
Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL Leukemia Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results and Discussion
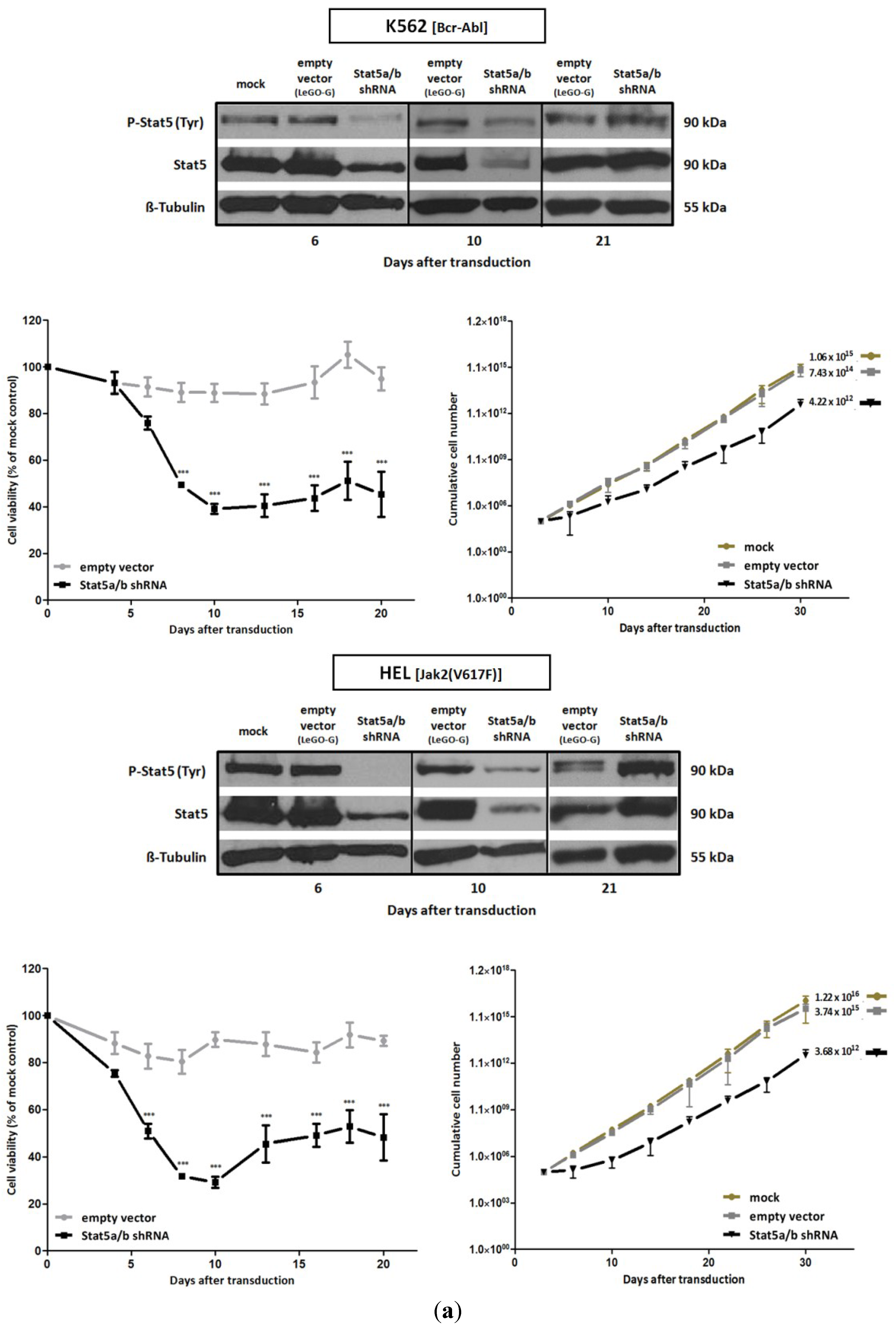
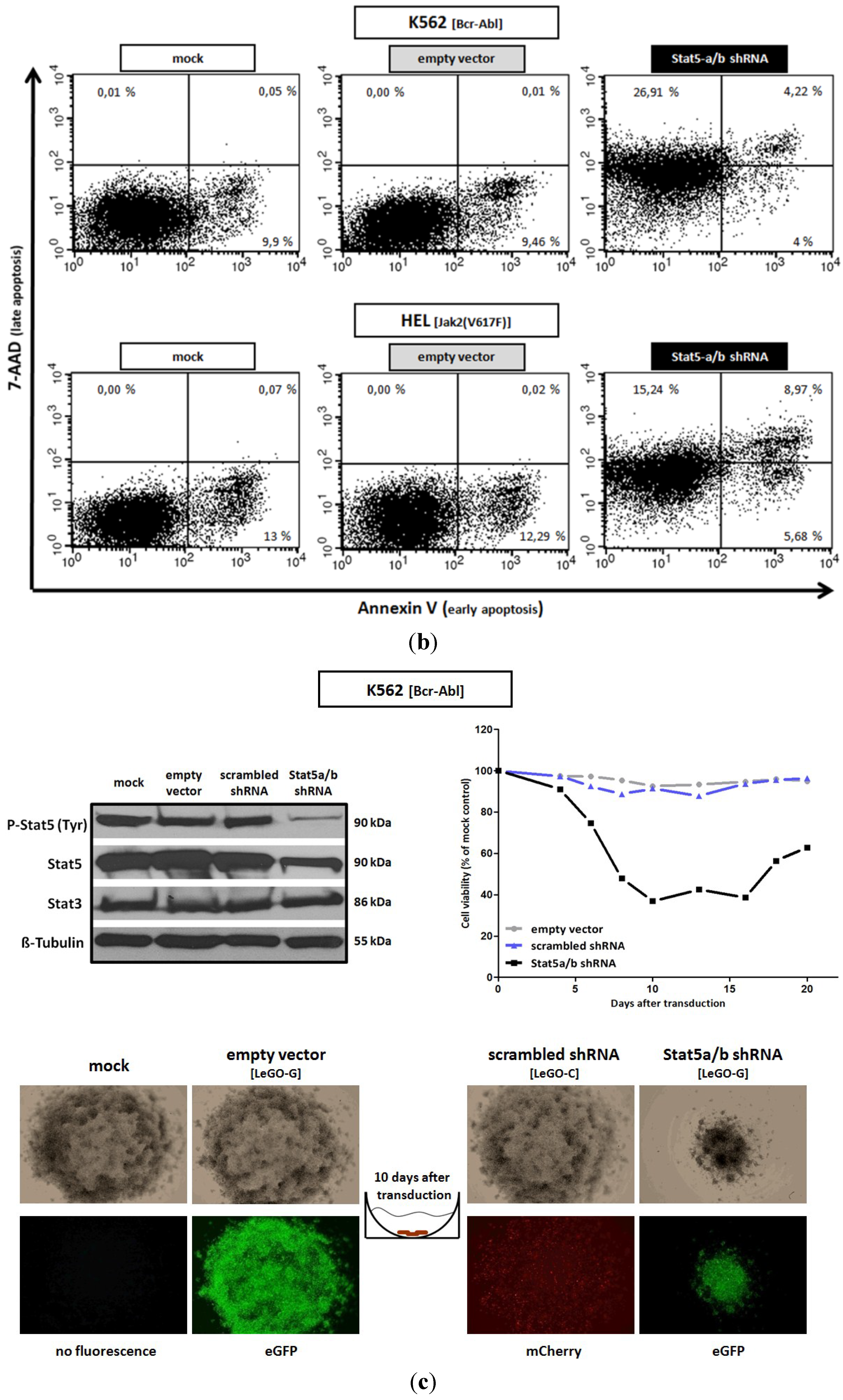
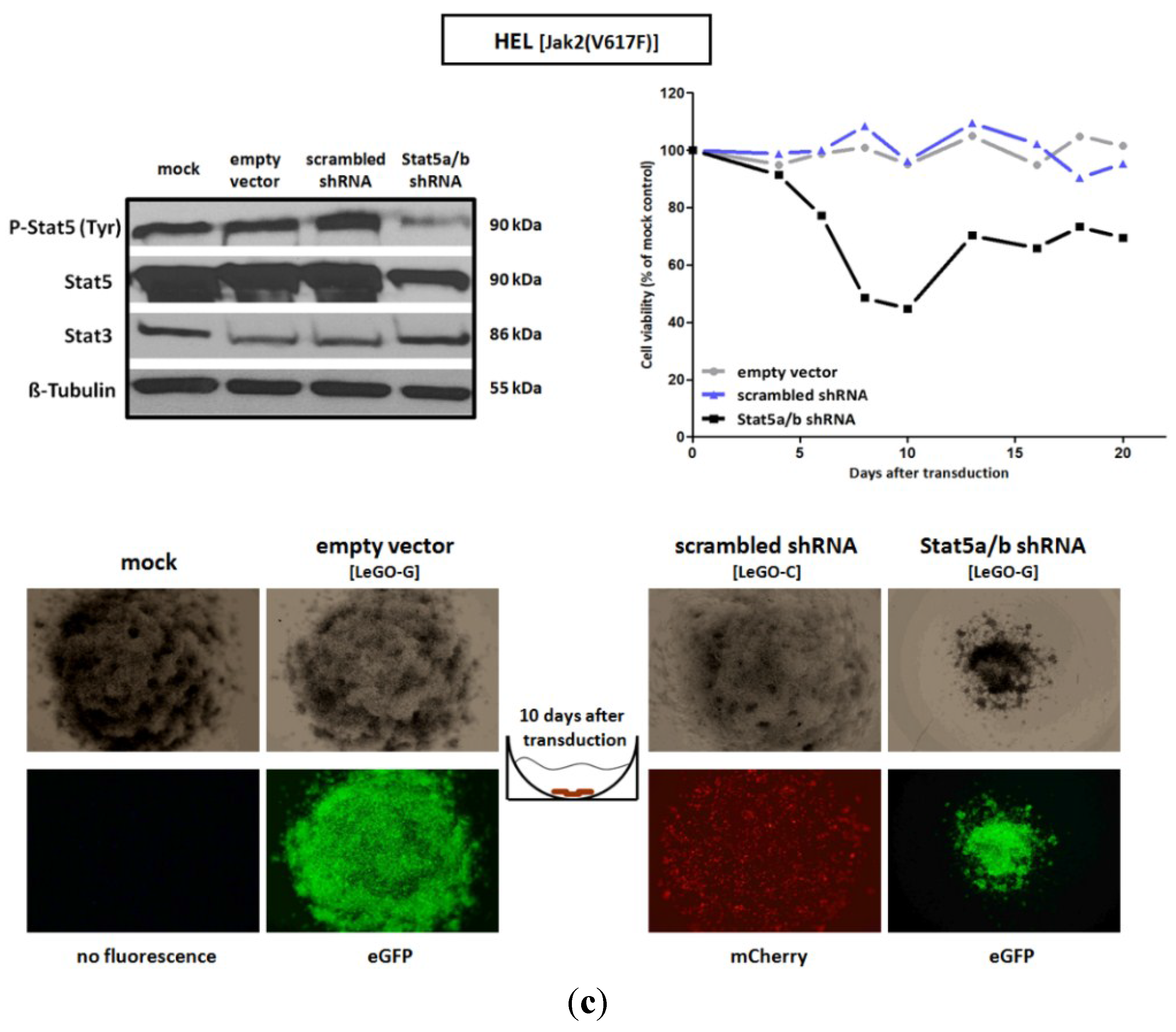
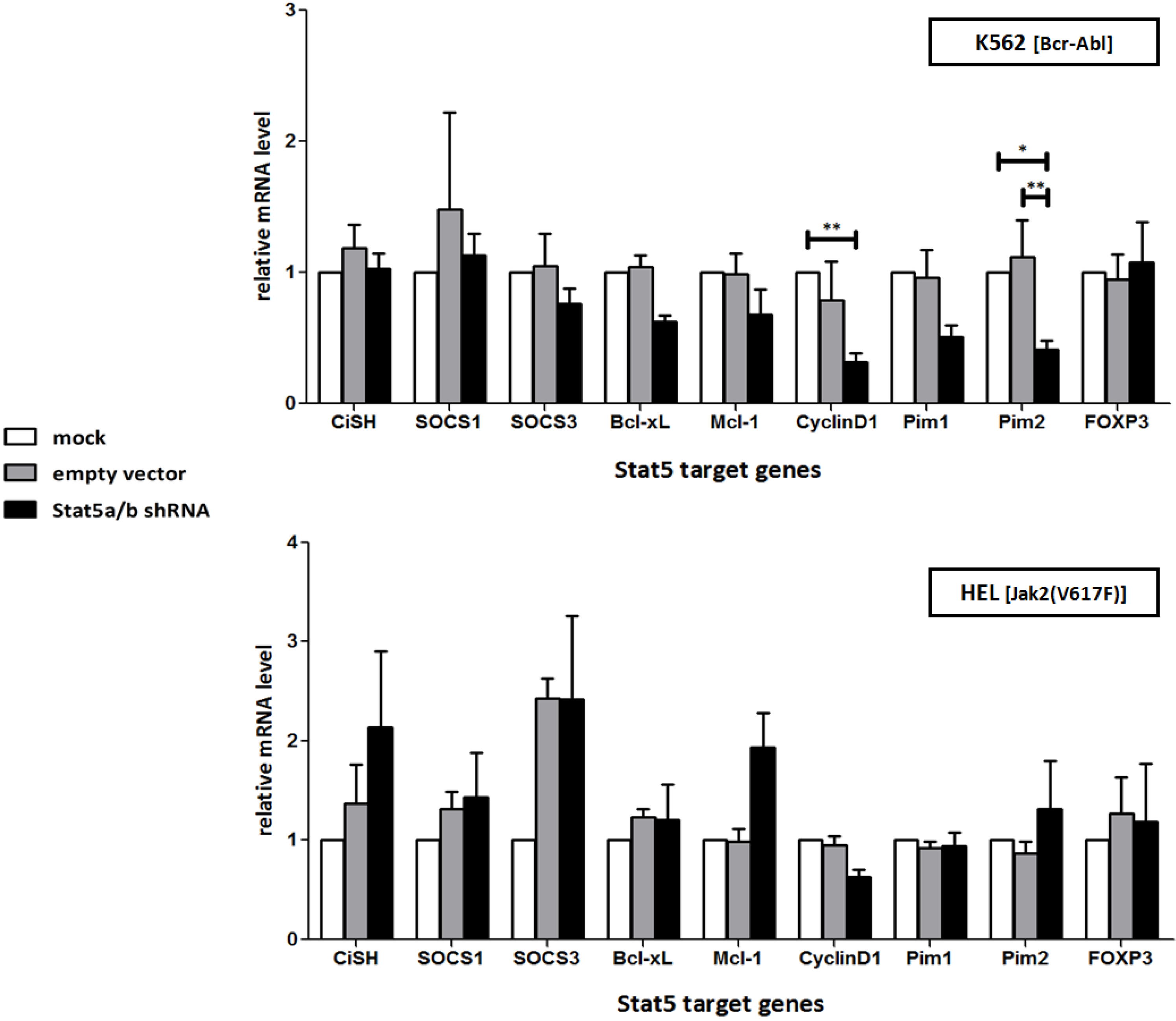
2.1. shRNA-Mediated Downregulation of Stat5 Causes Cell Death of Bcr-Abl Positive K562 and of Jak2(V617F) Positive HEL Leukemic Cells




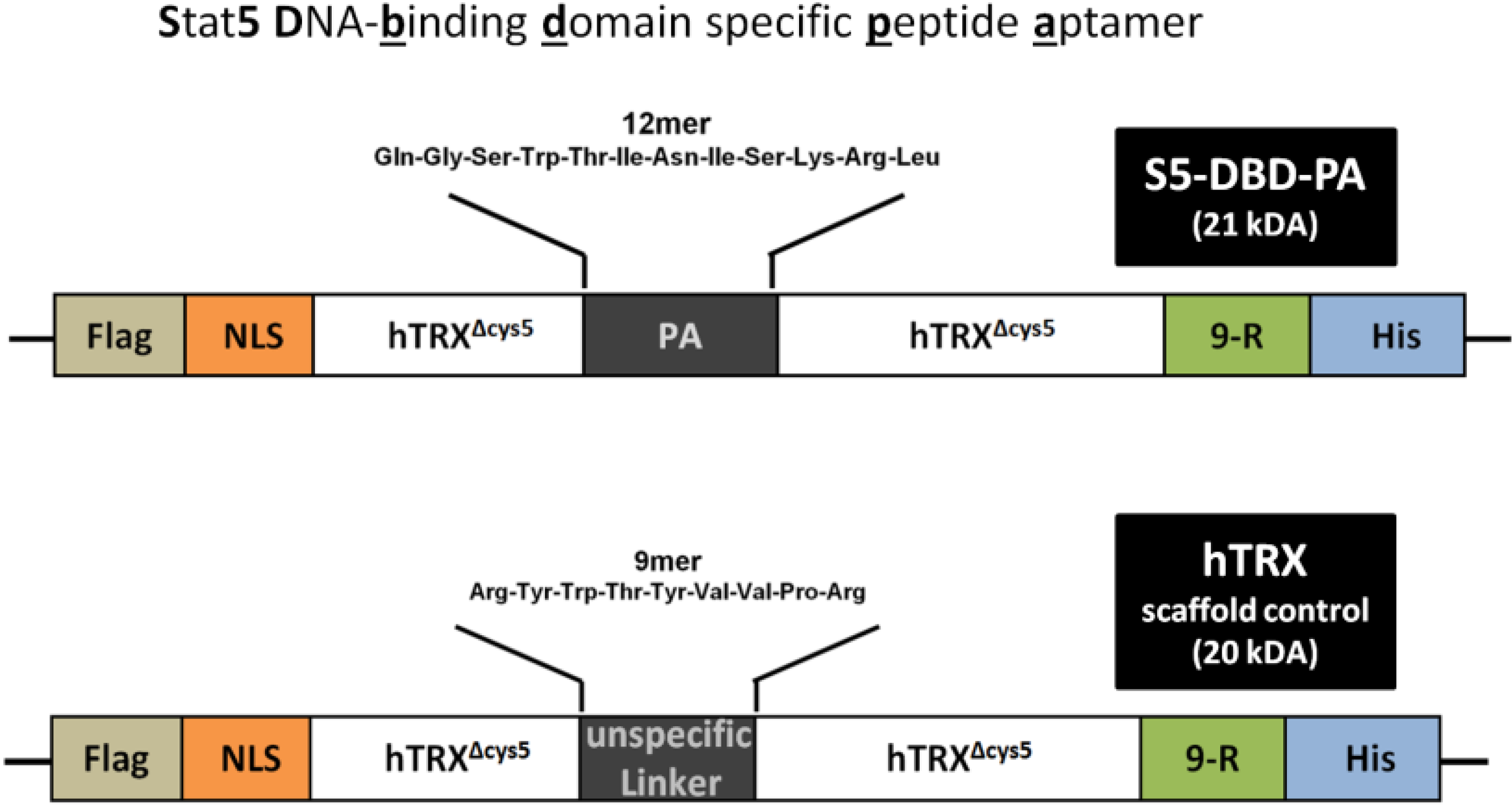
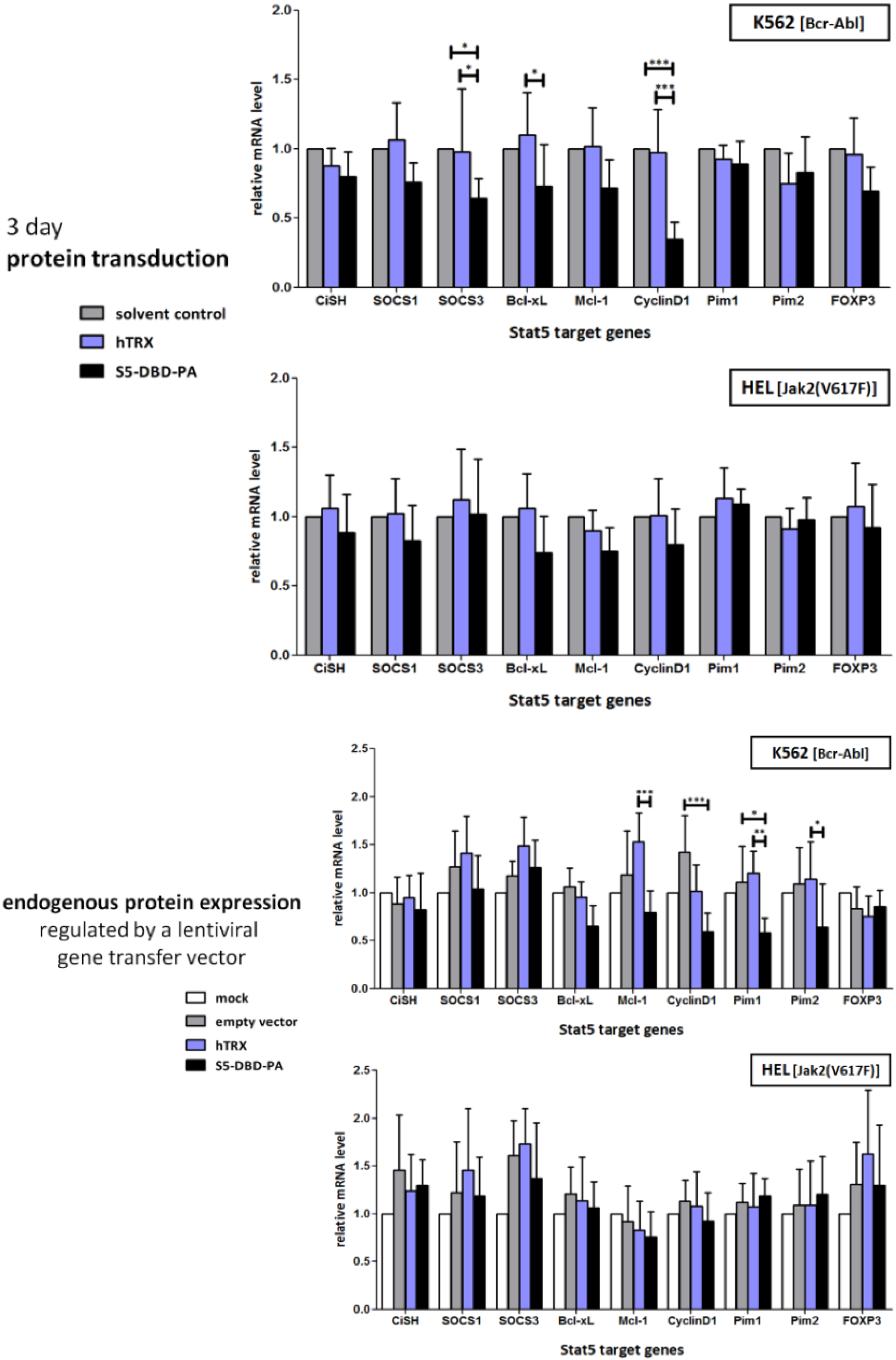
2.2. Protein Transduction of the Recombinant Stat5 Specific Ligand S5-DBD-PA and Intracellular Expression of a S5-DBD-PA Encoding gene Transfer Vector in K562 and HEL Cells


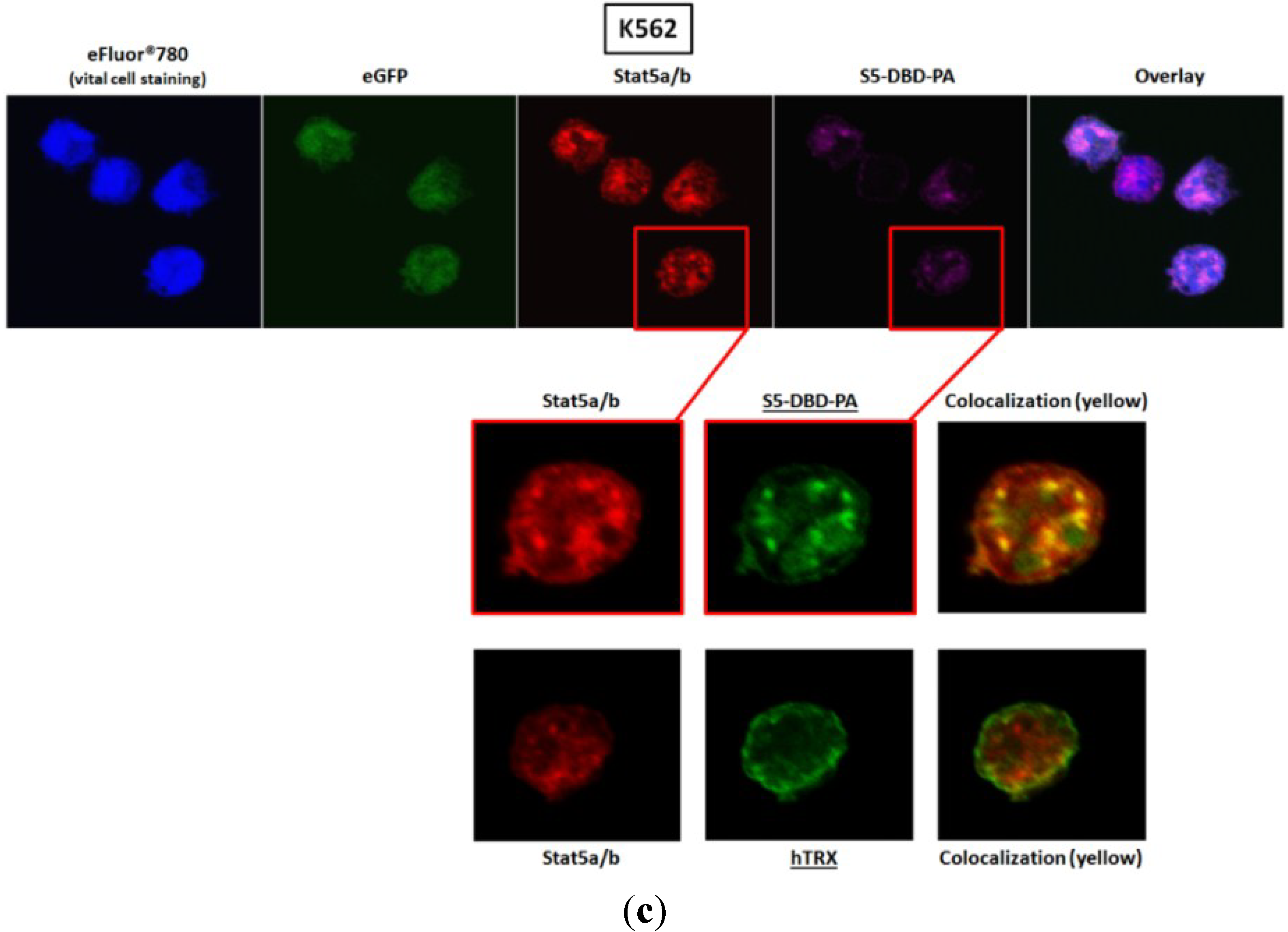
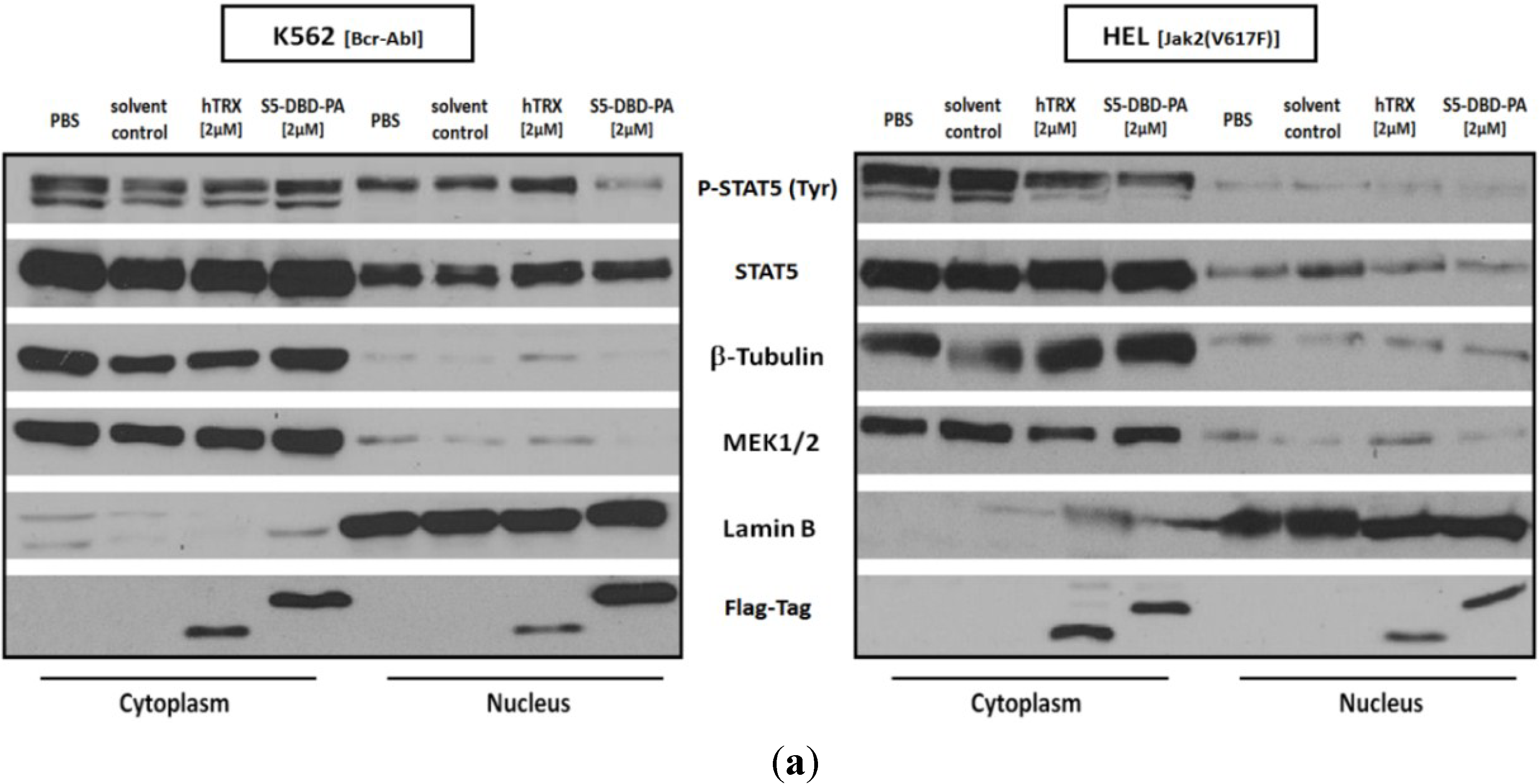
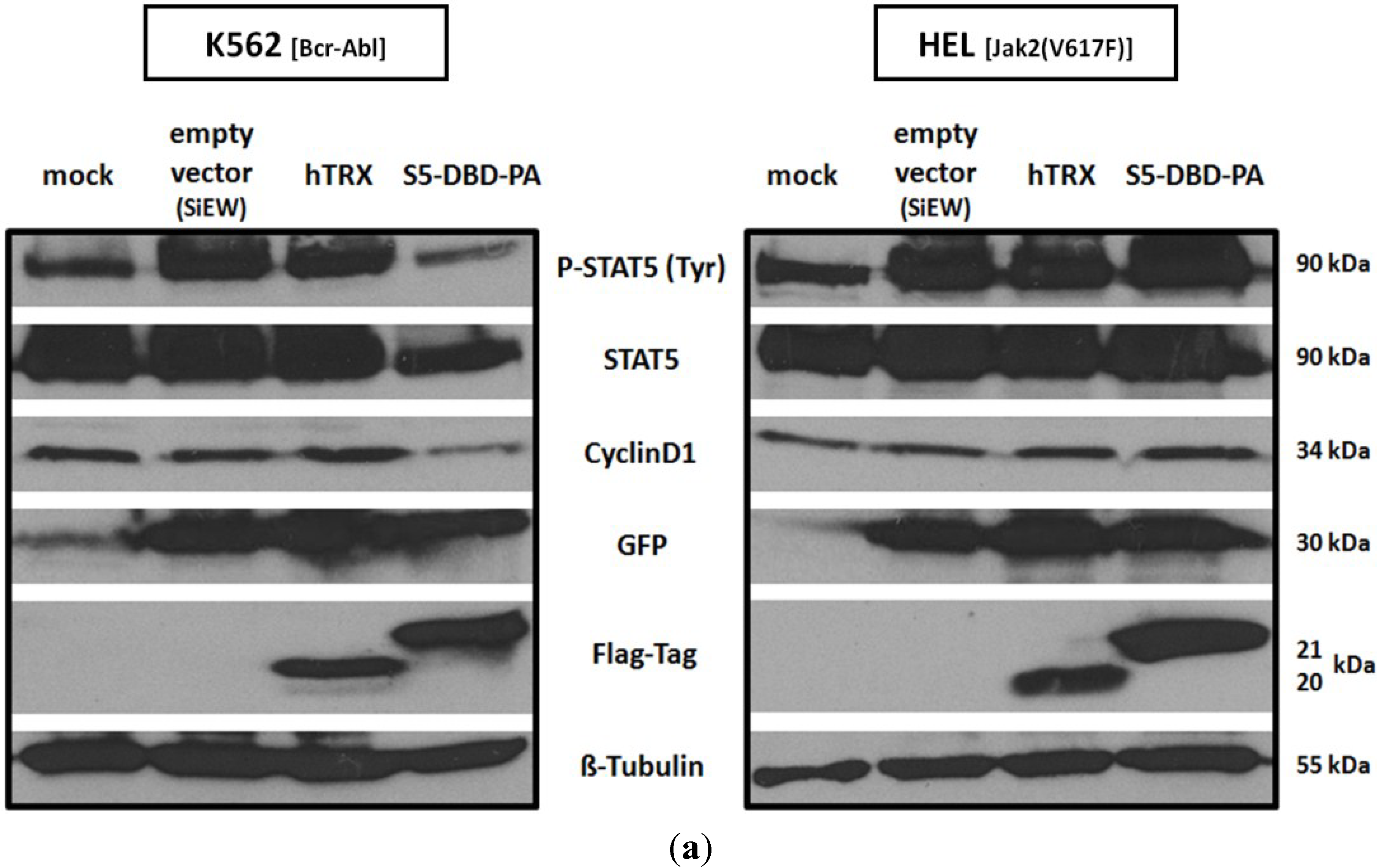
2.3. Activated Stat5 Is Present in the Nucleus of K652 Cells, but Nearly Absent from the Nucleus of HEL Cells; S5-DBD-PA Interferes with Nuclear Translocation, Target Gene Transactivation and Reduces the Level of the Transcription Factor in K562 and HEL Cells



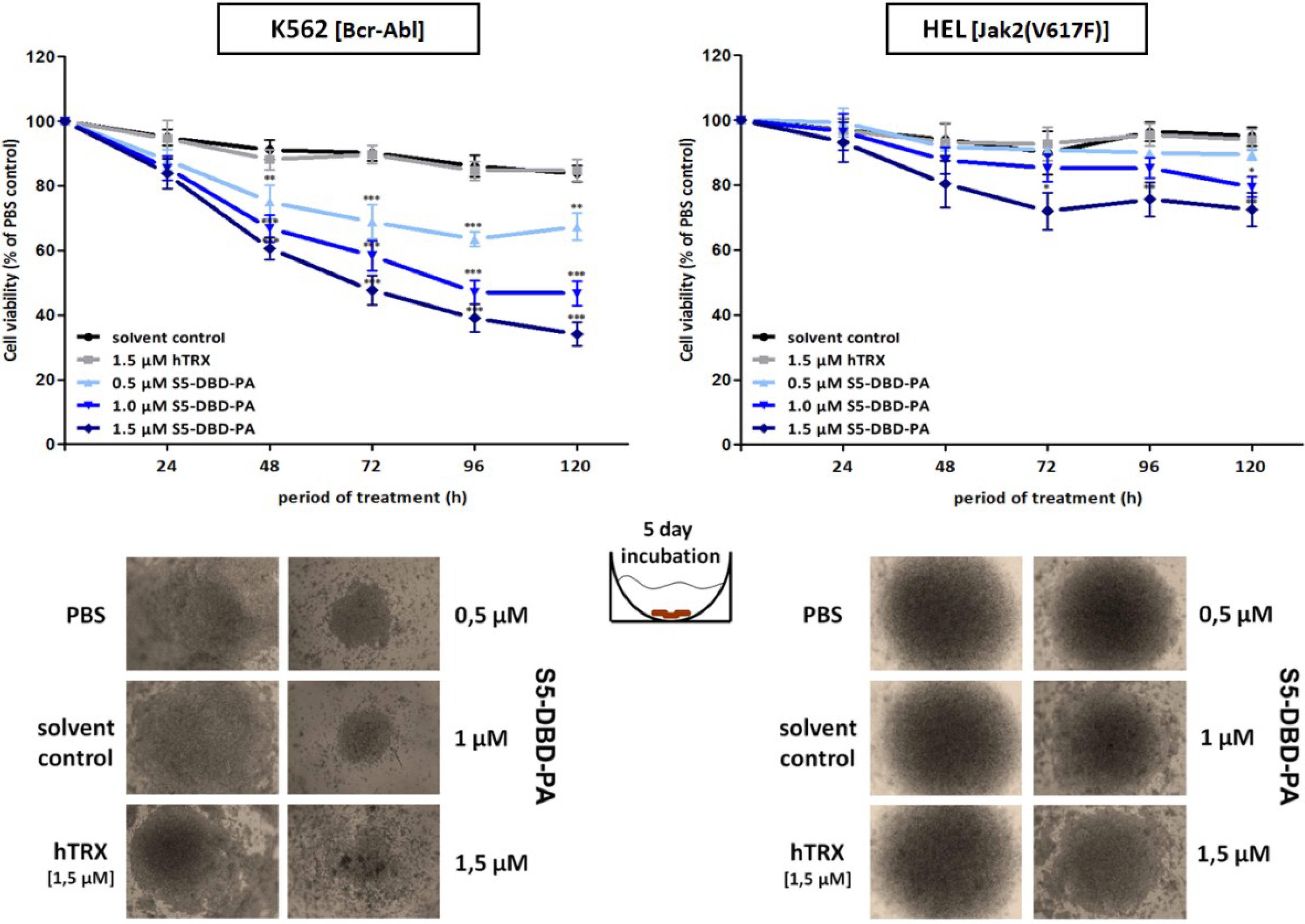
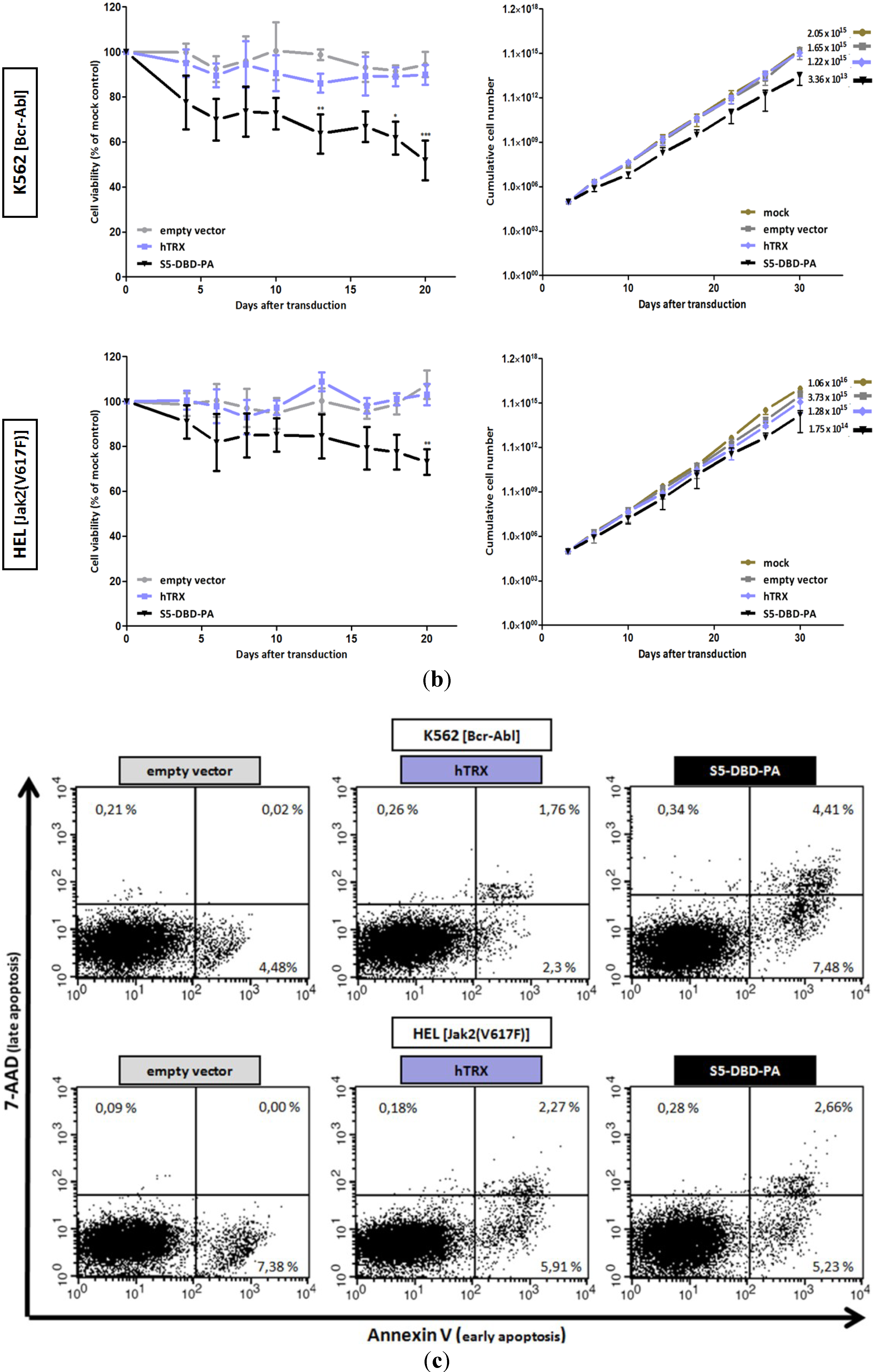
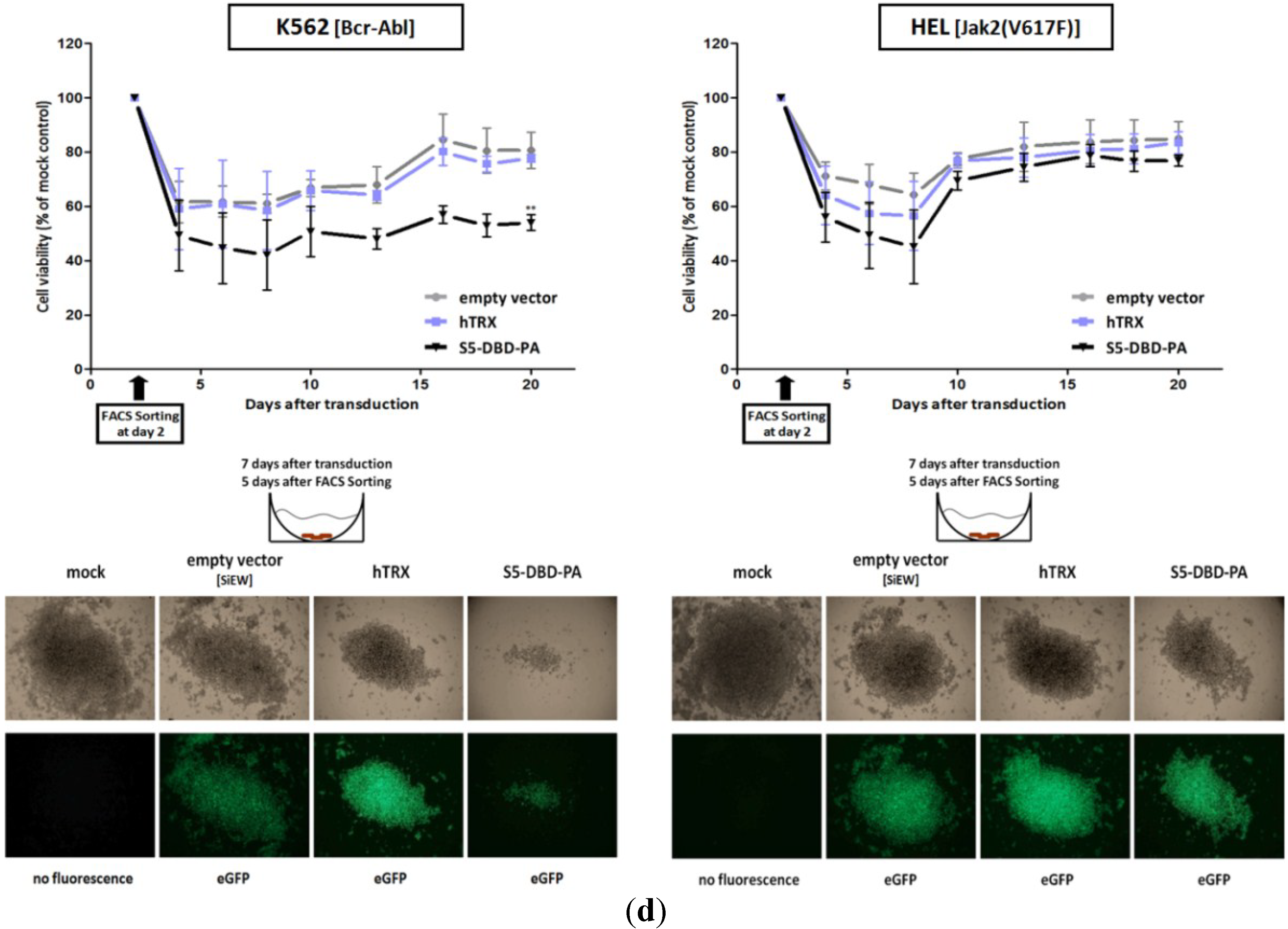
2.4. The Stat5 Inhibitor S5-DBD-PA Strongly Suppresses Growth and Viability in K562 Cells and, to A Lesser Extent, in HEL Cells




3. Experimental Section
3.1. Cell Lines and Culture Conditions
3.2. Reagents
3.3. Plasmid Construction
3.5. Identification of the 12mer Peptide Aptamer (PA) Sequence, which Mediates the Interaction of the S5-DBD-PA Construct with Stat5
3.6. Recombinant Expression, Protein Purification and Transduction
3.7. Western Blot Analysis
3.8. Cell Viability and Growth Measurements: XTT-Assay, Cumulative Cell Number (CCN) Determination and annexin V/7-AAD Apoptosis Staining
3.9. qRT-PCR Analysis
3.10. Immunofluorescence Staining
4. Conclusions


Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Groner, B.; Hennighausen, L. The versatile regulation of cellular events by jak-stat signaling: From transcriptional control to microtubule dynamics and energy metabolism. Horm. Mol. Biol. Clin. Investg. 2012, 10, 193–200. [Google Scholar]
- Groner, B.; Vafaizadeh, V. Cytokine regulation of mammary gland development and epithelial cell functions through discrete activities of stat proteins. Mol. Cell. Endocrinol. 2014, 382, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Xiang, M.; Frank, D.A. Distinct roles of stat3 and stat5 in the pathogenesis and targeted therapy of breast cancer. Mol. Cell. Endocrinol. 2014, 382, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Chan, K.S.; Sano, S.; Digiovanni, J. Signal transducer and activator of transcription 3 (stat3) in epithelial carcinogenesis. Mol. Carcinog. 2007, 46, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Vafaizadeh, V.; Klemmt, P.; Brendel, C.; Weber, K.; Doebele, C.; Britt, K.; Grez, M.; Fehse, B.; Desrivieres, S.; Groner, B. Mammary epithelial reconstitution with gene-modified stem cells assigns roles to stat5 in luminal alveolar cell fate decisions, differentiation, involution, and mammary tumor formation. Stem Cells 2010, 28, 928–938. [Google Scholar] [PubMed]
- Groner, B. Determinants of the extent and duration of stat3 signaling. JAKSTAT 2012, 1, 211–215. [Google Scholar] [PubMed]
