Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention



Abstract
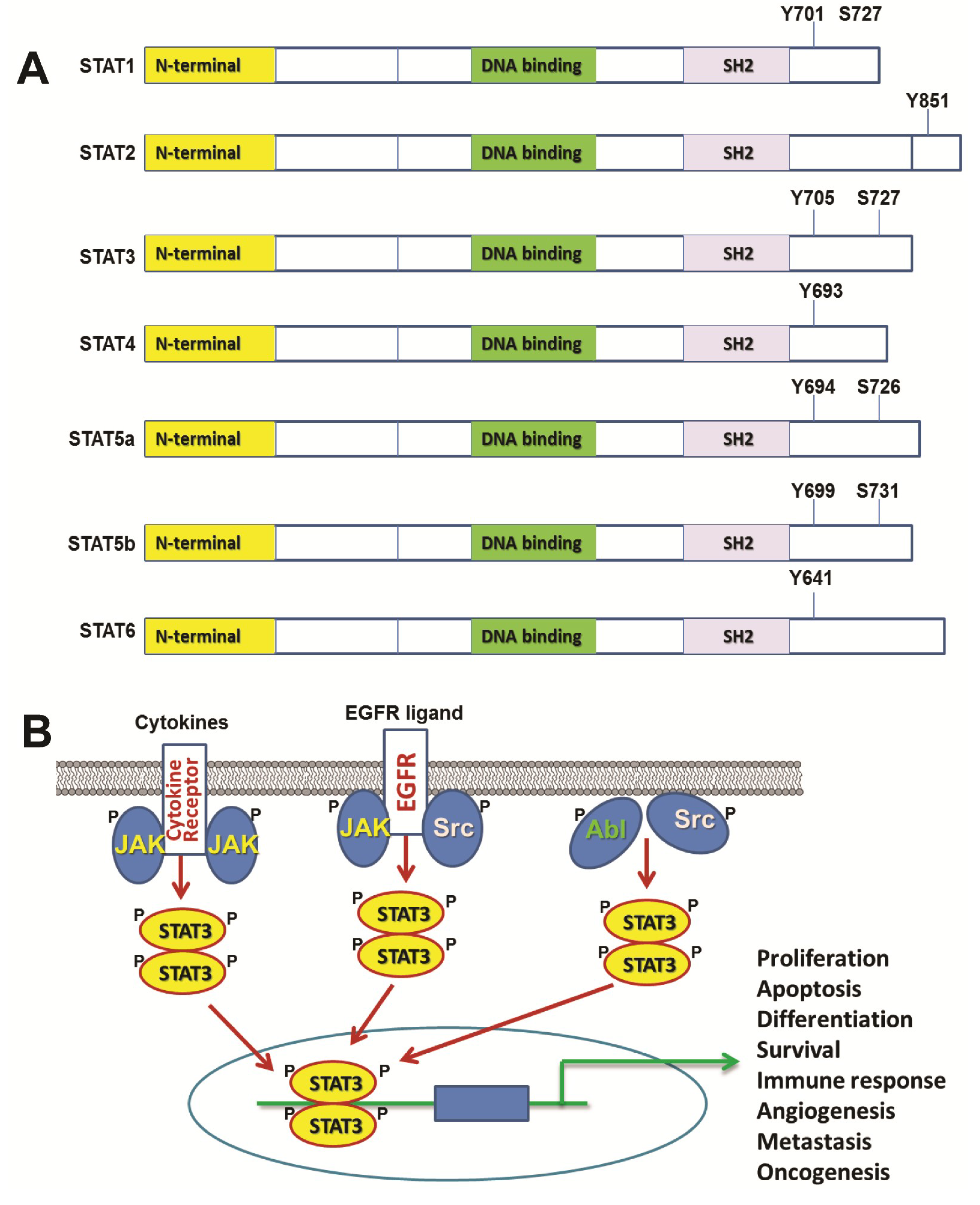
:1. Introduction of STAT Family of Transcription Factors and STAT3

2. STAT3 in Normal Cells and Development
2.1. STAT3 in Proliferation and Apoptosis
2.2. STAT3 and Differentiation
2.3. STAT3 and Immune System
2.4. STAT3 and Stem Cells
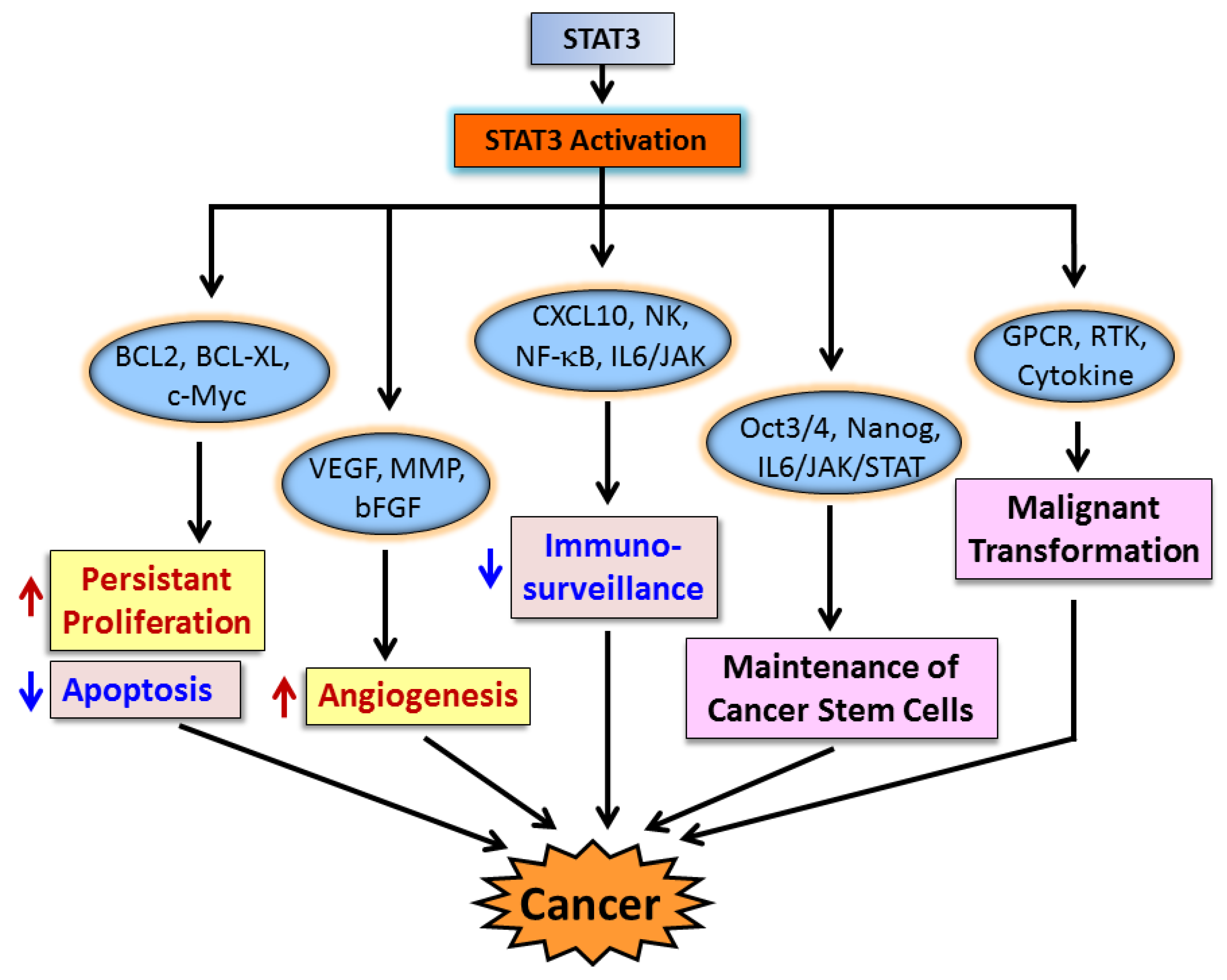
3. STAT3 in Cancer Cells and Cancer Progression
3.1. STAT3 in Cancer Cell Proliferation and Apoptosis
3.2. STAT3 in Angiogenesis
3.3. STAT3 and Immune Evasion
3.4. STAT3 and Cancer Stem Cells
4. STAT3 in Malignant Transformation
4.1. G-Protein Coupled Receptor Signaling Pathway
4.2. Receptor Tyrosine Kinase (RTK) Signaling Pathway
4.3. Cytokine/JAK/STAT3 Signaling Pathway
5. STAT3 Contributes to Early Carcinogenesis in Animal Models
6. Role of STAT3 in Cancer Prevention
6.1. STAT3 Regulates Stem Cell-Like Breast Cancer Cells
6.2. Targeting STAT3 for the Prevention of ER-Negative and Triple-Negative Breast Cancer
6.3. Targeting STAT3 for the Prevention of ER-Positive, SERM/Aromatase Inhibitor-Resistant Breast Cancer
6.4. Targeting STAT3 for the Prevention of Other Cancer Types
7. STAT3 Inhibitors as Potential Cancer Preventive Agents
7.1. SH2 Domain Inhibitors

{kind=link}
{kind=link}
| Inhibitor Name | Mechanism of Action | Selectivity | Cancer Type | Reference |
|---|---|---|---|---|
| PY*LKTK | SH2 domain inhibitor | STAT3 | NIH 3T3/v-Src fibroblasts | [139] |
| S31-M2001 | SH2 domain inhibitor | STAT3 | Breast cancer | [140] |
| S31-1757 | SH2 domain inhibitor | STAT3 | Breast and lung cancer | [141] |
| Curcumin-proline | SH2 domain inhibitor | STAT3 | [142] | |
| Cryptotashinone | SH2 domain inhibitor | STAT3 | Prostate cancer | [143] |
| STA-21 | SH2 domain inhibitor | Breast cancer | [144] | |
| Stattic | SH2 domain inhibitor | STAT3 | Breast cancer | [145] |
| S3I-201 | SH2 domain inhibitor | STAT3 | Breast cancer, prostate cancer, acute myeloid leukemia and human multiple myeloma | [146] |
| BP-1-102 | SH2 domain inhibitor | STAT3 | Breast and Lung cancer | [147] |
| Celecoxib | SH2 domain inhibitor | STAT3 | Human rhabdomyosarcoma | [148] |
| SPI | SH2 domain inhibitor | STAT3 | Breast, pancreatic, prostate, and non-small cell lung cancer cells | [149] |
| HIC 1 | DNA binding domain inhibitor | STAT3 | Breast cancer | [150] |
| IS3-295 | DNA binding domain inhibitor | STAT3 | Colon tumor | [151] |
| DBD-1 | DNA binding domain inhibitor | STAT3 | Melanoma | [152] |
| ST3-H2A2 | N-terminal domain inhibitor | STAT3 | Prostate cancer | [153] |
| G-quartet ODN | Oligonucleotide inhibitor | STAT3 | Head and neck, breast and prostate cancers | [154,155,156,157] |
| SiRNA to STAT3 | SiRNA | STAT3 | Laryngeal, breast, lymphoma, prostate and melanoma | [158,159,160,161,162] |
| KDI1 | RTK inhibitor | STAT3 | Vulval and breast cancer | [163] |
| PD153035 | RTK inhibitor | STAT3 | Oral squamous carcinoma | [164] |
| Ponatinib | FGFR inhibitor | STAT3 | Rhabdomyosarcoma | [165] |
| AG490 | JAK kinase inhibitor | STAT3 | Pancreatic cancer | [166] |
| SHP1 | STAT3 inhibitor | STAT3 | Multiple myeloma and head and neck squamous carcinoma cells | [167] |
| WP1066 | JAK kinase inhibitor | STAT3 | Acute myelogenous leukemia | [168,169] |
| TG101209 | JAK2 kinase inhibitor | STAT3,5 | Acute myeloid leukemia | [170] |
| AZD1480 | JAK kinase inhibitor | STAT3 | Myeloma,Neuroblastoma and Pediatric Sarcomas | [171,172] |
| Dasatinib | Src and PDGF inhibitor | STAT3 | Synovial sarcoma, hepatocellular carcinoma, glioma, prostate cancer | [173,174,175,176,177] |
| PP2 | Src inhibitor | STAT3 and Src | Intestinal epithelial cell | [178] |
| KX2-391 | Src inhibitor | Prostate cancer | [179] | |
| AZD0530 | Src inhibitor | STAT3 | Melanoma | [180] |
| E738 | Src and JAK inhibitor | STAT3 | Pancreatic cancer | [181] |
| MLS-2384 | Src and JAK inhibitor | STAT3 | Prostate, breast, skin, ovarian, lung, and liver cancer | [182] |
| Sophoraflavanone G | Src and JAK inhibitor | STAT3,5 | Breast, prostate, lymphoma,human multiple myeloma,large cell lung cancer, colorectal carcinoma | [183] |
| SHP2 | STAT3 inhibitor | STAT3 | Chronic myeloid leukemia | [184] |
| HJC0152 | STAT3 inhibitor | STAT3 | Breast cancer | [54,55] |
| HJC0123 | STAT3 inhibitor | STAT3 | Breast cancer | [54,55] |
| Xanthohumol | STAT3 and EGFR inhibitor | STAT3 | Breast cancer | [185] |
| Brevilin A | JAKs inhibitor | STAT3 | Breast cancer | [186] |
| Benzyl isothiocyanate | STAT3 inhibitor | STAT3 | Breast and pancreatic cancer | [187] |
7.2. Inhibitors for STAT3 DNA Binding Domain
7.3. Inhibitors Blocking STAT3 N-terminal Domain
7.4. Oligonucleotide Inhibitors for STAT3
7.5. Inhibitors of Receptor Tyrosine Kinase Activity
7.6. Inhibitors for JAK and Src Kinases
7.7. Natural Products that Inhibit STAT3 Activity
7.8. Orally Bioavailable STAT3 Inhibitors through Fragment-Based Drug Design Approach
8. Conclusions: Future Direction and Perspective

Acknowledgments
Conflicts of Interest
References
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar]
- Ihle, J.N. The Stat family in cytokine signaling. Curr. Opin. Cell Biol. 2001, 13, 211–217. [Google Scholar]
- Dimberg, L.Y.; Dimberg, A.; Ivarsson, K.; Fryknas, M.; Rickardson, L.; Tobin, G.; Ekman, S.; Larsson, R.; Gullberg, U.; Nilsson, K.; et al. Stat1 activation attenuates IL-6 induced Stat3 activity but does not alter apoptosis sensitivity in multiple myeloma. BMC Cancer 2012, 12, 318. [Google Scholar]
- Grandis, J.R.; Drenning, S.D.; Chakraborty, A.; Zhou, M.Y.; Zeng, Q.; Pitt, A.S.; Tweardy, D.J. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth In vitro. J. Clin. Investig. 1998, 102, 1385–1392. [Google Scholar] [CrossRef]
