang, J.; David, S.; Abraham, J.M.; Yu, W.; Kwon, J.H.; Lazarev, M.; Brant, S.R.; et al. Unique patterns of CpG island methylation in inflammatory bowel disease-associated colorectal cancers. Inflamm. Bowel Dis. 2012, 18, 641–648. [Google Scholar] [CrossRef]
- Konishi, K.; Shen, L.; Wang, S.; Meltzer, S.J.; Harpaz, N.; Issa, J.P. Rare CpG island methylator phenotype in ulcerative colitis-associated neoplasias. Gastroenterology 2007, 132, 1254–1260. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Esteller, M.; Harpaz, N.; Leytin, A.; Rashid, A.; Xu, Y.; Liang, J.; Stine, O.C.; Yin, J.; Zou, T.T.; et al. Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res. 2000, 60, 4864–4868. [Google Scholar]
- Antelo, M.; Balaguer, F.; Shia, J.; Shen, Y.; Hur, K.; Moreira, L.; Cuatrecasas, M.; Bujanda, L.; Giraldez, M.D.; Takahashi, M.; et al. A high degree of LINE-1 hypomethylation is a unique feature of early-onset colorectal cancer. PLoS One 2012, 7, e45357. [Google Scholar]
- Estecio, M.R.; Gharibyan, V.; Shen, L.; Ibrahim, A.E.; Doshi, K.; He, R.; Jelinek, J.; Yang, A.S.; Yan, P.S.; Huang, T.H.; et al. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS One 2007, 2, e399. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst. 2008, 100, 1734–1738. [Google Scholar] [CrossRef]
- Baba, Y.; Huttenhower, C.; Nosho, K.; Tanaka, N.; Shima, K.; Hazra, A.; Schernhammer, E.S.; Hunter, D.J.; Giovannucci, E.L.; Fuchs, C.S.; et al. Epigenomic diversity of colorectal cancer indicated by LINE-1 methylation in a database of 869 tumors. Mol. Cancer 2010, 9, 125. [Google Scholar] [CrossRef]
- Ahn, J.B.; Chung, W.B.; Maeda, O.; Shin, S.J.; Kim, H.S.; Chung, H.C.; Kim, N.K.; Issa, J.P. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer 2011, 117, 1847–1854. [Google Scholar] [CrossRef]
- Rhee, Y.Y.; Kim, M.J.; Bae, J.M.; Koh, J.M.; Cho, N.Y.; Juhnn, Y.S.; Kim, D.; Kang, G.H. Clinical outcomes of patients with microsatellite-unstable colorectal carcinomas depend on L1 methylation level. Ann. Surg. Oncol. 2012, 19, 3441–3448. [Google Scholar] [CrossRef]
- Ogino, S.; Nishihara, R.; Lochhead, P.; Imamura, Y.; Kuchiba, A.; Morikawa, T.; Yamauchi, M.; Liao, X.; Qian, Z.R.; Sun, R.; et al. Prospective study of family history and colorectal cancer risk by tumor LINE-1 methylation level. J. Natl. Cancer Inst. 2013, 105, 130–140. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Nakazawa, T.; Kondo, T.; Ma, D.; Niu, D.; Mochizuki, K.; Kawasaki, T.; Yamane, T.; Iino, H.; Fujii, H.; Katoh, R. Global histone modification of histone H3 in colorectal cancer and its precursor lesions. Hum. Pathol. 2012, 43, 834–842. [Google Scholar] [CrossRef]
- Li, Q.; Chen, H. Silencing of Wnt5a during colon cancer metastasis involves histone modifications. Epigenetics 2012, 7, 551–558. [Google Scholar] [CrossRef]
- Binder, H.; Steiner, L.; Przybilla, J.; Rohlf, T.; Prohaska, S.; Galle, J. Transcriptional regulation by histone modifications: Towards a theory of chromatin re-organization during stem cell differentiation. Phys. Biol. 2013, 10, 026006. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, K.; Grunstein, M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 2005, 121, 375–385. [Google Scholar] [CrossRef]
