
Targeting the Hedgehog Pathway in Pediatric Medulloblastoma

Abstract
:1. Clinical Description of Medulloblastoma
2. Hedgehog Pathway
3. SHH in Medulloblastoma

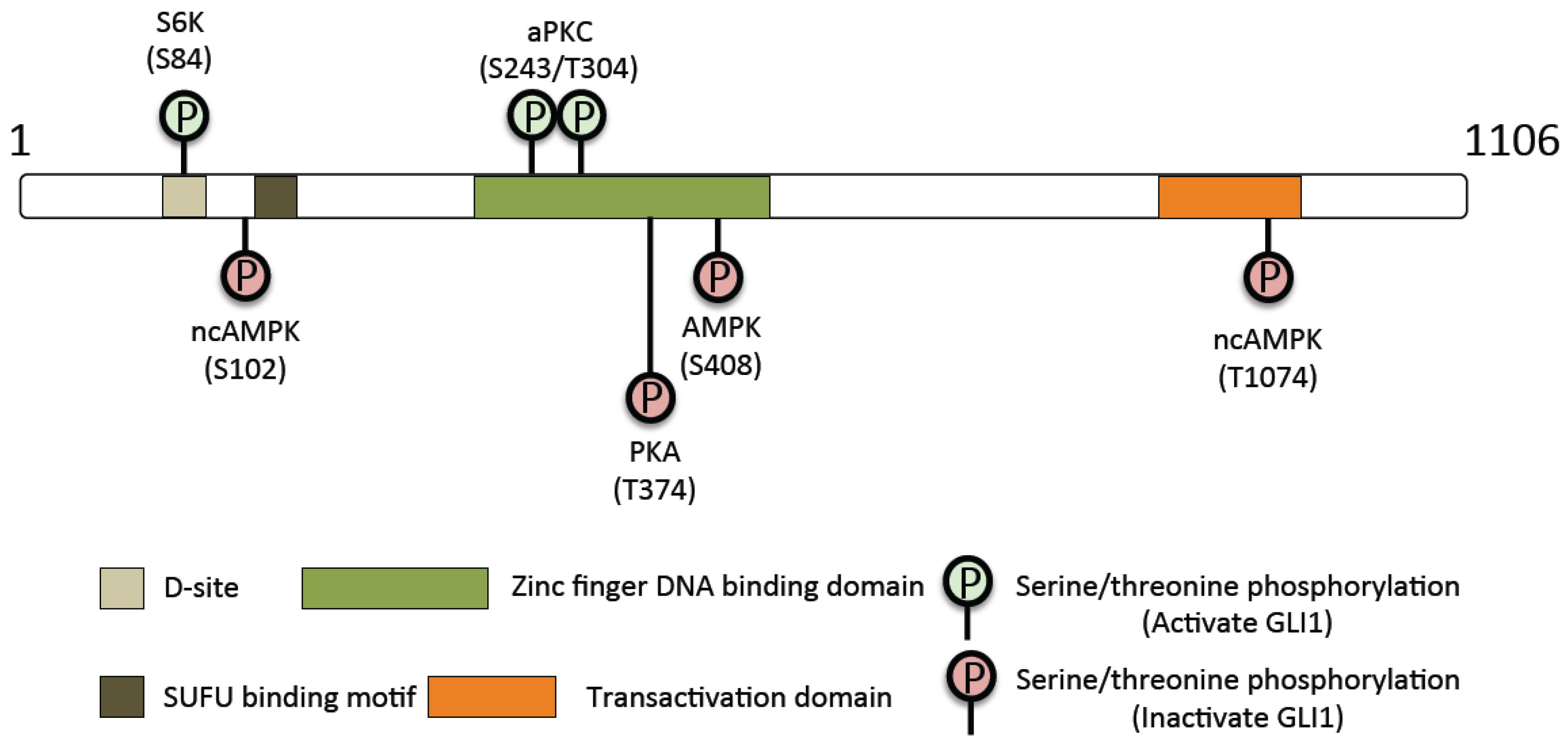{kind=link}
| Current and Potential Pipeline Medulloblastoma Drugs | |||
|---|---|---|---|
| Drug | Target | Pathway Affected | Limitations |
| Alkylating agents | DNA | DNA repair | Range of side effects |
| Cyclopamine | SMO | Hh | Low oral solubility |
| Vismodegib (GDC-0449) | SMO | Hh | Possible limited efficacy; mutations in SMO contributes to resistance |
| LDE-225 | SMO | Hh | In mouse model, tumors originally shrunk but then regrew [54] |
| GANT61 [52] | Gli1 and Gli2 | Hh | Tested in prostate cancer cells; Remains to be tested with medulloblastoma |
| HPI-1 [53] | Gli1 and Gli2 | Hh | Preclinical, remains to be tested with medulloblastoma |
| BMS-833923 [55] | SMO | Hh | Clinical trials with gastric and esophageal cancers; remains to be tested with medulloblastoma |
| Saridegib (IPI-926) [56] | SMO | Hh | Preclinical |
4. Genes Linked to Treat Hedgehog Inhibitors Resistance
5. Considerations for Targeting the SHH Pathway in MB
6. Pathways Interacting with SHH Signaling in MB
7. Non-MB Tumors: Pathway Crosstalk with SHH
8. Direct Regulation of SHH Signaling
| GLI1 Regulatory Kinases | ||
|---|---|---|
| Kinase | Residue | Activating or Inhibitory |
| Protein Kinase A | Thr-374 | Inhibitory |
| Protein Kinase A | Ser-640 | Inhibitory |
| Ribosomal protein S6 kinase 1 | Ser-84 | Activating |
| Atypical Protein Kinase C | Ser-243 | Activating |
| Atypical Protein Kinase C | Thr-304 | Activating |
| AMPK | Ser-102 | Inhibitory |
| AMPK | Ser-408 | Inhibitory |
| AMPK | Thr-1074 | Inhibitory |

9. Conclusions
- (1)
- Clinically, the identification and assessment of novel Gli inhibitors for Hh-mediated cancers should be evaluated in the context of medulloblastoma.
- (2)
- Clinically, given the unique genetic backgrounds of adult versus pediatric MB, assessment of novel MB treatments should be conducted with stratification of patient groups in terms of age.
- (3)
- Understanding of the resistance that arises from treatment of Hh-mediated cancers should be assessed in the context of Hh-mediated MB.
- (4)
- Non-transcriptional mechanisms by which SHH signaling mediates tumorigenesis in MB should be further studied.
- (5)
- Pathways such as the RAS/RAF/MEK, NF-κB, autophagy, and glucose sensing signaling have been shown to modulate SHH signaling. The effects of the crosstalk of these pathways on MB tumorigenesis should be further studied.
- (6)
- Gli1 represents a downstream signaling point of SHH driving SHH-mediated MB, and knowledge about the regulation points on Gli1 will prove essential to developing therapeutics targeting this transcription factor.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pui, C.H.; Gajjar, A.J.; Kane, J.R.; Qaddoumi, I.A.; Pappo, A.S. Challenging issues in pediatric oncology. Nat. Rev. Clin. Oncol. 2011, 8, 540–549. [Google Scholar] [CrossRef] [PubMed]