- Mertens, C.; Darnell, J.E., Jr. Snapshot: Jak-stat signaling. Cell 2007, 131, 612. [Google Scholar] [CrossRef] [PubMed]
- Wakao, H.; Schmitt-Ney, M.; Groner, B. Mammary gland-specific nuclear factor is present in lactating rodent and bovine mammary tissue and composed of a single polypeptide of 89 kda. J. Biol. Chem. 1992, 267, 16365–16370. [Google Scholar] [PubMed]
- Wakao, H.; Gouilleux, F.; Groner, B. Mammary gland factor (mgf) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 1994, 13, 2182–2191. [Google Scholar] [PubMed]
- Liu, X.; Robinson, G.W.; Gouilleux, F.; Groner, B.; Hennighausen, L. Cloning and expression of stat5 and an additional homologue (stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc. Natl. Acad. Sci. USA 1995, 92, 8831–8835. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Robinson, G.W. Interpretation of cytokine signaling through the transcription factors stat5a and stat5b. Genes Dev. 2008, 22, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M. Stat proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 2000, 25, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Herrington, J.; Rui, L.; Luo, G.; Yu-Lee, L.Y.; Carter-Su, C. A functional DNA binding domain is required for growth hormone-induced nuclear accumulation of stat5b. J. Biol. Chem. 1999, 274, 5138–5145. [Google Scholar] [CrossRef] [PubMed]
- Reich, N.C.; Liu, L. Tracking stat nuclear traffic. Nat. Rev. Immunol. 2006, 6, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Heltemes-Harris, L.M.; Farrar, M.A. The role of stat5 in lymphocyte development and transformation. Curr. Opin. Immunol. 2012, 24, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, J.W.; Grebien, F.; Kerenyi, M.A.; Friedbichler, K.; Kovacic, B.; Zankl, B.; Hoelbl, A.; Nivarti, H.; Beug, H.; Sexl, V.; et al. The different functions of stat5 and chromatin alteration through stat5 proteins. Front. Biosci. 2008, 13, 6237–6254. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Powers, S.E.; Maienschein-Cline, M.; Bartom, E.T.; Hamel, K.M.; Kee, B.L.; Dinner, A.R.; Clark, M.R. Epigenetic repression of the igk locus by stat5-mediated recruitment of the histone methyltransferase ezh2. Nat. Immunol. 2011, 12, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Hosui, A.; Sun, R.; Shen, K.; Gavrilova, O.; Chen, W.; Cam, M.C.; Gao, B.; Robinson, G.W.; Hennighausen, L. Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology 2007, 46, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Engblom, D.; Kornfeld, J.W.; Schwake, L.; Tronche, F.; Reimann, A.; Beug, H.; Hennighausen, L.; Moriggl, R.; Schutz, G. Direct glucocorticoid receptor-stat5 interaction in hepatocytes controls body size and maturation-related gene expression. Genes Dev. 2007, 21, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, M.T.; Ahonen, T.J.; Yamashita, H.; Chandrashekar, V.; Bartke, A.; Grimley, P.M.; Robinson, G.W.; Hennighausen, L.; Rui, H. Epithelial defect in prostates of stat5a-null mice. Lab. Investig. 2000, 80, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, D.; Na, R.; Feuermann, Y.; Pechhold, S.; Chen, W.; Robinson, G.W.; Hennighausen, L. Development of mammary luminal progenitor cells is controlled by the transcription factor stat5a. Genes Dev. 2009, 23, 2382–2387. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, G.; Tse, W.; Bunting, K.D. Conditional deletion of stat5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood 2009, 113, 4856–4865. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, A.T.; Vellenga, E.; Schuringa, J.J. Maximal stat5-induced proliferation and self-renewal at intermediate stat5 activity levels. Mol. Cell. Biol. 2008, 28, 6668–6680. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Cui, Y.; Watford, W.T.; Bream, J.H.; Yamaoka, K.; Hissong, B.D.; Li, D.; Durum, S.K.; Jiang, Q.; Bhandoola, A.; et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1000–1005. [Google Scholar] [CrossRef]
- Sitrin, J.; Ring, A.; Garcia, K.C.; Benoist, C.; Mathis, D. Regulatory t cells control nk cells in an insulitic lesion by depriving them of il-2. J. Exp. Med. 2013, 210, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Grebien, F.; Kerenyi, M.A.; Kovacic, B.; Kolbe, T.; Becker, V.; Dolznig, H.; Pfeffer, K.; Klingmuller, U.; Muller, M.; Beug, H.; et al. Stat5 activation enables erythropoiesis in the absence of epor and jak2. Blood 2008, 111, 4511–4522. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, M.A.; Grebien, F.; Gehart, H.; Schifrer, M.; Artaker, M.; Kovacic, B.; Beug, H.; Moriggl, R.; Mullner, E.W. Stat5 regulates cellular iron uptake of erythroid cells via irp-2 and tfr-1. Blood 2008, 112, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S.; Ward, A.C. Stat5 as a diagnostic marker for leukemia. Expert Rev. Mol. Diagn. 2008, 8, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Walker, S.R.; Nelson, E.A.; Cerulli, R.; Xiang, M.; Toniolo, P.A.; Qi, J.; Stone, R.M.; Wadleigh, M.; Bradner, J.E.; et al. Targeting stat5 in hematologic malignancies through inhibition of the bromodomain and extra-terminal (bet) bromodomain protein brd2. Mol. Cancer Ther. 2014, 13, 1194–1205. [Google Scholar] [CrossRef]
- Sternberg, D.W.; Gilliland, D.G. The role of signal transducer and activator of transcription factors in leukemogenesis. J. Clin. Oncol. 2004, 22, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Scheeren, F.A.; Diehl, S.A.; Smit, L.A.; Beaumont, T.; Naspetti, M.; Bende, R.J.; Blom, B.; Karube, K.; Ohshima, K.; van Noesel, C.J.; et al. Il-21 is expressed in hodgkin lymphoma and activates stat5: Evidence that activated stat5 is required for hodgkin lymphomagenesis. Blood 2008, 111, 4706–4715. [Google Scholar] [CrossRef] [PubMed]
- Hoelbl, A.; Kovacic, B.; Kerenyi, M.A.; Simma, O.; Warsch, W.; Cui, Y.; Beug, H.; Hennighausen, L.; Moriggl, R.; Sexl, V. Clarifying the role of stat5 in lymphoid development and abelson-induced transformation. Blood 2006, 107, 4898–4906. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.; Ahmed, W.; Lazarides, K.; Betancur, M.; Patel, N.; Hennighausen, L.; Zaleskas, V.M.; van Etten, R.A. Essential role for stat5a/b in myeloproliferative neoplasms induced by bcr-abl1 and jak2(v617f) in mice. Blood 2012, 119, 3550–3560. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.M.; Melo, J.V. Bcr-abl in chronic myelogenous leukemia—How does it work? Acta Haematol. 2008, 119, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase jak2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Funakoshi-Tago, M.; Tago, K.; Abe, M.; Sonoda, Y.; Kasahara, T. Stat5 activation is critical for the transformation mediated by myeloproliferative disorder-associated jak2 v617f mutant. J. Biol. Chem. 2010, 285, 5296–5307. [Google Scholar] [CrossRef] [PubMed]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Constantinescu, S.N. Jak/stat signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Holbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High stat5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar]