- Rane, S.G.; Reddy, E.P. Janus kinases: Components of multiple signaling pathways. Oncogene 2000, 19, 5662–5679. [Google Scholar] [CrossRef]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Choi, J.Y.; Li, W.L.; Kouri, R.E.; Yu, J.; Kao, F.T.; Ruano, G. Assignment of the acute phase response factor (APRF) gene to 17q21 by microdissection clone sequencing and fluorescence in situ hybridization of a P1 clone. Genomics 1996, 37, 264–265. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Wang, L.; Wei, W.; Kane, A.B.; Chin, Y.E. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol. Cell Biol. 2004, 24, 9390–9400. [Google Scholar] [CrossRef]
- Silva, C.M. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–8023. [Google Scholar] [CrossRef]
- Frank, D.A.; Mahajan, S.; Ritz, J. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J. Clin. Investig. 1997, 100, 3140–3148. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Wen, Z.; Darnell, J.E., Jr. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997, 25, 2062–2067. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef]
- Lim, C.P.; Cao, X. Serine phosphorylation and negative regulation of Stat3 by JNK. J. Biol. Chem. 1999, 274, 31055–31061. [Google Scholar] [CrossRef]
- Chung, J.; Uchida, E.; Grammer, T.C.; Blenis, J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell Biol. 1997, 17, 6508–6516. [Google Scholar]
- Zhang, W.; Chan, R.J.; Chen, H.; Yang, Z.; He, Y.; Zhang, X.; Luo, Y.; Yin, F.; Moh, A.; Miller, L.C.; et al. Negative regulation of Stat3 by activating PTPN11 mutants contributes to the pathogenesis of Noonan syndrome and juvenile myelomonocytic leukemia. J. Biol. Chem. 2009, 284, 22353–22363. [Google Scholar] [CrossRef]
- Hong, F.; Jaruga, B.; Kim, W.H.; Radaeva, S.; El-Assal, O.N.; Tian, Z.; Nguyen, V.A.; Gao, B. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: Regulation by SOCS. J. Clin. Investig. 2002, 110, 1503–1513. [Google Scholar] [CrossRef]
- Flowers, L.O.; Subramaniam, P.S.; Johnson, H.M. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene 2005, 24, 2114–2120. [Google Scholar] [CrossRef]
- Groner, B.; Lucks, P.; Borghouts, C. The function of Stat3 in tumor cells and their microenvironment. Semin. Cell Dev. Biol. 2008, 19, 341–350. [Google Scholar] [CrossRef]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar]
- Gronowski, A.M.; Zhong, Z.; Wen, Z.; Thomas, M.J.; Darnell, J.E., Jr.; Rotwein, P. In vivo growth hormone treatment rapidly stimulates the tyrosine phosphorylation and activation of Stat3. Mol. Endocrinol. 1995, 9, 171–177. [Google Scholar]
- Campbell, G.S.; Meyer, D.J.; Raz, R.; Levy, D.E.; Schwartz, J.; Carter-Su, C. Activation of acute phase response factor (APRF)/Stat3 transcription factor by growth hormone. J. Biol. Chem. 1995, 270, 3974–3979. [Google Scholar]
- Zhang, S.S.; Liu, M.G.; Kano, A.; Zhang, C.; Fu, X.Y.; Barnstable, C.J. STAT3 activation in response to growth factors or cytokines participates in retina precursor proliferation. Exp. Eye Res. 2005, 81, 103–115. [Google Scholar] [CrossRef]
- Hilfiker-Kleiner, D.; Limbourg, A.; Drexler, H. STAT3-mediated activation of myocardial capillary growth. Trends Cardiovasc. Med. 2005, 15, 152–157. [Google Scholar] [CrossRef]
- Fukada, T.; Hibi, M.; Yamanaka, Y.; Takahashi-Tezuka, M.; Fujitani, Y.; Yamaguchi, T.; Nakajima, K.; Hirano, T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: Involvement of STAT3 in anti-apoptosis. Immunity 1996, 5, 449–460. [Google Scholar] [CrossRef]
- Daino, H.; Matsumura, I.; Takada, K.; Odajima, J.; Tanaka, H.; Ueda, S.; Shibayama, H.; Ikeda, H.; Hibi, M.; Machii, T.; et al. Induction of apoptosis by extracellular ubiquitin in human hematopoietic cells: Possible involvement of STAT3 degradation by proteasome pathway in interleukin 6-dependent hematopoietic cells. Blood 2000, 95, 2577–2585. [Google Scholar]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.M.; Yang-Yen, H.F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar]
- Shirogane, T.; Fukada, T.; Muller, J.M.; Shima, D.T.; Hibi, M.; Hirano, T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity 1999, 11, 709–719. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, Y.; Li, W.; Wang, G.; Song, Y.; Yang, G.; Han, X.; Du, Z.; Sun, L.; Ma, K. STAT3 induces muscle stem cell differentiation by interaction with myoD. Cytokine 2009, 46, 137–141. [Google Scholar] [CrossRef]
- Wang, K.; Wang, C.; Xiao, F.; Wang, H.; Wu, Z. JAK2/STAT2/STAT3 are required for myogenic differentiation. J. Biol. Chem. 2008, 283, 34029–34036. [Google Scholar]
- Yanagisawa, M.; Nakashima, K.; Arakawa, H.; Ikenaka, K.; Yoshida, K.; Kishimoto, T.; Hisatsune, T.; Taga, T. Astrocyte differentiation of fetal neuroepithelial cells by interleukin-11 via activation of a common cytokine signal transducer, gp130, and a transcription factor, STAT3. J. Neurochem. 2000, 74, 1498–1504. [Google Scholar]
- Lee, S.; Shen, R.; Cho, H.H.; Kwon, R.J.; Seo, S.Y.; Lee, J.W.; Lee, S.K. STAT3 promotes motor neuron differentiation by collaborating with motor neuron-specific LIM complex. Proc. Natl. Acad. Sci. USA 2013, 110, 11445–11450. [Google Scholar] [CrossRef]
- Itoh, S.; Udagawa, N.; Takahashi, N.; Yoshitake, F.; Narita, H.; Ebisu, S.; Ishihara, K. A critical role for interleukin-6 family-mediated Stat3 activation in osteoblast differentiation and bone formation. Bone 2006, 39, 505–512. [Google Scholar] [CrossRef]