- Han, J.; Zhou, H.; Horazdovsky, B.; Zhang, K.; Xu, R.M.; Zhang, Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 2007, 315, 653–655. [Google Scholar] [CrossRef]
- Driscoll, R.; Hudson, A.; Jackson, S.P. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 2007, 315, 649–652. [Google Scholar] [CrossRef]
- Schneider, J.; Bajwa, P.; Johnson, F.C.; Bhaumik, S.R.; Shilatifard, A. Rtt109 is required for proper H3K56 acetylation: A chromatin mark associated with the elongating RNA polymerase II. J. Biol. Chem. 2006, 281, 37270–37274. [Google Scholar]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef]
- Ashktorab, H.; Belgrave, K.; Hosseinkhah, F.; Brim, H.; Nouraie, M.; Takkikto, M.; Hewitt, S.; Lee, E.L.; Dashwood, R.H.; Smoot, D. Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Dig. Dis. Sci. 2009, 54, 2109–2117. [Google Scholar]
- Ishihama, K.; Yamakawa, M.; Semba, S.; Takeda, H.; Kawata, S.; Kimura, S.; Kimura, W. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J. Clin. Pathol. 2007, 60, 1205–1210. [Google Scholar] [CrossRef]
- Nosho, K.; Shima, K.; Irahara, N.; Kure, S.; Firestein, R.; Baba, Y.; Toyoda, S.; Chen, L.; Hazra, A.; Giovannucci, E.L.; et al. SIRT1 histone deacetylase expression is associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod. Pathol. 2009, 22, 922–932. [Google Scholar]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol. Cell Biol. 2006, 26, 9185–9195. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Mahmoudi, T.; Boj, S.F.; Hatzis, P.; Li, V.S.; Taouatas, N.; Vries, R.G.; Teunissen, H.; Begthel, H.; Korving, J.; Mohammed, S.; et al. The leukemia-associated Mllt10/Af10-Dot1l are Tcf4/beta-catenin coactivators essential for intestinal homeostasis. PLoS Biol. 2010, 8, e1000539. [Google Scholar] [CrossRef]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; Li, X.; Toy, K.; DuPrie, M.; Ventura, A.C.; Banerjee, M.; Ljungman, M.; Merajver, S.D.; Kleer, C.G. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 2009, 28, 843–853. [Google Scholar] [CrossRef]
- Yu, J.; Cao, Q.; Mehra, R.; Laxman, B.; Yu, J.; Tomlins, S.A.; Creighton, C.J.; Dhanasekaran, S.M.; Shen, R.; Chen, G.; et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 2007, 12, 419–431. [Google Scholar]
- Natarajan, T.G.; Kallakury, B.V.; Sheehan, C.E.; Bartlett, M.B.; Ganesan, N.; Preet, A.; Ross, J.S.; Fitzgerald, K.T. Epigenetic regulator MLL2 shows altered expression in cancer cell lines and tumors from human breast and colon. Cancer Cell Int. 2010, 10, 13. [Google Scholar]
- Kang, M.Y.; Lee, B.B.; Kim, Y.H.; Chang, D.K.; Kyu Park, S.; Chun, H.K.; Song, S.Y.; Park, J.; Kim, D.H. Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer. Int. J. Cancer 2007, 121, 2192–2197. [Google Scholar] [CrossRef]
- Tiwari, V.K.; McGarvey, K.M.; Licchesi, J.D.; Ohm, J.E.; Herman, J.G.; Schubeler, D.; Baylin, S.B. PcG proteins, DNA methylation, and gene repression by chromatin looping. PLoS Biol. 2008, 6, 2911–2927. [Google Scholar]
- Bandres, E.; Cubedo, E.; Agirre, X.; Malumbres, R.; Zarate, R.; Ramirez, N.; Abajo, A.; Navarro, A.; Moreno, I.; Monzo, M.; et al. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol. Cancer 2006, 5, 29. [Google Scholar]