- Hantschel, O.; Warsch, W.; Eckelhart, E.; Kaupe, I.; Grebien, F.; Wagner, K.U.; Superti-Furga, G.; Sexl, V. Bcr-abl uncouples canonical jak2-stat5 signaling in chronic myeloid leukemia. Nature Chem. Biol. 2012, 8, 285–293. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Nelson, E.A.; Walker, S.R.; Kuang, Y.; Distel, R.J.; Frank, D.A. Dual inhibition of jak2 and stat5 enhances killing of myeloproliferative neoplasia cells. Leukemia 2012, 26, 1407–1410. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The stat5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Verdine, G.L.; Walensky, L.D. The challenge of drugging undruggable targets in cancer: Lessons learned from targeting bcl-2 family members. Clin. Cancer Res. 2007, 13, 7264–7270. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. The inhibition of stat5 by a peptide aptamer ligand specific for the DNA binding domain prevents target gene transactivation and the growth of breast and prostate tumor cells. Pharmaceuticals 2013, 6, 960–987. [Google Scholar] [CrossRef] [PubMed]
- Borghouts, C.; Kunz, C.; Delis, N.; Groner, B. Monomeric recombinant peptide aptamers are required for efficient intracellular uptake and target inhibition. Mol. Cancer Res. MCR 2008, 6, 267–281. [Google Scholar] [CrossRef]
- Heuser, M.; Sly, L.M.; Argiropoulos, B.; Kuchenbauer, F.; Lai, C.; Weng, A.; Leung, M.; Lin, G.; Brookes, C.; Fung, S.; et al. Modeling the functional heterogeneity of leukemia stem cells: Role of stat5 in leukemia stem cell self-renewal. Blood 2009, 114, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Brandts, C.; Schwable, J.; Tickenbrock, L.; Sargin, B.; Ueker, A.; Bohmer, F.D.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. Activation mechanisms of stat5 by oncogenic flt3-itd. Blood 2007, 110, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Harir, N.; Pecquet, C.; Kerenyi, M.; Sonneck, K.; Kovacic, B.; Nyga, R.; Brevet, M.; Dhennin, I.; Gouilleux-Gruart, V.; Beug, H.; et al. Constitutive activation of stat5 promotes its cytoplasmic localization and association with pi3-kinase in myeloid leukemias. Blood 2007, 109, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in alk provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Gesbert, F.; Griffin, J.D. Bcr/abl activates transcription of the bcl-x gene through stat5. Blood 2000, 96, 2269–2276. [Google Scholar] [PubMed]
- Lacronique, V.; Boureux, A.; Monni, R.; Dumon, S.; Mauchauffe, M.; Mayeux, P.; Gouilleux, F.; Berger, R.; Gisselbrecht, S.; Ghysdael, J.; et al. Transforming properties of chimeric tel-jak proteins in ba/f3 cells. Blood 2000, 95, 2076–2083. [Google Scholar] [PubMed]
- Mayerhofer, M.; Gleixner, K.V.; Hoelbl, A.; Florian, S.; Hoermann, G.; Aichberger, K.J.; Bilban, M.; Esterbauer, H.; Krauth, M.T.; Sperr, W.R.; et al. Unique effects of kit d816v in baf3 cells: Induction of cluster formation, histamine synthesis, and early mast cell differentiation antigens. J. Immunol. 2008, 180, 5466–5476. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Nosaka, T.; Misawa, K.; Mui, A.L.; Gorman, D.; McMahon, M.; Miyajima, A.; Kitamura, T. Identification and characterization of a constitutively active stat5 mutant that promotes cell proliferation. Mol. Cell. Biol. 1998, 18, 3871–3879. [Google Scholar] [PubMed]