- Wu, R.; Sun, S.; Steinberg, B.M. Requirement of STAT3 activation for differentiation of mucosal stratified squamous epithelium. Mol. Med. 2003, 9, 77–84. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Ernst, M.; Novak, U.; Nicholson, S.E.; Layton, J.E.; Dunn, A.R. The carboxyl-terminal domains of gp130-related cytokine receptors are necessary for suppressing embryonic stem cell differentiation. Involvement of STAT3. J. Biol. Chem. 1999, 274, 9729–9737. [Google Scholar]
- Suzuki, R.; Sakamoto, H.; Yasukawa, H.; Masuhara, M.; Wakioka, T.; Sasaki, A.; Yuge, K.; Komiya, S.; Inoue, A.; Yoshimura, A. CIS3 and JAB have different regulatory roles in interleukin-6 mediated differentiation and STAT3 activation in M1 leukemia cells. Oncogene 1998, 17, 2271–2278. [Google Scholar]
- Tomida, M.; Heike, T.; Yokota, T. Cytoplasmic domains of the leukemia inhibitory factor receptor required for STAT3 activation, differentiation, and growth arrest of myeloid leukemic cells. Blood 1999, 93, 1934–1941. [Google Scholar]
- Chakraborty, A.; Tweardy, D.J. Stat3 and G-CSF-induced myeloid differentiation. Leuk. Lymphoma 1998, 30, 433–442. [Google Scholar]
- Shimozaki, K.; Nakajima, K.; Hirano, T.; Nagata, S. Involvement of STAT3 in the granulocyte colony-stimulating factor-induced differentiation of myeloid cells. J. Biol. Chem. 1997, 272, 25184–25189. [Google Scholar] [CrossRef]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef]
- Park, S.J.; Nakagawa, T.; Kitamura, H.; Atsumi, T.; Kamon, H.; Sawa, S.; Kamimura, D.; Ueda, N.; Iwakura, Y.; Ishihara, K.; et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 2004, 173, 3844–3854. [Google Scholar]
- Rajasingh, J.; Bord, E.; Hamada, H.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. STAT3-dependent mouse embryonic stem cell differentiation into cardiomyocytes: Analysis of molecular signaling and therapeutic efficacy of cardiomyocyte precommitted mES transplantation in a mouse model of myocardial infarction. Circ. Res. 2007, 101, 910–918. [Google Scholar] [CrossRef]
- You, W.; Tang, Q.; Zhang, C.; Wu, J.; Gu, C.; Wu, Z.; Li, X. IL-26 promotes the proliferation and survival of human gastric cancer cells by regulating the balance of STAT1 and STAT3 activation. PLoS One 2013, 8, e63588. [Google Scholar]
- Liu, Y.; Lv, L.; Xiao, W.; Gong, C.; Yin, J.; Wang, D.; Sheng, H. Leptin activates STAT3 and ERK1/2 pathways and induces endometrial cancer cell proliferation. J. Huazhong. Univ. Sci. Technolog. Med. Sci. 2011, 31, 365–370. [Google Scholar] [CrossRef]
- Chen, R.J.; Ho, Y.S.; Guo, H.R.; Wang, Y.J. Rapid activation of Stat3 and ERK1/2 by nicotine modulates cell proliferation in human bladder cancer cells. Toxicol. Sci. 2008, 104, 283–293. [Google Scholar] [CrossRef]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufmann, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef]
- Horiguchi, A.; Oya, M.; Marumo, K.; Murai, M. STAT3, but not ERKs, mediates the IL-6-induced proliferation of renal cancer cells, ACHN and 769P. Kidney Int. 2002, 61, 926–938. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, X.L.; Wang, C.J.; Lin, D.C.; Ruan, X.; Feng, Y.B.; Huo, Y.Q.; Peng, H.; Cui, J.L.; Zhang, T.T.; et al. Reciprocal activation between PLK1 and Stat3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology 2012, 142, 521–530.e3. [Google Scholar] [CrossRef]
- Lin, L.; Liu, A.; Peng, Z.; Lin, H.J.; Li, P.K.; Li, C.; Lin, J. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011, 71, 7226–7237. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, J.G.; Shi, Y.; Qin, Q.; Liu, Y.; Wang, B.; Tian, K.; Deng, S.C.; Li, X.; Zhu, S.; et al. MiR-130b is a prognostic marker and inhibits cell proliferation and invasion in pancreatic cancer through targeting STAT3. PLoS One 2013, 8, e73803. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Z.; Ding, C.; Chu, L.; Zhang, Y.; Terry, K.; Liu, H.; Shen, Q.; Zhou, J. Discovery of O-Alkylamino tethered niclosamide derivatives as potent and orally bioavailable anticancer agents. ACS Med. Chem. Lett. 2013, 4, 180–185. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Z.; Ding, C.; Chu, L.; Zhang, Y.; Terry, K.; Liu, H.; Shen, Q.; Zhou, J. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur. J. Med. Chem. 2013, 62, 498–507. [Google Scholar] [CrossRef]
- Lin, W.; Zheng, L.; Zhuang, Q.; Zhao, J.; Cao, Z.; Zeng, J.; Lin, S.; Xu, W.; Peng, J. Spica prunellae promotes cancer cell apoptosis, inhibits cell proliferation and tumor angiogenesis in a mouse model of colorectal cancer via suppression of stat3 pathway. BMC Complement. Altern. Med. 2013, 13, 144. [Google Scholar] [CrossRef]
- Zhuang, Q.; Hong, F.; Shen, A.; Zheng, L.; Zeng, J.; Lin, W.; Chen, Y.; Sferra, T.J.; Hong, Z.; Peng, J. Pien Tze Huang inhibits tumor cell proliferation and promotes apoptosis via suppressing the STAT3 pathway in a colorectal cancer mouse model. Int. J. Oncol. 2012, 40, 1569–1574. [Google Scholar]
- Kanai, M.; Konda, Y.; Nakajima, T.; Izumi, Y.; Kanda, N.; Nanakin, A.; Kubohara, Y.; Chiba, T. Differentiation-inducing factor-1 (DIF-1) inhibits STAT3 activity involved in gastric cancer cell proliferation via MEK-ERK-dependent pathway. Oncogene 2003, 22, 548–554. [Google Scholar] [CrossRef]
- Pancotti, F.; Roncuzzi, L.; Maggiolini, M.; Gasperi-Campani, A. Caveolin-1 silencing arrests the proliferation of metastatic lung cancer cells through the inhibition of STAT3 signaling. Cell. Signal. 2012, 24, 1390–1397. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- McDougall, S.R.; Anderson, A.R.; Chaplain, M.A. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: Clinical implications and therapeutic targeting strategies. J. Theor. Biol. 2006, 241, 564–589. [Google Scholar] [CrossRef]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef]
