Next-Generation Sequencing in Clinical Oncology: Next Steps Towards Clinical Validation

Abstract
:1. Introduction
2. Recommendations and Guidelines for Clinical NGS Applications

{kind=link}
| Group | NGS Document | References | Current Guideline Status |
|---|---|---|---|
| 1. Royal College of Pathologists of Australasia (RCPA) | Standards for Massively Parallel Sequencing in Diagnostic Genetic Testing (in development) | [4] | The recommendations presented in this document do not carry regulatory authority. |
| 2. National Association of Testing Authorities (NATA), Australia | Using recommendations in RCPA document for best laboratory practices | [4] | |
| 3. US Centres for Disease Control and Prevention (CDC). Next-generation Sequencing: Standardization of Clinical Testing (Nex-StoCT) workgroup. (USA) | Next-generation Sequencing: Standardization of Clinical Testing (Nex-StoCT) Workgroup Principles and Guidelines. | [5] | Principles and guidelines designed for NGS incorporation into clinical practice while meeting necessary quality assurance standards [1] |
| 4. U.S. Food and Drug Administration (FDA) | Ultra High Throughput Sequencing for Clinical Diagnostic Applications—Approaches to Assess Analytical Validity, June 23, 2011 | [6] | The purpose of the meeting was to discuss challenges in assessing analytical performance for ultra high throughput genomic sequencing-based clinical applications. |
| 5. Dutch Society for Clinical Genetic Laboratory Diagnostics (VKGL) | Best Practice Guidelines for the Use of Next-Generation Sequencing Applications in Genome Diagnostics: A National Collaborative Study of Dutch Genome Diagnostic Laboratories | [7] | |
| 6. Whole Genome Analysis group of the Association for Molecular Pathology | Opportunities and Challenges Associated with Clinical Diagnostic Genome Sequencing: A Report of the Association for Molecular Pathology | [8] | |
| 7. American College of Medical Genetics (ACMG) | ACMG clinical laboratory standards for next-generation sequencing | [8] | |
| 8. Wadsworth Centre; New York State Department of Health | “Next Generation” Sequencing (NGS) guidelines for somatic genetic variant detection | [2] | Governed by the Official Compilation of Codes, Rules and Regulations of the State of New York |
| 9. Association for Clinical Genetic Science (ACGS) | Practice guidelines for targeted Next Generation Sequencing analysis and interpretation. | [9] | |
| 10. Clinical and Laboratory Standards Institute (CLSI) | Nucleic Acid Sequencing Methods in Diagnostic Laboratory Medicine; Approved Guideline—Second Edition | [10] |