- De Bont, J.M.; Packer, R.J.; Michiels, E.M.; den Boer, M.L.; Pieters, R. Biological background of pediatric medulloblastoma and ependymoma: A review from a translational research perspective. Neuro Oncol. 2008, 10, 1040–1060. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.R.; Renowden, S.A.; Lowis, S.P.; Kurian, K.M. Early morning headache with vomiting in a 5 year old boy. BMJ 2013, 346, f3315. [Google Scholar] [CrossRef] [PubMed]
- Amene, C.S.; Yeh-Nayre, L.A.; Crawford, J.R. Isolated sensorineural hearing loss as initial presentation of recurrent medulloblastoma: Neuroimaging and audiologic correlates. Clin. Neuroradiol. 2013, 23, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, F.H.; Scheithauer, B.W.; Meyer, F.B.; Smithson, W.A.; Shaw, E.G.; Miller, G.M.; Groover, R.V. Medulloblastoma: I. Clinical, diagnostic, and therapeutic overview. J. Child Neurol. 1992, 7, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Dobrovoljac, M.; Hengartner, H.; Boltshauser, E.; Grotzer, M.A. Delay in the diagnosis of paediatric brain tumours. Eur. J. Pediatr. 2002, 161, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Shirsat, N.; Jalali, R. Molecular subgrouping of medulloblastoma: Impact upon research and clinical practice. Curr. Pediatr. Rev. 2015, 11, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic prognostication within medulloblastoma subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Ekker, S.C.; von Kessler, D.P.; Porter, J.A.; Sun, B.I.; Beachy, P.A. Autoproteolysis in hedgehog protein biogenesis. Science 1994, 266, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.Y.; Resh, M.D. Identification of N-terminal residues of Sonic Hedgehog important for palmitoylation by Hedgehog acyltransferase. J. Biol. Chem. 2012, 287, 42881–42889. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Eugster, C.; Panakova, D.; Mahmoud, A.; Eaton, S. Lipoprotein-heparan sulfate interactions in the Hh pathway. Dev. Cell 2007, 13, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Vajdos, F.F.; Ultsch, M.; Maun, H.R.; Wendt, U.; Cannon, J.; Desmarais, W.; Lazarus, R.A.; de Vos, A.M.; de Sauvage, F.J. Suppressor of fused regulates Gli activity through a dual binding mechanism. Mol. Cell. Biol. 2004, 24, 8627–8641. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ren, X.R.; Nelson, C.D.; Barak, L.S.; Chen, J.K.; Beachy, P.A.; de Sauvage, F.; Lefkowitz, R.J. Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science 2004, 306, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Rahnama, F.; Shimokawa, T.; Lauth, M.; Finta, C.; Kogerman, P.; Teglund, S.; Toftgard, R.; Zaphiropoulos, P.G. Inhibition of GLI1 gene activation by Patched1. Biochem. J. 2006, 394, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Spassky, N.; Han, Y.G.; Aguilar, A.; Strehl, L.; Besse, L.; Laclef, C.; Ros, M.R.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev. Biol. 2008, 317, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Dahmane, N.; Ruiz i Altaba, A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 1999, 126, 3089–3100. [Google Scholar] [PubMed]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [PubMed]
- Dey, J.; Ditzler, S.; Knoblaugh, S.E.; Hatton, B.A.; Schelter, J.M.; Cleary, M.A.; Mecham, B.; Rorke-Adams, L.B.; Olson, J.M. A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol. Cell. Biol. 2012, 32, 4104–4115. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Villavicencio, E.H.; Tsuchiya, K.D.; Pritchard, J.I.; Ditzler, S.; Pullar, B.; Hansen, S.; Knoblaugh, S.E.; Lee, D.; Eberhart, C.G.; et al. The Smo/Smo model: Hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008, 68, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar] [PubMed]