- Bandres, E.; Cubedo, E.; Agirre, X.; Malumbres, R.; Zarate, R.; Ramirez, N.; Abajo, A.; Navarro, A.; Moreno, I.; Monzo, M.; et al. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol. Cancer 2006, 5, 29. [Google Scholar]
- Schepeler, T.; Reinert, J.T.; Ostenfeld, M.S.; Christensen, L.L.; Silahtaroglu, A.N.; Dyrskjot, L.; Wiuf, C.; Sorensen, F.J.; Kruhoffer, M.; Laurberg, S.; et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008, 68, 6416–6424. [Google Scholar] [CrossRef]
- Wang, C.J.; Zhou, Z.G.; Wang, L.; Yang, L.; Zhou, B.; Gu, J.; Chen, H.Y.; Sun, X.F. Clinicopathological significance of microRNA-31, -143 and -145 expression in colorectal cancer. Dis. Markers 2009, 26, 27–34. [Google Scholar]
- Yan, H.; Choi, A.J.; Lee, B.H.; Ting, A.H. Identification and functional analysis of epigenetically silenced microRNAs in colorectal cancer cells. PLoS One 2011, 6, e20628. [Google Scholar]
- Motoyama, K.; Inoue, H.; Takatsuno, Y.; Tanaka, F.; Mimori, K.; Uetake, H.; Sugihara, K.; Mori, M. Over- and under-expressed microRNAs in human colorectal cancer. Int. J. Oncol. 2009, 34, 1069–1075. [Google Scholar]
- Sarver, A.L.; French, A.J.; Borralho, P.M.; Thayanithy, V.; Oberg, A.L.; Silverstein, K.A.; Morlan, B.W.; Riska, S.M.; Boardman, L.A.; Cunningham, J.M.; et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer 2009, 9, 401. [Google Scholar] [CrossRef]
- Nagel, R.; le Sage, C.; Diosdado, B.; van der Waal, M.; Oude Vrielink, J.A.; Bolijn, A.; Meijer, G.A.; Agami, R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008, 68, 5795–5802. [Google Scholar] [CrossRef]
- Arndt, G.M.; Dossey, L.; Cullen, L.M.; Lai, A.; Druker, R.; Eisbacher, M.; Zhang, C.; Tran, N.; Fan, H.; Retzlaff, K.; et al. Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC Cancer 2009, 9, 374. [Google Scholar] [CrossRef]
- Monzo, M.; Navarro, A.; Bandres, E.; Artells, R.; Moreno, I.; Gel, B.; Ibeas, R.; Moreno, J.; Martinez, F.; Diaz, T.; et al. Overlapping expression of microRNAs in human embryonic colon and colorectal cancer. Cell Res. 2008, 18, 823–833. [Google Scholar] [CrossRef]
- Oberg, A.L.; French, A.J.; Sarver, A.L.; Subramanian, S.; Morlan, B.W.; Riska, S.M.; Borralho, P.M.; Cunningham, J.M.; Boardman, L.A.; Wang, L.; et al. miRNA expression in colon polyps provides evidence for a multihit model of colon cancer. PLoS One 2011, 6, e20465. [Google Scholar] [CrossRef]
- Earle, J.S.; Luthra, R.; Romans, A.; Abraham, R.; Ensor, J.; Yao, H.; Hamilton, S.R. Association of microRNA expression with microsatellite instability status in colorectal adenocarcinoma. J. Mol. Diagn. 2010, 12, 433–440. [Google Scholar] [CrossRef]
- Vogt, M.; Munding, J.; Gruner, M.; Liffers, S.T.; Verdoodt, B.; Hauk, J.; Steinstraesser, L.; Tannapfel, A.; Hermeking, H. Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch. 2011, 458, 313–322. [Google Scholar] [CrossRef]
- Stratmann, J.; Wang, C.J.; Gnosa, S.; Wallin, A.; Hinselwood, D.; Sun, X.F.; Zhang, H. Dicer and miRNA in relation to clinicopathological variables in colorectal cancer patients. BMC Cancer 2011, 11, 345. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, L.; Cogdell, D.E.; Zheng, H.; Schetter, A.J.; Nykter, M.; Harris, C.C.; Chen, K.; Hamilton, S.R.; Zhang, W. Circulating plasma MiR-141 is a novel biomarker for metastatic colon cancer and predicts poor prognosis. PLoS One 2011, 6, e17745. [Google Scholar] [CrossRef]