- Baskiewicz-Masiuk, M.; Masiuk, M.; Machalinski, B. The influence of stat5 antisense oligonucleotides on the proliferation and apoptosis of selected human leukaemic cell lines. Cell Prolif. 2003, 36, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Kosova, B.; Tezcanli, B.; Ekiz, H.A.; Cakir, Z.; Selvi, N.; Dalmizrak, A.; Kartal, M.; Gunduz, U.; Baran, Y. Suppression of stat5a increases chemotherapeutic sensitivity in imatinib-resistant and imatinib-sensitive k562 cells. Leuk. Lymphoma 2010, 51, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zeng, J.; Shi, M.; Zhao, S.; Bai, W.; Cao, W.; Tu, Z.; Huang, Z.; Feng, W. Targeted blockage of signal transducer and activator of transcription 5 signaling pathway with decoy oligodeoxynucleotides suppresses leukemic k562 cell growth. DNA Cell Biol. 2011, 30, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Chueh, F.Y.; Leong, K.F.; Yu, C.L. Mitochondrial translocation of signal transducer and activator of transcription 5 (stat5) in leukemic t cells and cytokine-stimulated cells. Biochem. Biophys. Res. Commun. 2010, 402, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Yang, Y.M.; Liang, F.X.; Gough, D.J.; Levy, D.E.; Sehgal, P.B. Nongenomic stat5-dependent effects on golgi apparatus and endoplasmic reticulum structure and function. Am. J. Physiol. Cell Physiol. 2012, 302, C804–C820. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Chatain, N.; Domoszlai, T.; Rinis, N.; Sommerauer, M.; Vogt, M.; Muller-Newen, G. Dynamics and non-canonical aspects of jak/stat signalling. Eur. J. Cell Biol. 2012, 91, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Basham, B.; Sathe, M.; Grein, J.; McClanahan, T.; D’Andrea, A.; Lees, E.; Rascle, A. In vivo identification of novel stat5 target genes. Nucl. Acids Res. 2008, 36, 3802–3818. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.P.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L. Stat5-dependent cyclind1 and bcl-xl expression in bcr-abl-transformed cells. Mol. Cell Biol. Res. Commun. MCBRC 2000, 3, 299–305. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Zhong, B.; Bai, F.; Jiang, K.; Bailey, R.D.; Garcia, R.; Jove, R.; Djeu, J.Y.; Loughran, T.P., Jr.; Wei, S. Cooperative regulation of mcl-1 by janus kinase/stat and phosphatidylinositol 3-kinase contribute to granulocyte-macrophage colony-stimulating factor-delayed apoptosis in human neutrophils. J. Immunol. 2001, 166, 7486–7495. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, I.; Kitamura, T.; Wakao, H.; Tanaka, H.; Hashimoto, K.; Albanese, C.; Downward, J.; Pestell, R.G.; Kanakura, Y. Transcriptional regulation of the cyclin d1 promoter by stat5: Its involvement in cytokine-dependent growth of hematopoietic cells. EMBO J. 1999, 18, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Paukku, K.; Silvennoinen, O. Stats as critical mediators of signal transduction and transcription: Lessons learned from stat5. Cytokine Growth Factor Rev. 2004, 15, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Irino, T.; Uemura, M.; Yamane, H.; Umemura, S.; Utsumi, T.; Kakazu, N.; Shirakawa, T.; Ito, M.; Suzuki, T.; Kinoshita, K. Jak2 v617f-dependent upregulation of pu.1 expression in the peripheral blood of myeloproliferative neoplasm patients. PLOS ONE 2011, 6, e22148. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Ito, M.; Raina, D.; Wu, Z.; Rosenblatt, J.; Avigan, D.; Stone, R.; Kufe, D. Muc1 oncoprotein regulates bcr-abl stability and pathogenesis in chronic myelogenous leukemia cells. Cancer Res. 2007, 67, 11576–11584. [Google Scholar] [CrossRef] [PubMed]
- Salas, E.M.; Garcia-Barchino, M.J.; Labiano, S.; Shugay, M.; Perez-Encinas, M.; Quinteiro, C.; Garcia-Delgado, M.; Vizmanos, J.L.; Novo, F.J. Lif, a novel stat5-regulated gene, is aberrantly expressed in myeloproliferative neoplasms. Genes Cancer 2011, 2, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Ezoe, S.; Tanaka, H.; Satoh, Y.; Fukushima, K.; Matsui, K.; Shibata, M.; Tanimura, A.; Oritani, K.; Matsumura, I.; et al. Bcr-abl but not jak2 v617f inhibits erythropoiesis through the ras signal by inducing p21cip1/waf1. J. Biol. Chem. 2010, 285, 31774–31782. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Kanno, Y.; Kerenyi, M.; Stephens, G.; Durant, L.; Watford, W.T.; Laurence, A.; Robinson, G.W.; Shevach, E.M.; Moriggl, R.; et al. Nonredundant roles for stat5a/b in directly regulating foxp3. Blood 2007, 109, 4368–4375. [Google Scholar] [CrossRef] [PubMed]
- Schwemmers, S.; Will, B.; Waller, C.F.; Abdulkarim, K.; Johansson, P.; Andreasson, B.; Pahl, H.L. Jak2v617f-negative et patients do not display constitutively active jak/stat signaling. Exp. Hematol. 2007, 35, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Von Manstein, V.; Yang, C.M.; Richter, D.; Delis, N.; Vafaizadeh, V.; Groner, B. Resistance of cancer cells to targeted therapies through the activation of compensating signaling loops. Curr. Signal Transduct. Ther. 2013, 8, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Chatain, N.; Ziegler, P.; Fahrenkamp, D.; Jost, E.; Moriggl, R.; Schmitz-Van de Leur, H.; Muller-Newen, G. Src family kinases mediate cytoplasmic retention of activated stat5 in bcr-abl-positive cells. Oncogene 2013, 32, 3587–3597. [Google Scholar] [CrossRef] [PubMed]
- Kosan, C.; Ginter, T.; Heinzel, T.; Krämer, O.H. Stat5 acetylation - mechanisms and consequences for immunological control and leukemogenesis. JAK-STAT 2013, 3, e26102. [Google Scholar] [CrossRef]
- Li, G.; Miskimen, K.L.; Wang, Z.; Xie, X.Y.; Brenzovich, J.; Ryan, J.J.; Tse, W.; Moriggl, R.; Bunting, K.D. Stat5 requires the n-domain for suppression of mir15/16, induction of bcl-2, and survival signaling in myeloproliferative disease. Blood 2010, 115, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Lockyer, H.M.; Tran, E.; Nelson, B.H. Stat5 is essential for akt/p70s6 kinase activity during il-2-induced lymphocyte proliferation. J. Immunol. 2007, 179, 5301–5308. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. Il-7 promotes glut1 trafficking and glucose uptake via stat5-mediated activation of akt to support t-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Diaz-Flores, E.; Li, G.; Wang, Z.; Kang, Z.; Haviernikova, E.; Rowe, S.; Qu, C.K.; Tse, W.; Shannon, K.M.; et al. Abnormal hematopoiesis in gab2 mutant mice. Blood 2007, 110, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Eulenfeld, R.; Gäbler, K.; Rolvering, C.; Haan, S.; Behrmann, I.; Denecke, B.; Haan, C.; Schaper, F. Jak2-v617f-induced mapk activity is regulated by pi3k and acts synergistically with pi3k on the proliferation of jak2-v617f-positive cells. JAK-STAT 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Borghouts, C.; Delis, N.; Brill, B.; Weiss, A.; Mack, L.; Lucks, P.; Groner, B. A membrane penetrating aptamer inhibits STAT3 function and suppresses the growth of STAT3 addicted tumor cells. JAK-STAT 2012, 1, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Williams, A.; Vultur, A.; List, A.; Bhalla, K.; Smith, D.; Lee, F.Y.; Jove, R. Dasatinib (bms-354825) inhibits STAT5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol. Cancer Ther. 2007, 6, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Chien, C.M.; Su, J.C.; Chen, Y.L.; Chang, L.S.; Lin, S.R. Novel indoloquinoline derivative, iqdma, inhibits stat5 signaling associated with apoptosis in k562 cells. J. Biochem. Mol. Toxicol. 2008, 22, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Quentmeier, H.; MacLeod, R.A.; Zaborski, M.; Drexler, H.G. Jak2 v617f tyrosine kinase mutation in cell lines derived from myeloproliferative disorders. Leukemia 2006, 20, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Demaison, C.; Parsley, K.; Brouns, G.; Scherr, M.; Battmer, K.; Kinnon, C.; Grez, M.; Thrasher, A.J. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of imunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 2002, 13, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Koptyra, M.; Gupta, S.; Talati, P.; Nevalainen, M.T. Signal transducer and activator of transcription 5a/b: Biomarker and therapeutic target in prostate and breast cancer. Int. J. Biochem. Cell Biol. 2011, 43, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Warsch, W.; Walz, C.; Sexl, V. Jak of all trades: Jak2-stat5 as novel therapeutic targets in bcr-abl1+ chronic myeloid leukemia. Blood 2013, 122, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Birkbak, N.J.; Vafaizadeh, V.; Walker, S.R.; Yeh, J.E.; Liu, S.; Kroll, Y.; Boldin, M.; Taganov, K.; Groner, B.; et al. Stat3 induction of mir-146b forms a feedback loop to inhibit the nf-kappab to il-6 signaling axis and stat3-driven cancer phenotypes. Sci. Signal. 2014, 7, ra11. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Xiang, M.; Frank, D.A. Stat3 activity and function in cancer: Modulation by stat5 and mir-146b. Cancers 2014, 6, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.A. Stat3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007, 251, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Dagvadorj, A.; Kirken, R.A.; Leiby, B.; Karras, J.; Nevalainen, M.T. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin. Cancer Res. 2008, 14, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Miskimen, K.L.; Wang, Z.; Xie, X.Y.; Tse, W.; Gouilleux, F.; Moriggl, R.; Bunting, K.D. Effective targeting of stat5-mediated survival in myeloproliferative neoplasms using abt-737 combined with rapamycin. Leukemia 2010, 24, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Dagvadorj, A.; Tan, S.H.; Liao, Z.; Xie, J.; Nurmi, M.; Alanen, K.; Rui, H.; Mirtti, T.; Nevalainen, M.T. N-terminal truncation of stat5a/b circumvents pias3-mediated transcriptional