3. Summary of Information Available within Table 1
4. NGS Use in Clinical Oncology
| Oncology Organisation | Guideline Statement | References |
|---|---|---|
| The European Society for Medical Oncology (ESMO) | Clinical Practice Guidelines relating to many cancers. Each guideline includes information related to incidence, diagnostic criteria, staging, risk and treatment plans designed to help oncologists deliver an appropriate quality of patient care. | [16] |
| The National Comprehensive Cancer Network (NCCN) | Treatment guidelines, for most cancers, to improve the quality, effectiveness, and efficiency of cancer care. NCCN offers a number of programs to help guide clinicians through the decision-making process of cancer management. | [17] |
| The Cancer Council Australia (CCA) | Clinical practice guidelines that bring together the best available evidence to underpin scientifically-valid recommendations for the prevention and diagnosis of cancer and treatment of cancer patients | [18] |
| American Society of Clinical Oncology (ASCO) | Guidelines can address specific clinical situations (disease-oriented) or use of approved medical products, procedures, or tests (modality-oriented). Using the best available evidence, ASCO expert panels identify and develop practice recommendations for specific areas of cancer care that would benefit from using practice guidelines. | [19] |
4.1. Clinical Utility
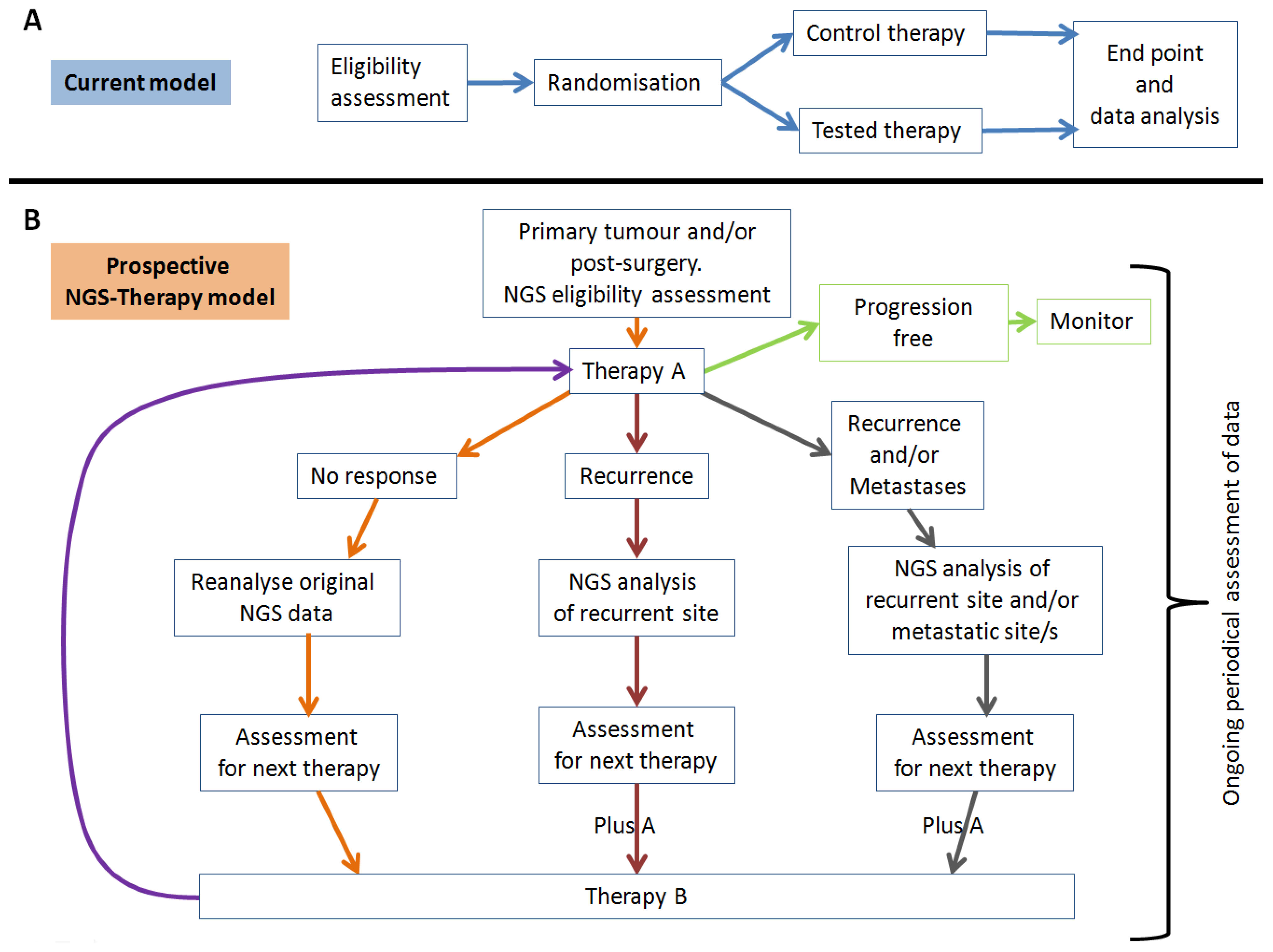
4.2. Tumour Heterogeneity
4.3. Variant Confirmation
4.4. Reporting and Structure
4.5. Reporting: “Incidental Findings”
4.6. Reporting: Pertinent Negatives
4.7. Clinical Bioinformatics
5. Conclusions
Conflicts of Interest
References
- Sheridan, C. Milestone approval lifts Illumina’s NGS from research into clinic. Nat. Biotechnol. 2014, 32, 111–112. [Google Scholar] [CrossRef] [PubMed]
- New York State Department of Health. “Next Generation” Sequencing (NGS) guidelines for somatic genetic variant detection. Available online: http://www.wadsworth.org/labcert/TestApproval/forms/NextGenSeq_ONCO_Guidelines.pdf (accessed on 19 May 2014).
- New York State Department of Health. Test Approval Policy. Available online: http://www.wadsworth.org/labcert/TestApproval/ (accessed on 19 May 2014).
- Royal College of Pathologists of Australasia (RCPA). Implementation of Massively Parallel Sequencing in Diagnostic Medical Genetic Testing. Available online: http://pathwiki.rcpaqap.com.au/pathwiki/index.php/Introduction (accessed 20 May 2014).