- Fahrner, J.A.; Eguchi, S.; Herman, J.G.; Baylin, S.B. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002, 62, 7213–7218. [Google Scholar]
- Slaby, O.; Svoboda, M.; Michalek, J.; Vyzula, R. MicroRNAs in colorectal cancer: Translation of molecular biology into clinical application. Mol. Cancer 2009, 8, 102. [Google Scholar] [CrossRef]
- Ferracin, M.; Gafa, R.; Miotto, E.; Veronese, A.; Pultrone, C.; Sabbioni, S.; Lanza, G.; Negrini, M. The methylator phenotype in microsatellite stable colorectal cancers is characterized by a distinct gene expression profile. J. Pathol. 2008, 214, 594–602. [Google Scholar]
- Nagaraju, G.P.; El-Rayes, B.F. SPARC and DNA methylation: Possible diagnostic and therapeutic implications in gastrointestinal cancers. Cancer Lett. 2013, 328, 10–17. [Google Scholar] [CrossRef]
- Tai, I.T.; Dai, M.; Owen, D.A.; Chen, L.B. Genome-wide expression analysis of therapy-resistant tumors reveals SPARC as a novel target for cancer therapy. J. Clin. Invest. 2005, 115, 1492–1502. [Google Scholar] [CrossRef]
- Tai, I.T.; Tang, M.J. SPARC in cancer biology: Its role in cancer progression and potential for therapy. Drug Resist. Updat. 2008, 11, 231–246. [Google Scholar] [CrossRef]
- Crea, F.; Nobili, S.; Paolicchi, E.; Perrone, G.; Napoli, C.; Landini, I.; Danesi, R.; Mini, E. Epigenetics and chemoresistance in colorectal cancer: An opportunity for treatment tailoring and novel therapeutic strategies. Drug Resist. Updat. 2011, 14, 280–296. [Google Scholar] [CrossRef]
- Greiner, D.; Bonaldi, T.; Eskeland, R.; Roemer, E.; Imhof, A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat. Chem. Biol. 2005, 1, 143–145. [Google Scholar] [CrossRef]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef]
- Vedadi, M.; Barsyte-Lovejoy, D.; Liu, F.; Rival-Gervier, S.; Allali-Hassani, A.; Labrie, V.; Wigle, T.J.; Dimaggio, P.A.; Wasney, G.A.; Siarheyeva, A.; Dong, A.; et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011, 7, 566–574. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar]
- Ferguson, A.D.; Larsen, N.A.; Howard, T.; Pollard, H.; Green, I.; Grande, C.; Cheung, T.; Garcia-Arenas, R.; Cowen, S.; Wu, J.; et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [Google Scholar] [CrossRef]
- Chang, Y.; Zhang, X.; Horton, J.R.; Upadhyay, A.K.; Spannhoff, A.; Liu, J.; Snyder, J.P.; Bedford, M.T.; Cheng, X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009, 16, 312–317. [Google Scholar]
- Foulks, J.M.; Parnell, K.M.; Nix, R.N.; Chau, S.; Swierczek, K.; Saunders, M.; Wright, K.; Hendrickson, T.F.; Ho, K.K.; McCullar, M.V.; et al. Epigenetic drug discovery: Targeting DNA methyltransferases. J. Biomol. Screen. 2012, 17, 2–17. [Google Scholar] [CrossRef]
- McGovern, A.P.; Powell, B.E.; Chevassut, T.J. A dynamic multi-compartmental model of DNA methylation with demonstrable predictive value in hematological malignancies. J. Theor. Biol. 2012, 310, 14–20. [Google Scholar] [CrossRef]
- Szyf, M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 243–263. [Google Scholar] [CrossRef]