- Kang, S.H.; Yu, M.O.; Park, K.J.; Chi, S.G.; Park, D.H.; Chung, Y.G. Activated STAT3 regulates hypoxia-induced angiogenesis and cell migration in human glioblastoma. Neurosurgery 2010, 67, 1386–1395. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Jee, S.H.; Chu, C.Y.; Chiu, H.C.; Huang, Y.L.; Tsai, W.L.; Liao, Y.H.; Kuo, M.L. Interleukin-6 induced basic fibroblast growth factor-dependent angiogenesis in basal cell carcinoma cell line via JAK/STAT3 and PI3-kinase/Akt pathways. J. Investig. Dermatol. 2004, 123, 1169–1175. [Google Scholar] [CrossRef]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Saudemont, A.; Jouy, N.; Hetuin, D.; Quesnel, B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood 2005, 105, 2428–2435. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Gibbs, C.P.; Kukekov, V.G.; Reith, J.D.; Tchigrinova, O.; Suslov, O.N.; Scott, E.W.; Ghivizzani, S.C.; Ignatova, T.N.; Steindler, D.A. Stem-like cells in bone sarcomas: Implications for tumorigenesis. Neoplasia 2005, 7, 967–976. [Google Scholar] [CrossRef]
- Cao, Y.; Lathia, J.D.; Eyler, C.E.; Wu, Q.; Li, Z.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Erythropoietin receptor signaling through stat3 is required for glioma stem cell maintenance. Genes Cancer 2010, 1, 50–61. [Google Scholar] [CrossRef]
- Villalva, C.; Martin-Lanneree, S.; Cortes, U.; Dkhissi, F.; Wager, M.; le Corf, A.; Tourani, J.M.; Dusanter-Fourt, I.; Turhan, A.G.; Karayan-Tapon, L. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: A potential for targeted therapy? Int. J. Cancer 2011, 128, 826–838. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro. Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Zhao, Y.; Liu, X.; Ding, Q.; Shi, J.; Ding, Y.; Wang, S. STAT3 mediates resistance of CD44(+)CD24(−/low) breast cancer stem cells to tamoxifen in vitro. J. Biomed. Res. 2012, 26, 325–335. [Google Scholar] [CrossRef]
- Tseng, L.M.; Huang, P.I.; Chen, Y.R.; Chen, Y.C.; Chou, Y.C.; Chen, Y.W.; Chang, Y.L.; Hsu, H.S.; Lan, Y.T.; Chen, K.H.; et al. Targeting signal transducer and activator of transcription 3 pathway by cucurbitacin I diminishes self-renewing and radiochemoresistant abilities in thyroid cancer-derived CD133+ cells. J. Pharmacol. Exp. Ther. 2012, 341, 410–423. [Google Scholar] [CrossRef]
- Hellsten, R.; Johansson, M.; Dahlman, A.; Sterner, O.; Bjartell, A. Galiellalactone inhibits stem cell-like ALDH-positive prostate cancer cells. PLoS One 2011, 6, e22118. [Google Scholar]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Liang, H.; Venema, V.J.; Wang, X.; Ju, H.; Venema, R.C.; Marrero, M.B. Regulation of angiotensin II-induced phosphorylation of STAT3 in vascular smooth muscle cells. J. Biol. Chem 1999, 274, 19846–19851. [Google Scholar]
- Arvanitakis, L.; Geras-Raaka, E.; Varma, A.; Gershengorn, M.C.; Cesarman, E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 1997, 385, 347–350. [Google Scholar] [CrossRef]
- Cesarman, E.; Nador, R.G.; Bai, F.; Bohenzky, R.A.; Russo, J.J.; Moore, P.S.; Chang, Y.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi’s sarcoma and malignant lymphoma. J. Virol. 1996, 70, 8218–8223. [Google Scholar]
- Jiang, H.; Wu, D.; Simon, M.I. The transforming activity of activated G alpha 12. FEBS Lett. 1993, 330, 319–322. [Google Scholar] [CrossRef]
- Kalinec, G.; Nazarali, A.J.; Hermouet, S.; Xu, N.; Gutkind, J.S. Mutated alpha subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol. Cell Biol. 1992, 12, 4687–4693. [Google Scholar]
- Vara Prasad, M.V.; Shore, S.K.; Dhanasekaran, N. Activated mutant of G alpha 13 induces Egr-1, c-fos, and transformation in NIH 3T3 cells. Oncogene 1994, 9, 2425–2429. [Google Scholar]
- Corre, I.; Baumann, H.; Hermouet, S. Regulation by Gi2 proteins of v-fms-induced proliferation and transformation via Src-kinase and STAT3. Oncogene 1999, 18, 6335–6342. [Google Scholar] [CrossRef]
- Ram, P.T.; Iyengar, R. G protein coupled receptor signaling through the Src and Stat3 pathway: Role in proliferation and transformation. Oncogene 2001, 20, 1601–1606. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef]
- Brown, M.T.; Cooper, J.A. Regulation, substrates and functions of src. Biochim. Biophys. Acta 1996, 1287, 121–149. [Google Scholar]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell Biol. 1998, 18, 2553–2558. [Google Scholar]
- Turkson, J.; Bowman, T.; Adnane, J.; Zhang, Y.; Djeu, J.Y.; Sekharam, M.; Frank, D.A.; Holzman, L.B.; Wu, J.; Sebti, S.; et al. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol. Cell Biol. 1999, 19, 7519–7528. [Google Scholar]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar]
- Ceresa, B.P.; Horvath, C.M.; Pessin, J.E. Signal transducer and activator of transcription-3 serine phosphorylation by insulin is mediated by a Ras/Raf/MEK-dependent pathway. Endocrinology 1997, 138, 4131–4137. [Google Scholar]
- Goi, T.; Shipitsin, M.; Lu, Z.; Foster, D.A.; Klinz, S.G.; Feig, L.A. An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. EMBO J. 2000, 19, 623–630. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Yarden, Y.; Ullrich, A. Growth factor receptor tyrosine kinases. Annu. Rev. Biochem. 1988, 57, 443–478. [Google Scholar] [CrossRef]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef]
- Kaptein, A.; Paillard, V.; Saunders, M. Dominant negative stat3 mutant inhibits interleukin-6-induced Jak-STAT signal transduction. J. Biol. Chem. 1996, 271, 5961–5964. [Google Scholar]
- Stahl, N.; Farruggella, T.J.; Boulton, T.G.; Zhong, Z.; Darnell, J.E., Jr.; Yancopoulos, G.D. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995, 267, 1349–1353. [Google Scholar] [CrossRef]