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Ultra High Throughput Sequencing for Clinical Diagnostic Applications—Approaches to Assess Analytical Validity. Silver Spring, MD, USA, 23 June 2011. [Google Scholar]
- Weiss, M.M.; van der Zwaag, B.; Jongbloed, J.D.; Vogel, M.J.; Bruggenwirth, H.T.; Lekanne Deprez, R.H.; Mook, O.; Ruivenkamp, C.A.; van Slegtenhorst, M.A.; van den Wijngaard, A.; et al. Best practice guidelines for the use of next-generation sequencing applications in genome diagnostics: A national collaborative study of Dutch genome diagnostic laboratories. Hum. Mutat. 2013, 34, 1313–1321. [Google Scholar] [CrossRef]
- Schrijver, I.; Aziz, N.; Farkas, D.H.; Furtado, M.; Gonzalez, A.F.; Greiner, T.C.; Grody, W.W.; Hambuch, T.; Kalman, L.; Kant, J.A.; et al. Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology. J. Mol. Diagn. 2012, 14, 525–540. [Google Scholar] [CrossRef]
- Ellard, S.; Lindsay, H.; Camm, N.; Watson, C.; Abbs, S.; Mattocks, C.; Taylor, G.R.; Charlton, R. Practice guidelines for targeted Next Generation Sequencing analysis and interpretation. Association for Clinical Genetic Science (ACGS). Available online: http://www.acgs.uk.com/media/815227/bpg_for_targeted_next_generation_sequencing_201113__4_.pdf (accessed on 2 June 2014).
- Clinical and Laboratory Standards Institute. Nucleic Acid Sequencing Methods in Diagnostic Laboratory Medicine; Approved Guideline—Second Edition. Available online: http://shopping.netsuite.com/s.nl/c.1253739/it.A/id.1787/.f (accessed on 3 June 2014).
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef] [PubMed]
- The Provision of Direct to Consumer Genetic Tests, Guiding Principles for Providers: National Pathology Accreditation Advisory Council, Australia. Available online: http://www.health.gov.au/internet/main/publishing.nsf/Content/health-npaac-path-bestpractice (accessed on 23 July 2014).
- Human Genome Variation Society: Guidelines for Sequence Variation (“mutation”) Nomenclature. Available online: http://www.hgvs.org/content/guidelines (accessed 19 May 2014).
- Park, J.Y.; Kricka, L.J.; Fortina, P. Next-generation sequencing in the clinic. Nat. Biotechnol. 2013, 31, 990–992. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wendl, M.C.; McMichael, J.F.; Raphael, B.J. Expanding the computational toolbox for mining cancer genomes. Nat. Rev. Genet. 2014, 15, 556–570. [Google Scholar] [CrossRef] [PubMed]
- European Society for Medical Oncology. Clinical Practice Guidelines by Tumour Type. Available online: http://www.esmo.org/Guidelines-Practice/Clinical-Practice-Guidelines (accessed 28 May 2014).
- National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology. Available online: http://www.nccn.org/professionals/physician_gls/f_guidelines.asp (accessed 28 May 2014).
- Cancer Council, Australia. Clinical Practice Guidelines. Available online: http://www.cancer.org.au/health-professionals/clinical-guidelines/ (accessed 28 May 2014).
- American Society of Clinical Oncology. Clinical Practice Guidelines. Available online: http://www.asco.org/quality-guidelines/guidelines (accessed 28 May 2014).
- Parkinson, D.R.; Johnson, B.E.; Sledge, G.W. Making personalized cancer medicine a reality: Challenges and opportunities in the development of biomarkers and companion diagnostics. Clin. Cancer Res. 2012, 18, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Shibata, D.; Curtis, C.; Tavare, S. Single-molecule genomic data delineate patient-specific tumor profiles and cancer stem cell organization. Cancer Res. 2013, 73, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Bloemena, E.; Leemans, C.R.; Brakenhoff, R.H. Molecular analysis of surgical margins in head and neck cancer: more than a marginal issue. Oral Oncol. 2010, 46, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Catto, J.W.; Orntoft, T.F.; Real, F.X.; Zwarthoff, E.C.; Swanton, C. Intratumour Heterogeneity in Urologic Cancers: From Molecular Evidence to Clinical Implications. Eur. Urol. 2014. [Google Scholar] [CrossRef]
- McWhinney, S.R.; McLeod, H.L. Using germline genotype in cancer pharmacogenetic studies. Pharmacogenomics 2009, 10, 489–493. [Google Scholar] [CrossRef]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Brownstein, C.A.; Beggs, A.H.; Homer, N.; Merriman, B.; Yu, T.W.; Flannery, K.C.; Dechene, E.T.; Towne, M.C.; Savage, S.K.; Price, E.N.; et al. An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY Challenge. Genome Biol. 2014, 15, R53. [Google Scholar] [CrossRef] [Green Version]
- Salto-Tellez, M.; Gonzalez de Castro, D. Next generation sequencing: A change of paradigm in molecular diagnostic validation. J. Pathol. 2013. [Google Scholar] [CrossRef]