- Glover, A.B.; Leyland-Jones, B.R.; Chun, H.G.; Davies, B.; Hoth, D.F. Azacitidine: 10 years later. Cancer Treat. Rep. 1987, 71, 737–746. [Google Scholar]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar]
- Lee, W.J.; Zhu, B.T. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis 2006, 27, 269–277. [Google Scholar] [CrossRef]
- Lee, W.J.; Shim, J.Y.; Zhu, B.T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 2005, 68, 1018–1030. [Google Scholar] [CrossRef]
- Link, A.; Balaguer, F.; Shen, Y.; Lozano, J.J.; Leung, H.C.; Boland, C.R.; Goel, A. Curcumin modulates DNA methylation in colorectal cancer cells. PLoS One 2013, 8, e57709. [Google Scholar]
- Fang, M.; Chen, D.; Yang, C.S. Dietary polyphenols may affect DNA methylation. J. Nutr. 2007, 137, 223S–228S. [Google Scholar]
- Howman, R.A.; Prince, H.M. New drug therapies in peripheral T-cell lymphoma. Expert Rev. Anticancer Ther. 2011, 11, 457–472. [Google Scholar] [CrossRef]
- Park, M.A.; Reinehr, R.; Haussinger, D.; Voelkel-Johnson, C.; Ogretmen, B.; Yacoub, A.; Grant, S.; Dent, P. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol. Cancer Ther. 2010, 9, 2220–2231. [Google Scholar] [CrossRef]
- Park, M.A.; Zhang, G.; Martin, A.P.; Hamed, H.; Mitchell, C.; Hylemon, P.B.; Graf, M.; Rahmani, M.; Ryan, K.; Liu, X.; et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol. Ther. 2008, 7, 1648–1662. [Google Scholar] [CrossRef]
- Spannhoff, A.; Hauser, A.T.; Heinke, R.; Sippl, W.; Jung, M. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. Chem. Med. Chem. 2009, 4, 1568–1582. [Google Scholar]
- Benoit, Y.D.; Laursen, K.B.; Witherspoon, M.S.; Lipkin, S.M.; Gudas, L.J. Inhibition of PRC2 histone methyltransferase activity increases TRAIL-mediated apoptosis sensitivity in human colon cancer cells. J. Cell Physiol. 2013, 228, 764–772. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; van Cutsem, E.; Dumez, H.; Chen, C.; Ricker, J.L.; Randolph, S.S.; Schoffski, P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest. New Drugs 2008, 26, 483–488. [Google Scholar] [CrossRef]
- Yang, D.; Torres, C.M.; Bardhan, K.; Zimmerman, M.; McGaha, T.L.; Liu, K. Decitabine and vorinostat cooperate to sensitize colon carcinoma cells to fas ligand-induced apoptosis in vitro and tumor suppression in vivo. J. Immunol. 2012, 188, 4441–4449. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Strater, J.; Hinz, U.; Hasel, C.; Bhanot, U.; Mechtersheimer, G.; Lehnert, T.; Moller, P. Impaired CD95 expression predisposes for recurrence in curatively resected colon carcinoma: Clinical evidence for immunoselection and CD95L mediated control of minimal residual disease. Gut 2005, 54, 661–665. [Google Scholar]
- Owen-Schaub, L.B.; van Golen, K.L.; Hill, L.L.; Price, J.E. Fas and Fas ligand interactions suppress melanoma lung metastasis. J. Exp. Med. 1998, 188, 1717–1723. [Google Scholar] [CrossRef]
- Moller, P.; Koretz, K.; Leithauser, F.; Bruderlein, S.; Henne, C.; Quentmeier, A.; Krammer, P.H. Expression of APO-1 (CD95), a member of the NGF/TNF receptor superfamily, in normal and neoplastic colon epithelium. Int. J. Cancer 1994, 57, 371–377. [Google Scholar] [CrossRef]