- Shuai, K.; Ziemiecki, A.; Wilks, A.F.; Harpur, A.G.; Sadowski, H.B.; Gilman, M.Z.; Darnell, J.E. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature 1993, 366, 580–583. [Google Scholar] [CrossRef]
- Wright, J.D.; Reuter, C.W.; Weber, M.J. An incomplete program of cellular tyrosine phosphorylations induced by kinase-defective epidermal growth factor receptors. J. Biol. Chem. 1995, 270, 12085–12093. [Google Scholar] [CrossRef]
- Ihle, J.N.; Kerr, I.M. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995, 11, 69–74. [Google Scholar]
- Ihle, J.N.; Witthuhn, B.A.; Quelle, F.W.; Yamamoto, K.; Thierfelder, W.E.; Kreider, B.; Silvennoinen, O. Signaling by the cytokine receptor superfamily: JAKs and STATs. Trends Biochem. Sci. 1994, 19, 222–227. [Google Scholar] [CrossRef]
- Muller, M.; Briscoe, J.; Laxton, C.; Guschin, D.; Ziemiecki, A.; Silvennoinen, O.; Harpur, A.G.; Barbieri, G.; Witthuhn, B.A.; Schindler, C.; et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature 1993, 366, 129–135. [Google Scholar] [CrossRef]
- Wilks, A.F. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1989, 86, 1603–1607. [Google Scholar] [CrossRef]
- Heim, M.H.; Kerr, I.M.; Stark, G.R.; Darnell, J.E., Jr. Contribution of STAT SH2 groups to specific interferon signaling by the Jak-STAT pathway. Science 1995, 267, 1347–1349. [Google Scholar] [CrossRef]
- Luo, F.; Xu, Y.; Ling, M.; Zhao, Y.; Xu, W.; Liang, X.; Jiang, R.; Wang, B.; Bian, Q.; Liu, Q. Arsenite evokes IL-6 secretion, autocrine regulation of STAT3 signaling, and miR-21 expression, processes involved in the EMT and malignant transformation of human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 2013, 273, 27–34. [Google Scholar] [CrossRef]
- Chan, K.S.; Sano, S.; Kiguchi, K.; Anders, J.; Komazawa, N.; Takeda, J.; DiGiovanni, J. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Investig. 2004, 114, 720–728. [Google Scholar] [CrossRef]
- Chan, K.S.; Sano, S.; Kataoka, K.; Abel, E.; Carbajal, S.; Beltran, L.; Clifford, J.; Peavey, M.; Shen, J.; Digiovanni, J. Forced expression of a constitutively active form of Stat3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene 2008, 27, 1087–1094. [Google Scholar] [CrossRef]
- Kataoka, K.; Kim, D.J.; Carbajal, S.; Clifford, J.L.; DiGiovanni, J. Stage-specific disruption of Stat3 demonstrates a direct requirement during both the initiation and promotion stages of mouse skin tumorigenesis. Carcinogenesis 2008, 29, 1108–1114. [Google Scholar] [CrossRef]
- De Andrea, M.; Ritta, M.; Landini, M.M.; Borgogna, C.; Mondini, M.; Kern, F.; Ehrenreiter, K.; Baccarini, M.; Marcuzzi, G.P.; Smola, S.; et al. Keratinocyte-specific stat3 heterozygosity impairs development of skin tumors in human papillomavirus 8 transgenic mice. Cancer Res. 2010, 70, 7938–7948. [Google Scholar] [CrossRef]
- Blando, J.M.; Carbajal, S.; Abel, E.; Beltran, L.; Conti, C.; Fischer, S.; DiGiovanni, J. Cooperation between Stat3 and Akt signaling leads to prostate tumor development in transgenic mice. Neoplasia 2011, 13, 254–265. [Google Scholar]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmaki, H.; Andersson, E.I.; Lagstrom, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell. Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Ferriere, F.; Habauzit, D.; Pakdel, F.; Saligaut, C.; Flouriot, G. Unliganded estrogen receptor alpha promotes PC12 survival during serum starvation. PLoS One 2013, 8, e69081. [Google Scholar]
- De Miguel, F.; Lee, S.O.; Onate, S.A.; Gao, A.C. Stat3 enhances transactivation of steroid hormone receptors. Nucl. Recept. 2003, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.T.; Ou-Yang, F.; Chen, I.F.; Yang, S.F.; Wang, Y.Y.; Chuang, H.Y.; Su, J.H.; Hou, M.F.; Yuan, S.S. STAT3 ser727 phosphorylation and its association with negative estrogen receptor status in breast infiltrating ductal carcinoma. Int. J. Cancer 2006, 118, 2943–2947. [Google Scholar] [CrossRef]
- Yamamoto, T.; Matsuda, T.; Junicho, A.; Kishi, H.; Saatcioglu, F.; Muraguchi, A. Cross-talk between signal transducer and activator of transcription 3 and estrogen receptor signaling. FEBS Lett. 2000, 486, 143–148. [Google Scholar] [CrossRef]
- Wang, L.H.; Yang, X.Y.; Mihalic, K.; Xiao, W.; Li, D.; Farrar, W.L. Activation of estrogen receptor blocks interleukin-6-inducible cell growth of human multiple myeloma involving molecular cross-talk between estrogen receptor and STAT3 mediated by co-regulator PIAS3. J. Biol. Chem. 2001, 276, 31839–31844. [Google Scholar]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Suba, Z. Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: Preventive and therapeutic implications. Onco Targets Ther. 2014, 7, 147–164. [Google Scholar] [CrossRef]
- Schuringa, J.J.; Schepers, H.; Vellenga, E.; Kruijer, W. Ser727-dependent transcriptional activation by association of p300 with STAT3 upon IL-6 stimulation. FEBS Lett. 2001, 495, 71–76. [Google Scholar] [CrossRef]
- Lewis, J.S.; Jordan, V.C. Selective estrogen receptor modulators (SERMs): Mechanisms of anticarcinogenesis and drug resistance. Mutat. Res. 2005, 591, 247–263. [Google Scholar] [CrossRef]
- Neuman, E.; Ladha, M.H.; Lin, N.; Upton, T.M.; Miller, S.J.; DiRenzo, J.; Pestell, R.G.; Hinds, P.W.; Dowdy, S.F.; Brown, M.; et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol. Cell Biol. 1997, 17, 5338–5347. [Google Scholar]
- Zwijsen, R.M.; Wientjens, E.; Klompmaker, R.; van der Sman, J.; Bernards, R.; Michalides, R.J. CDK-independent activation of estrogen receptor by cyclin D1. Cell 1997, 88, 405–415. [Google Scholar] [CrossRef]