- Clark, M.J.; Chen, R.; Lam, H.Y.; Karczewski, K.J.; Chen, R.; Euskirchen, G.; Butte, A.J.; Snyder, M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011, 29, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Speed, T.P. Comparing somatic mutation-callers: Beyond Venn diagrams. BMC Bioinformatics 2013, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Goode, D.L.; Hunter, S.M.; Doyle, M.A.; Ma, T.; Rowley, S.M.; Choong, D.; Ryland, G.L.; Campbell, I.G. A simple consensus approach improves somatic mutation prediction accuracy. Genome Med. 2013, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Roychowdhury, S. Implementing personalized cancer genomics in clinical trials. Nat. Rev. Drug Discov. 2013, 12, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Jessri, M.; Farah, C.S. Next generation sequencing and its application in deciphering head and neck cancer. Oral Oncol. 2014, 50, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Jessri, M.; Farah, C.S. Harnessing massively parallel sequencing in personalized head and neck oncology. J. Dent. Res. 2014, 93, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.A.; Yates, B.; Seal, R.L.; Wright, M.W.; Bruford, E.A. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef]
- Chang, F.; Li, M.M. Clinical application of amplicon-based next-generation sequencing in cancer. Cancer Genet. 2013, 206, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Dong, F.; Borger, D.; Dias-Santagata, D.; Ellisen, L.W.; Le, L.P.; Iafrate, A.J. Standardized decision support in next generation sequencing reports of somatic cancer variants. Mol. Oncol. 2014, 8, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Kassahn, K.S.; Scott, H.S.; Caramins, M.C. Integrating massively parallel sequencing into diagnostic workflows and managing the annotation and clinical interpretation challenge. Hum. Mutat. 2014, 35, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Tarczy-Hornoch, P.; Amendola, L.; Aronson, S.J.; Garraway, L.; Gray, S.; Grundmeier, R.W.; Hindorff, L.A.; Jarvik, G.; Karavite, D.; Lebo, M.; et al. A survey of informatics approaches to whole-exome and whole-genome clinical reporting in the electronic health record. Genet. Med. 2013, 15, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, L.; Sincan, M.; Markello, T.; Adams, D.R.; Gill, F.; Godfrey, R.; Golas, G.; Groden, C.; Landis, D.; Nehrebecky, M.; et al. The implications of familial incidental findings from exome sequencing: the NIH Undiagnosed Diseases Program experience. Genet. Med. 2014, 16, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Rubinstein, W.S.; Facio, F.M.; Ng, D.; Singh, L.N.; Teer, J.K.; Mullikin, J.C.; Biesecker, L.G. Secondary variants in individuals undergoing exome sequencing: Screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am. J. Hum. Genet. 2012, 91, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Dorschner, M.O.; Amendola, L.M.; Turner, E.H.; Robertson, P.D.; Shirts, B.H.; Gallego, C.J.; Bennett, R.L.; Jones, K.L.; Tokita, M.J.; Bennett, J.T.; et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am. J. Hum. Genet. 2013, 93, 631–640. [Google Scholar] [CrossRef] [PubMed]
- O’Rawe, J.; Jiang, T.; Sun, G.; Wu, Y.; Wang, W.; Hu, J.; Bodily, P.; Tian, L.; Hakonarson, H.; Johnson, W.E.; et al. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guo, Y.; Li, J.; Long, J.; Zhang, B.; Shyr, Y. Steps to ensure accuracy in genotype and SNP calling from Illumina sequencing data. BMC Genomics 2012, 13, S8. [Google Scholar]
- Tarabeux, J.; Zeitouni, B.; Moncoutier, V.; Tenreiro, H.; Abidallah, K.; Lair, S.; Legoix-Ne, P.; Leroy, Q.; Rouleau, E.; Golmard, L.; et al. Streamlined ion torrent PGM-based diagnostics: BRCA1 and BRCA2 genes as a model. Eur. J. Hum. Genet. 2014, 22, 535–541. [Google Scholar] [CrossRef] [PubMed]
- American Society of Clinical Oncology. The state of cancer care in america, 2014: A report by the American Society of Clinical Oncology. J. Oncol. Pract. 2014, 10, 119–142. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennett, N.C.; Farah, C.S. Next-Generation Sequencing in Clinical Oncology: Next Steps Towards Clinical Validation. Cancers 2014, 6, 2296-2312. https://doi.org/10.3390/cancers6042296
Bennett NC, Farah CS. Next-Generation Sequencing in Clinical Oncology: Next Steps Towards Clinical Validation. Cancers. 2014; 6(4):2296-2312. https://doi.org/10.3390/cancers6042296
Chicago/Turabian StyleBennett, Nigel C., and Camile S. Farah. 2014. "Next-Generation Sequencing in Clinical Oncology: Next Steps Towards Clinical Validation" Cancers 6, no. 4: 2296-2312. https://doi.org/10.3390/cancers6042296