- June, C.H. Adoptive T cell therapy for cancer in the clinic. J. Clin. Invest. 2007, 117, 1466–1476. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Yang, D.; Wang, S.; Brooks, C.; Dong, Z.; Schoenlein, P.V.; Kumar, V.; Ouyang, X.; Xiong, H.; Lahat, G.; Hayes-Jordan, A.; et al. IFN regulatory factor 8 sensitizes soft tissue sarcoma cells to death receptor-initiated apoptosis via repression of FLICE-like protein expression. Cancer Res. 2009, 69, 1080–1088. [Google Scholar] [CrossRef]
- Yang, D.; Ud Din, N.; Browning, D.D.; Abrams, S.I.; Liu, K. Targeting lymphotoxin beta receptor with tumor-specific T lymphocytes for tumor regression. Clin. Cancer Res. 2007, 13, 5202–5210. [Google Scholar] [CrossRef]
- Ocker, M.; Alajati, A.; Ganslmayer, M.; Zopf, S.; Luders, M.; Neureiter, D.; Hahn, E.G.; Schuppan, D.; Herold, C. The histone-deacetylase inhibitor SAHA potentiates proapoptotic effects of 5-fluorouracil and irinotecan in hepatoma cells. J. Cancer Res. Clin. Oncol. 2005, 131, 385–394. [Google Scholar] [CrossRef]
- Arnold, C.N.; Goel, A.; Boland, C.R. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int. J. Cancer 2003, 106, 66–73. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Wohlhueter, R.M.; McIvor, R.S.; Plagemann, P.G. Facilitated transport of uracil and 5-fluorouracil, and permeation of orotic acid into cultured mammalian cells. J. Cell Physiol. 1980, 104, 309–319. [Google Scholar] [CrossRef]
- Noordhuis, P.; Holwerda, U.; van der Wilt, C.L.; van Groeningen, C.J.; Smid, K.; Meijer, S.; Pinedo, H.M.; Peters, G.J. 5-Fluorouracil incorporation into RNA and DNA in relation to thymidylate synthase inhibition of human colorectal cancers. Ann. Oncol. 2004, 15, 1025–1032. [Google Scholar] [CrossRef]
- Humeniuk, R.; Menon, L.G.; Mishra, P.J.; Gorlick, R.; Sowers, R.; Rode, W.; Pizzorno, G.; Cheng, Y.C.; Kemeny, N.; Bertino, J.R.; et al. Decreased levels of UMP kinase as a mechanism of fluoropyrimidine resistance. Mol. Cancer Ther. 2009, 8, 1037–1044. [Google Scholar] [CrossRef]
- Sobrero, A.; Guglielmi, A.; Grossi, F.; Puglisi, F.; Aschele, C. Mechanism of action of fluoropyrimidines: Relevance to the new developments in colorectal cancer chemotherapy. Semin. Oncol. 2000, 27, 72–77. [Google Scholar]
- Danenberg, P.V.; Langenbach, R.J.; Heidelberger, C. Structures of reversible and irreversible complexes of thymidylate synthetase and fluorinated pyrimidine nucleotides. Biochemistry 1974, 13, 926–933. [Google Scholar]
- Santi, D.V.; McHenry, C.S.; Sommer, H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 1974, 13, 471–481. [Google Scholar] [CrossRef]
- Houghton, J.A.; Torrance, P.M.; Radparvar, S.; Williams, L.G.; Houghton, P.J. Binding of 5-fluorodeoxyuridylate to thymidylate synthase in human colon adenocarcinoma xenografts. Eur. J. Cancer Clin. Oncol. 1986, 22, 505–510. [Google Scholar] [CrossRef]
- Houghton, J.A.; Tillman, D.M.; Harwood, F.G. Ratio of 2'-deoxyadenosine-5'-triphosphate/thymidine-5'-triphosphate influences the commitment of human colon carcinoma cells to thymineless death. Clin. Cancer Res. 1995, 1, 723–730. [Google Scholar]
- Yoshioka, A.; Tanaka, S.; Hiraoka, O.; Koyama, Y.; Hirota, Y.; Ayusawa, D.; Seno, T.; Garrett, C.; Wataya, Y. Deoxyribonucleoside triphosphate imbalance. 5-Fluorodeoxyuridine-induced DNA double strand breaks in mouse FM3A cells and the mechanism of cell death. J. Biol. Chem. 1987, 262, 8235–8241. [Google Scholar]