- Stendahl, M.; Kronblad, A.; Ryden, L.; Emdin, S.; Bengtsson, N.O.; Landberg, G. Cyclin D1 overexpression is a negative predictive factor for tamoxifen response in postmenopausal breast cancer patients. Br. J. Cancer 2004, 90, 1942–1948. [Google Scholar] [CrossRef]
- Jirstrom, K.; Stendahl, M.; Ryden, L.; Kronblad, A.; Bendahl, P.O.; Stal, O.; Landberg, G. Adverse effect of adjuvant tamoxifen in premenopausal breast cancer with cyclin D1 gene amplification. Cancer Res. 2005, 65, 8009–8016. [Google Scholar]
- Ishii, Y.; Waxman, S.; Germain, D. Tamoxifen stimulates the growth of cyclin D1-overexpressingbreast cancer cells by promoting the activation of signal transducer and activator of transcription 3. Cancer Ress 2008, 68, 852–860. [Google Scholar] [CrossRef]
- Klampfer, L. The role of signal transducers and activators of transcription in colon cancer. Front. Biosci. 2008, 13, 2888–2899. [Google Scholar] [CrossRef]
- Alexandrow, M.G.; Song, L.J.; Altiok, S.; Gray, J.; Haura, E.B.; Kumar, N.B. Curcumin: A novel Stat3 pathway inhibitor for chemoprevention of lung cancer. Eur. J. Cancer Prev. 2012, 21, 407–412. [Google Scholar] [CrossRef]
- Kroon, P.; Berry, P.A.; Stower, M.J.; Rodrigues, G.; Mann, V.M.; Simms, M.; Bhasin, D.; Chettiar, S.; Li, C.; Li, P.K.; et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013, 73, 5288–5298. [Google Scholar] [CrossRef]
- Hossain, D.M.; Dos Santos, C.; Zhang, Q.; Kozlowska, A.; Liu, H.; Gao, C.; Moreira, D.; Swiderski, P.; Jozwiak, A.; Kline, J.; et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood 2014, 123, 15–25. [Google Scholar] [CrossRef]
- Haura, E.B.; Turkson, J.; Jove, R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat. Clin. Pract. Oncol. 2005, 2, 315–324. [Google Scholar] [CrossRef]
- Debnath, B.; Xu, S.; Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef]
- Deng, J.; Grande, F.; Neamati, N. Small molecule inhibitors of Stat3 signaling pathway. Curr. Cancer Drug Targets 2007, 7, 91–107. [Google Scholar] [CrossRef]
- Dhanik, A.; McMurray, J.S.; Kavraki, L.E. Binding modes of peptidomimetics designed to inhibit STAT3. PLoS One 2012, 7, e51603. [Google Scholar]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef]
- Siddiquee, K.A.; Gunning, P.T.; Glenn, M.; Katt, W.P.; Zhang, S.; Schrock, C.; Sebti, S.M.; Jove, R.; Hamilton, A.D.; Turkson, J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007, 2, 787–798. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Pireddu, R.; Yang, H.; Urlam, M.K.; Lawrence, H.R.; Guida, W.C.; Lawrence, N.J.; Sebti, S.M. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933. [Google Scholar] [CrossRef]
- Kumar, A.; Bora, U. Molecular docking studies on inhibition of Stat3 dimerization by curcumin natural derivatives and its conjugates with amino acids. Bioinformation 2012, 8, 988–993. [Google Scholar] [CrossRef]
- Shin, D.S.; Kim, H.N.; Shin, K.D.; Yoon, Y.J.; Kim, S.J.; Han, D.C.; Kwon, B.M. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer Res. 2009, 69, 193–202. [Google Scholar] [CrossRef]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef]
- Fletcher, S.; Page, B.D.; Zhang, X.; Yue, P.; Li, Z.H.; Sharmeen, S.; Singh, J.; Zhao, W.; Schimmer, A.D.; Trudel, S.; et al. Antagonism of the Stat3-Stat3 protein dimer with salicylic acid based small molecules. Chem. Med. Chem. 2011, 6, 1459–1470. [Google Scholar] [CrossRef]
- Zhang, X.; Yue, P.; Page, B.D.; Li, T.; Zhao, W.; Namanja, A.T.; Paladino, D.; Zhao, J.; Chen, Y.; Gunning, P.T.; et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl. Acad. Sci. USA 2012, 109, 9623–9628. [Google Scholar] [CrossRef]
- Reed, S.; Li, H.; Li, C.; Lin, J. Celecoxib inhibits STAT3 phosphorylation and suppresses cell migration and colony forming ability in rhabdomyosarcoma cells. Biochem. Biophys. Res. Commun. 2011, 407, 450–455. [Google Scholar] [CrossRef]
- Zhao, W.; Jaganathan, S.; Turkson, J. A cell-permeable Stat3 SH2 domain mimetic inhibits Stat3 activation and induces antitumor cell effects in vitro. J. Biol. Chem. 2010, 285, 35855–35865. [Google Scholar] [CrossRef]
- Lin, Y.M.; Wang, C.M.; Jeng, J.C.; Leprince, D.; Shih, H.M. HIC1 interacts with and modulates the activity of STAT3. Cell Cycle 2013, 12, 2266–2276. [Google Scholar] [CrossRef]
- Turkson, J.; Zhang, S.; Palmer, J.; Kay, H.; Stanko, J.; Mora, L.B.; Sebti, S.; Yu, H.; Jove, R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004, 3, 1533–1542. [Google Scholar]
- Nagel-Wolfrum, K.; Buerger, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerization domain of the transcription factor Stat3 inhibits transactivation and induces apoptosis in tumor cells. Mol. Cancer Res. 2004, 2, 170–182. [Google Scholar]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef]
- Jing, N.; Li, Y.; Xu, X.; Sha, W.; Li, P.; Feng, L.; Tweardy, D.J. Targeting Stat3 with G-quartet oligodeoxynucleotides in human cancer cells. DNA Cell Biol. 2003, 22, 685–696. [Google Scholar] [CrossRef]
- Zhu, Q.; Jing, N. Computational study on mechanism of G-quartet oligonucleotide T40214 selectively targeting Stat3. J. Comput. Aided Mol. Des. 2007, 21, 641–648. [Google Scholar] [CrossRef]
- Jing, N.; Zhu, Q.; Yuan, P.; Li, Y.; Mao, L.; Tweardy, D.J. Targeting signal transducer and activator of transcription 3 with G-quartet oligonucleotides: A potential novel therapy for head and neck cancer. Mol. Cancer Ther. 2006, 5, 279–286. [Google Scholar] [CrossRef]
- Jing, N.; Li, Y.; Xiong, W.; Sha, W.; Jing, L.; Tweardy, D.J. G-quartet oligonucleotides: A new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004, 64, 6603–6609. [Google Scholar] [CrossRef]
- Gao, L.F.; Xu, D.Q.; Wen, L.J.; Zhang, X.Y.; Shao, Y.T.; Zhao, X.J. Inhibition of STAT3 expression by siRNA suppresses growth and induces apoptosis in laryngeal cancer cells. Acta Pharmacol. Sin. 2005, 26, 377–383. [Google Scholar]