inhibition of stat5 in prostate cancer cells. Int. J. Biochem. Cell Biol. 2010, 42, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Hoelbl-Kovacic, A.; Bourgeais, J.; Hoefling, L.; Warsch, W.; Grundschober, E.; Uras, I.Z.; Menzl, I.; Putz, E.M.; Hoermann, G.; et al. Pak-dependent stat5 serine phosphorylation is required for bcr-abl-induced leukemogenesis. Leukemia 2014, 28, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Friedbichler, K.; Kerenyi, M.A.; Kovacic, B.; Li, G.; Hoelbl, A.; Yahiaoui, S.; Sexl, V.; Mullner, E.W.; Fajmann, S.; Cerny-Reiterer, S.; et al. Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood 2010, 116, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y.; Reich, N.C. Dynamic trafficking of stat5 depends on an unconventional nuclear localization signal. J. Cell Sci. 2013, 126, 3333–3343. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Yu, C.; Souza, R.F.; Theiss, A.L. Prohibitin 1 modulates mitochondrial function of stat3. Cell. Signal. 2014, 26, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Lee, J.E.; Yang, Y.M.; Liang, F.X.; Sehgal, P.B. Live-cell imaging of the association of stat6-gfp with mitochondria. PLOS ONE 2013, 8, e55426. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dutta, P.; Tsurumi, A.; Li, J.; Wang, J.; Land, H.; Li, W.X. Unphosphorylated stat5a stabilizes heterochromatin and suppresses tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10213–10218. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Liao, Z.; Hoang, D.T.; Dagvadorj, A.; Gupta, S.; Blackmon, S.; Ellsworth, E.; Talati, P.; Leiby, B.; Zinda, M.; et al. Pharmacologic inhibition of jak2-stat5 signaling by jak2 inhibitor azd1480 potently suppresses growth of both primary and castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 5658–5674. [Google Scholar] [CrossRef] [PubMed]
- Cumaraswamy, A.A.; Lewis, A.M.; Geletu, M.; Todic, A.; Diaz, D.B.; Cheng, X.R.; Brown, C.E.; Laister, R.C.; Muench, D.; Kerman, K.; et al. Nanomolar-potency small molecule inhibitor of stat5 protein. ACS Med. Chem. Lett. 2014, 5, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of bcr-abl activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on bcr-abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; Weber, A.; Mack, L. Increasing the range of drug targets: Interacting peptides provide leads for the development of oncoprotein inhibitors. Bioengineered 2012, 3, 320–325. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL Leukemia Cells. Cancers 2015, 7, 503-537. https://doi.org/10.3390/cancers7010503
Weber A, Borghouts C, Brendel C, Moriggl R, Delis N, Brill B, Vafaizadeh V, Groner B. Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL Leukemia Cells. Cancers. 2015; 7(1):503-537. https://doi.org/10.3390/cancers7010503
Chicago/Turabian StyleWeber, Axel, Corina Borghouts, Christian Brendel, Richard Moriggl, Natalia Delis, Boris Brill, Vida Vafaizadeh, and Bernd Groner. 2015. "Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL Leukemia Cells" Cancers 7, no. 1: 503-537. https://doi.org/10.3390/cancers7010503
APA StyleWeber, A., Borghouts, C., Brendel, C., Moriggl, R., Delis, N., Brill, B., Vafaizadeh, V., & Groner, B. (2015). Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL Leukemia Cells. Cancers, 7(1), 503-537. https://doi.org/10.3390/cancers7010503