- Wang, T.L.; Diaz, L.A., Jr.; Romans, K.; Bardelli, A.; Saha, S.; Galizia, G.; Choti, M.; Donehower, R.; Parmigiani, G.; Shih Ie, M.; et al. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3089–3094. [Google Scholar] [CrossRef]
- Yu, J.; Miller, R.; Zhang, W.; Sharma, M.; Holtschlag, V.; Watson, M.A.; McLeod, H.L. Copy-number analysis of topoisomerase and thymidylate synthase genes in frozen and FFPE DNAs of colorectal cancers. Pharmacogenomics 2008, 9, 1459–1466. [Google Scholar] [CrossRef]
- Watson, R.G.; Muhale, F.; Thorne, L.B.; Yu, J.; O’Neil, B.H.; Hoskins, J.M.; Meyers, M.O.; Deal, A.M.; Ibrahim, J.G.; Hudson, M.L.; et al. Amplification of thymidylate synthetase in metastatic colorectal cancer patients pretreated with 5-fluorouracil-based chemotherapy. Eur. J. Cancer 2010, 46, 3358–3364. [Google Scholar] [CrossRef]
- Brody, J.R.; Hucl, T.; Gallmeier, E.; Winter, J.M.; Kern, S.E.; Murphy, K.M. Genomic copy number changes affecting the thymidylate synthase (TYMS) gene in cancer: A model for patient classification to aid fluoropyrimidine therapy. Cancer Res. 2006, 66, 9369–9373. [Google Scholar]
- Nief, N.; Le Morvan, V.; Robert, J. Involvement of gene polymorphisms of thymidylate synthase in gene expression, protein activity and anticancer drug cytotoxicity using the NCI-60 panel. Eur. J. Cancer 2007, 43, 955–962. [Google Scholar] [CrossRef]
- Banerjee, D.; Gorlick, R.; Liefshitz, A.; Danenberg, K.; Danenberg, P.C.; Danenberg, P.V.; Klimstra, D.; Jhanwar, S.; Cordon-Cardo, C.; Fong, Y.; et al. Levels of E2F-1 expression are higher in lung metastasis of colon cancer as compared with hepatic metastasis and correlate with levels of thymidylate synthase. Cancer Res. 2000, 60, 2365–2367. [Google Scholar]
- Yoo, B.K.; Gredler, R.; Vozhilla, N.; Su, Z.Z.; Chen, D.; Forcier, T.; Shah, K.; Saxena, U.; Hansen, U.; Fisher, P.B.; et al. Identification of genes conferring resistance to 5-fluorouracil. Proc. Natl. Acad. Sci. USA 2009, 106, 12938–12943. [Google Scholar] [CrossRef]
- Rodriguez-Paredes, M.; Ceballos-Chavez, M.; Esteller, M.; Garcia-Dominguez, M.; Reyes, J.C. The chromatin remodeling factor CHD8 interacts with elongating RNA polymerase II and controls expression of the cyclin E2 gene. Nucleic Acids Res. 2009, 37, 2449–2460. [Google Scholar] [CrossRef]
- Dejligbjerg, M.; Grauslund, M.; Christensen, I.J.; Tjornelund, J.; Buhl Jensen, P.; Sehested, M. Identification of predictive biomarkers for the histone deacetylase inhibitor belinostat in a panel of human cancer cell lines. Cancer Biomark. 2008, 4, 101–109. [Google Scholar]
- Humeniuk, R.; Mishra, P.J.; Bertino, J.R.; Banerjee, D. Epigenetic reversal of acquired resistance to 5-fluorouracil treatment. Mol. Cancer Ther. 2009, 8, 1045–1054. [Google Scholar]
- Pasti, C.; Gallois-Montbrun, S.; Munier-Lehmann, H.; Veron, M.; Gilles, A.M.; Deville-Bonne, D. Reaction of human UMP-CMP kinase with natural and analog substrates. Eur. J. Biochem. 2003, 270, 1784–1790. [Google Scholar] [CrossRef]