- Kunigal, S.; Lakka, S.S.; Sodadasu, P.K.; Estes, N.; Rao, J.S. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int. J. Oncol. 2009, 34, 1209–1220. [Google Scholar]
- Verma, N.K.; Davies, A.M.; Long, A.; Kelleher, D.; Volkov, Y. STAT3 knockdown by siRNA induces apoptosis in human cutaneous T-cell lymphoma line Hut78 via downregulation of Bcl-xL. Cell Mol. Biol. Lett. 2010, 15, 342–355. [Google Scholar] [CrossRef]
- Liang, Z.W.; Guo, B.F.; Li, Y.; Li, X.J.; Li, X.; Zhao, L.J.; Gao, L.F.; Yu, H.; Zhao, X.J.; Zhang, L.; et al. Plasmid-based Stat3 siRNA delivered by hydroxyapatite nanoparticles suppresses mouse prostate tumour growth in vivo. Asian J. Androl. 2011, 13, 481–486. [Google Scholar] [CrossRef]
- Hong, J.; Zhao, Y.; Huang, W. Blocking c-myc and stat3 by E. coli expressed and enzyme digested siRNA in mouse melanoma. Biochem. Biophys. Res. Commun. 2006, 348, 600–605. [Google Scholar] [CrossRef]
- Buerger, C.; Nagel-Wolfrum, K.; Kunz, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. Sequence-specific peptide aptamers, interacting with the intracellular domain of the epidermal growth factor receptor, interfere with Stat3 activation and inhibit the growth of tumor cells. J. Biol. Chem. 2003, 278, 37610–37621. [Google Scholar]
- Ge, H.; Liu, H.; Fu, Z.; Sun, Z. Therapeutic and preventive effects of an epidermal growth factor receptor inhibitor on oral squamous cell carcinoma. J. Int. Med. Res. 2012, 40, 455–466. [Google Scholar] [CrossRef]
- Li, S.Q.; Cheuk, A.T.; Shern, J.F.; Song, Y.K.; Hurd, L.; Liao, H.; Wei, J.S.; Khan, J. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS One 2013, 8, e76551. [Google Scholar]
- Huang, C.; Cao, J.; Huang, K.J.; Zhang, F.; Jiang, T.; Zhu, L.; Qiu, Z.J. Inhibition of STAT3 activity with AG490 decreases the invasion of human pancreatic cancer cells in vitro. Cancer Sci. 2006, 97, 1417–1423. [Google Scholar] [CrossRef]
- Gupta, S.C.; Phromnoi, K.; Aggarwal, B.B. Morin inhibits STAT3 tyrosine 705 phosphorylation in tumor cells through activation of protein tyrosine phosphatase SHP1. Biochem. Pharmacol. 2013, 85, 898–912. [Google Scholar] [CrossRef]
- Ferrajoli, A.; Faderl, S.; Van, Q.; Koch, P.; Harris, D.; Liu, Z.; Hazan-Halevy, I.; Wang, Y.; Kantarjian, H.M.; Priebe, W.; et al. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007, 67, 11291–11299. [Google Scholar]
- Iwamaru, A.; Szymanski, S.; Iwado, E.; Aoki, H.; Yokoyama, T.; Fokt, I.; Hess, K.; Conrad, C.; Madden, T.; Sawaya, R.; et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 2007, 26, 2435–2444. [Google Scholar] [CrossRef]
- Pardanani, A.; Hood, J.; Lasho, T.; Levine, R.L.; Martin, M.B.; Noronha, G.; Finke, C.; Mak, C.C.; Mesa, R.; Zhu, H.; et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 2007, 21, 1658–1668. [Google Scholar] [CrossRef]
- Scuto, A.; Krejci, P.; Popplewell, L.; Wu, J.; Wang, Y.; Kujawski, M.; Kowolik, C.; Xin, H.; Chen, L.; Kretzner, L.; et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia 2011, 25, 538–550. [Google Scholar] [CrossRef]
- Yan, S.; Li, Z.; Thiele, C.J. Inhibition of STAT3 with orally active JAK inhibitor, AZD1480, decreases tumor growth in Neuroblastoma and Pediatric Sarcomas in vitro and in vivo. Oncotarget 2013, 4, 433–445. [Google Scholar]
- Chen, Z.; Lee, F.Y.; Bhalla, K.N.; Wu, J. Potent inhibition of platelet-derived growth factor-induced responses in vascular smooth muscle cells by BMS-354825 (dasatinib). Mol. Pharmacol. 2006, 69, 1527–1533. [Google Scholar] [CrossRef]
- Michels, S.; Trautmann, M.; Sievers, E.; Kindler, D.; Huss, S.; Renner, M.; Friedrichs, N.; Kirfel, J.; Steiner, S.; Endl, E.; et al. SRC signaling is crucial in the growth of synovial sarcoma cells. Cancer Res. 2013, 73, 2518–2528. [Google Scholar] [CrossRef]
- Chang, A.Y.; Wang, M. Molecular mechanisms of action and potential biomarkers of growth inhibition of dasatinib (BMS-354825) on hepatocellular carcinoma cells. BMC Cancer 2013, 13, 267. [Google Scholar] [CrossRef]
- Premkumar, D.R.; Jane, E.P.; Agostino, N.R.; Scialabba, J.L.; Pollack, I.F. Dasatinib synergizes with JSI-124 to inhibit growth and migration and induce apoptosis of malignant human glioma cells. J. Carcinog. 2010. [Google Scholar] [CrossRef]
- Rice, L.; Lepler, S.; Pampo, C.; Siemann, D.W. Impact of the SRC inhibitor dasatinib on the metastatic phenotype of human prostate cancer cells. Clin. Exp. Metastasis 2012, 29, 133–142. [Google Scholar] [CrossRef]
- Oyaizu, T.; Fung, S.Y.; Shiozaki, A.; Guan, Z.; Zhang, Q.; dos Santos, C.C.; Han, B.; Mura, M.; Keshavjee, S.; Liu, M. Src tyrosine kinase inhibition prevents pulmonary ischemia-reperfusion-induced acute lung injury. Intensive Care Med. 2012, 38, 894–905. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Heath, E.I.; Posadas, E.M.; Yu, E.Y.; Harrison, M.R.; Bruce, J.Y.; Cho, S.Y.; Wilding, G.E.; Fetterly, G.J.; Hangauer, D.G.; et al. A phase 2 study of KX2–391, an oral inhibitor of Src kinase and tubulin polymerization, in men with bone-metastatic castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 71, 883–892. [Google Scholar] [CrossRef]
- Gangadhar, T.C.; Clark, J.I.; Karrison, T.; Gajewski, T.F. Phase II study of the Src kinase inhibitor saracatinib (AZD0530) in metastatic melanoma. Investig. New Drugs 2013, 31, 769–773. [Google Scholar] [CrossRef]
- Nam, S.; Wen, W.; Schroeder, A.; Herrmann, A.; Yu, H.; Cheng, X.; Merz, K.H.; Eisenbrand, G.; Li, H.; Yuan, Y.C.; et al. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol. Oncol. 2013, 7, 369–378. [Google Scholar] [CrossRef]
- Liu, L.; Gaboriaud, N.; Vougogianopoulou, K.; Tian, Y.; Wu, J.; Wen, W.; Skaltsounis, A.L.; Jove, R. MLS-2384, a new 6-bromoindirubin derivative with dual JAK/Src kinase inhibitory activity, suppresses growth of diverse cancer cells. Cancer Biol. Ther. 2013, 15. [Google Scholar]
- Kim, B.H.; Won, C.; Lee, Y.H.; Choi, J.S.; Noh, K.H.; Han, S.; Lee, H.; Lee, C.S.; Lee, D.S.; Ye, S.K.; et al. Sophoraflavanone G induces apoptosis of human cancer cells by targeting upstream signals of STATs. Biochem. Pharmacol. 2013, 86, 950–959. [Google Scholar] [CrossRef]
- Jung, J.H.; Kwon, T.R.; Jeong, S.J.; Kim, E.O.; Sohn, E.J.; Yun, M.; Kim, S.H. Apoptosis induced by tanshinone IIA and cryptotanshinone is mediated by distinct JAK/STAT3/5 and SHP1/2 signaling in chronic myeloid leukemia K562 cells. Evid. Based Complement. Alternat. Med. 2013, 2013, 805639. [Google Scholar]
- Kang, Y.; Park, M.A.; Heo, S.W.; Park, S.Y.; Kang, K.W.; Park, P.H.; Kim, J.A. The radio-sensitizing effect of xanthohumol is mediated by STAT3 and EGFR suppression in doxorubicin-resistant MCF-7 human breast cancer cells. Biochim. Biophys. Acta 2013, 1830, 2638–2648. [Google Scholar]
- Chen, X.; Du, Y.; Nan, J.; Zhang, X.; Qin, X.; Wang, Y.; Hou, J.; Wang, Q.; Yang, J. Brevilin A, a novel natural product, inhibits janus kinase activity and blocks STAT3 signaling in cancer cells. PLoS One 2013, 8, e63697. [Google Scholar]
- Hutzen, B.; Willis, W.; Jones, S.; Cen, L.; Deangelis, S.; Fuh, B.; Lin, J. Dietary agent, benzyl isothiocyanate inhibits signal transducer and activator of transcription 3 phosphorylation and collaborates with sulforaphane in the growth suppression of PANC-1 cancer cells. Cancer Cell Int. 2009, 9, 24. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef]
- Page, B.D.; Fletcher, S.; Yue, P.; Li, Z.; Zhang, X.; Sharmeen, S.; Datti, A.; Wrana, J.L.; Trudel, S.; Schimmer, A.D.; et al. Identification of a non-phosphorylated, cell permeable, small molecule ligand for the Stat3 SH2 domain. Bioorg. Med. Chem. Lett. 2011, 21, 5605–5609. [Google Scholar] [CrossRef]
- Becker, S.; Groner, B.; Muller, C.W. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 1998, 394, 145–151. [Google Scholar] [CrossRef]
- Timofeeva, O.A.; Gaponenko, V.; Lockett, S.J.; Tarasov, S.G.; Jiang, S.; Michejda, C.J.; Perantoni, A.O.; Tarasova, N.I. Rationally designed inhibitors identify STAT3 N-domain as a promising anticancer drug target. ACS Chem. Biol. 2007, 2, 799–809. [Google Scholar] [CrossRef]
- Souissi, I.; Najjar, I.; Ah-Koon, L.; Schischmanoff, P.O.; Lesage, D.; Le Coquil, S.; Roger, C.; Dusanter-Fourt, I.; Varin-Blank, N.; Cao, A.; et al. A STAT3-decoy oligonucleotide induces cell death in a human colorectal carcinoma cell line by blocking nuclear transfer of STAT3 and STAT3-bound NF-kappaB. BMC Cell Biol. 2011, 12, 14. [Google Scholar] [CrossRef] [Green Version]
- Weerasinghe, P.; Garcia, G.E.; Zhu, Q.; Yuan, P.; Feng, L.; Mao, L.; Jing, N. Inhibition of Stat3 activation and tumor growth suppression of non-small cell lung cancer by G-quartet oligonucleotides. Int. J. Oncol. 2007, 31, 129–136. [Google Scholar]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, J.; Wang, L.; Tian, Z. Growth inhibition of human hepatocellular carcinoma cells by blocking STAT3 activation with decoy-ODN. Cancer Lett. 2008, 262, 201–213. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Wang, L.; Wei, H.; Tian, Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC Cancer 2007, 7, 149. [Google Scholar]
- Xi, S.; Gooding, W.E.; Grandis, J.R. In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: Implications for cancer therapy. Oncogene 2005, 24, 970–979. [Google Scholar] [CrossRef]
- Nielsen, M.; Kaltoft, K.; Nordahl, M.; Ropke, C.; Geisler, C.; Mustelin, T.; Dobson, P.; Svejgaard, A.; Odum, N. Constitutive activation of a slowly migrating isoform of Stat3 in mycosis fungoides: Tyrphostin AG490 inhibits Stat3 activation and growth of mycosis fungoides tumor cell lines. Proc. Natl. Acad. Sci. USA 1997, 94, 6764–6769. [Google Scholar] [CrossRef]
- Seltana, A.; Guezguez, A.; Lepage, M.; Basora, N.; Beaulieu, J.F. Src family kinase inhibitor PP2 accelerates differentiation in human intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2013, 430, 1195–1200. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer Res. 2003, 23, 363–398. [Google Scholar]
- Ren, X.M.; Duan, L.; He, Q.A.; Zhang, Z.; Zhou, Y.; Wu, D.H.; Pan, J.X.; Pei, D.Q.; Ding, K. Identification of Niclosamide as a New Small-Molecule Inhibitor of the STAT3 Signaling Pathway. ACS Med. Chem. Lett. 2010, 1, 454–459. [Google Scholar] [CrossRef]
- Lippman, S.M. Cancer prevention research: Back to the future. Cancer Prev. Res. 2009, 2, 503–513. [Google Scholar] [CrossRef]
- Uray, I.P.; Brown, P.H. Chemoprevention of hormone receptor-negative breast cancer: New approaches needed. Recent Results Cancer Res. 2011, 188, 147–162. [Google Scholar] [CrossRef]
- Den Hollander, P.; Savage, M.I.; Brown, P.H. Targeted therapy for breast cancer prevention. Front. Oncol. 2013, 3, 250. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 2014, 6, 926-957. https://doi.org/10.3390/cancers6020926
Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers. 2014; 6(2):926-957. https://doi.org/10.3390/cancers6020926
Chicago/Turabian StyleXiong, Ailian, Zhengduo Yang, Yicheng Shen, Jia Zhou, and Qiang Shen. 2014. "Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention" Cancers 6, no. 2: 926-957. https://doi.org/10.3390/cancers6020926
APA StyleXiong, A., Yang, Z., Shen, Y., Zhou, J., & Shen, Q. (2014). Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers, 6(2), 926-957. https://doi.org/10.3390/cancers6020926