- Cheetham, S.; Tang, M.J.; Mesak, F.; Kennecke, H.; Owen, D.; Tai, I.T. SPARC promoter hypermethylation in colorectal cancers can be reversed by 5-Aza-2'deoxycytidine to increase SPARC expression and improve therapy response. Br. J. Cancer 2008, 98, 1810–1819. [Google Scholar] [CrossRef]
- Liu, F.; Liu, Q.; Yang, D.; Bollag, W.B.; Robertson, K.; Wu, P.; Liu, K. Verticillin A overcomes apoptosis resistance in human colon carcinoma through DNA methylation-dependent upregulation of BNIP3. Cancer Res. 2011, 71, 6807–6816. [Google Scholar] [CrossRef]
- Sugita, H.; Iida, S.; Inokuchi, M.; Kato, K.; Ishiguro, M.; Ishikawa, T.; Takagi, Y.; Enjoji, M.; Yamada, H.; Uetake, H.; Kojima, K.; Sugihara, K. Methylation of BNIP3 and DAPK indicates lower response to chemotherapy and poor prognosis in gastric cancer. Oncol. Rep. 2011, 25, 513–518. [Google Scholar]
- Deng, Q.; Huang, C.M.; Chen, N.; Li, L.; Wang, X.D.; Zhang, W.; Bi, F.; Tang, Q.L.; Li, Z.P.; Wang, W. Chemotherapy and Radiotherapy Downregulate the Activity and Expression of DNA Methyltransferase and Enhance Bcl-2/E1B-19-kDa Interacting Protein-3-Induced Apoptosis in Human Colorectal Cancer Cells. Chemotherapy 2012, 58, 445–453. [Google Scholar] [CrossRef]
- Di Gennaro, E.; Bruzzese, F.; Pepe, S.; Leone, A.; Delrio, P.; Subbarayan, P.R.; Avallone, A.; Budillon, A. Modulation of thymidilate synthase and p53 expression by HDAC inhibitor vorinostat resulted in synergistic antitumor effect in combination with 5FU or raltitrexed. Cancer Biol. Ther. 2009, 8, 782–791. [Google Scholar] [CrossRef]
- Claerhout, S.; Lim, J.Y.; Choi, W.; Park, Y.Y.; Kim, K.; Kim, S.B.; Lee, J.S.; Mills, G.B.; Cho, J.Y. Gene expression signature analysis identifies vorinostat as a candidate therapy for gastric cancer. PLoS One 2011, 6, e24662. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, J.H.; Jung, Y.; Kim, J.H.; Jong, H.S.; Kim, T.Y.; Bang, Y.J. Histone deacetylase inhibitor enhances 5-fluorouracil cytotoxicity by down-regulating thymidylate synthase in human cancer cells. Mol. Cancer Ther. 2006, 5, 3085–3095. [Google Scholar] [CrossRef]
- Tumber, A.; Collins, L.S.; Petersen, K.D.; Thougaard, A.; Christiansen, S.J.; Dejligbjerg, M.; Jensen, P.B.; Sehested, M.; Ritchie, J.W. The histone deacetylase inhibitor PXD101 synergises with 5-fluorouracil to inhibit colon cancer cell growth in vitro and in vivo. Cancer Chemother. Pharmacol. 2007, 60, 275–283. [Google Scholar] [CrossRef]
- Fazzone, W.; Wilson, P.M.; Labonte, M.J.; Lenz, H.J.; Ladner, R.D. Histone deacetylase inhibitors suppress thymidylate synthase gene expression and synergize with the fluoropyrimidines in colon cancer cells. Int. J. Cancer 2009, 125, 463–473. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bardhan, K.; Liu, K. Epigenetics and Colorectal Cancer Pathogenesis. Cancers 2013, 5, 676-713. https://doi.org/10.3390/cancers5020676
Bardhan K, Liu K. Epigenetics and Colorectal Cancer Pathogenesis. Cancers. 2013; 5(2):676-713. https://doi.org/10.3390/cancers5020676
Chicago/Turabian StyleBardhan, Kankana, and Kebin Liu. 2013. "Epigenetics and Colorectal Cancer Pathogenesis" Cancers 5, no. 2: 676-713. https://doi.org/10.3390/cancers5020676
APA StyleBardhan, K., & Liu, K. (2013). Epigenetics and Colorectal Cancer Pathogenesis. Cancers, 5(2), 676-713. https://doi.org/10.3390/cancers5020676