- Zurawel, R.H.; Allen, C.; Wechsler-Reya, R.; Scott, M.P.; Raffel, C. Evidence that haploinsufficiency of Ptch leads to medulloblastoma in mice. Genes Chromosomes Cancer 2000, 28, 77–81. [Google Scholar] [CrossRef]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Stephen, D.; Joyner, A.; Curran, T. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene 2005, 24, 4026–4036. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Borensztajn, K.S.; Roelink, H.; Peppelenbosch, M.P.; Spek, C.A. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell. Signal. 2007, 19, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.; Jager, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.M.; Raabe, E.; Eberhart, C.; Cohen, K.J. Management of pediatric and adult patients with medulloblastoma. Curr. Treat. Options Oncol. 2014, 15, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Fossati, P.; Ricardi, U.; Orecchia, R. Pediatric medulloblastoma: Toxicity of current treatment and potential role of protontherapy. Cancer Treat. Rev. 2009, 35, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Michiels, E.M.; Schouten-Van Meeteren, A.Y.; Doz, F.; Janssens, G.O.; van Dalen, E.C. Chemotherapy for children with medulloblastoma. Cochrane Database Syst. Rev. 2015, 1, Cd006678. [Google Scholar] [PubMed]
- Keeler, R.F. Teratogenic effects of cyclopamine and jervine in rats, mice and hamsters. Proc. Soc. Exp. Biol. Med. 1975, 149, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar] [PubMed]
- Athar, M.; Li, C.; Tang, X.; Chi, S.; Zhang, X.; Kim, A.L.; Tyring, S.K.; Kopelovich, L.; Hebert, J.; Epstein, E.H., Jr.; et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 2004, 64, 7545–7552. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Ruiz i Altaba, A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech. Dev. 2005, 122, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, R.J.; Hutson, P.R.; Hannam, P.W.; Nydza, R.J.; Washington, I.M.; Moore, R.W.; Girdaukas, G.G.; Peterson, R.E.; Bushman, W. Dose- and route-dependent teratogenicity, toxicity, and pharmacokinetic profiles of the hedgehog signaling antagonist cyclopamine in the mouse. Toxicol. Sci. 2008, 104, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Fellner, C. Vismodegib (erivedge) for advanced Basal cell carcinoma. P & T: Peer-Rev. J. Formul. Manag. 2012, 37, 670–682. [Google Scholar]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Matsui, W. Hedgehog pathway as a drug target: Smoothened inhibitors in development. Onco Targets Ther. 2012, 5, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Hatton, B.A.; Villavicencio, E.H.; Khanna, P.C.; Friedman, S.D.; Ditzler, S.; Pullar, B.; Robison, K.; White, K.F.; Tunkey, C.; et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. USA 2012, 109, 7859–7864. [Google Scholar] [CrossRef] [PubMed]
- Ingram, W.J.; Crowther, L.M.; Little, E.B.; Freeman, R.; Harliwong, I.; Veleva, D.; Hassall, T.E.; Remke, M.; Taylor, M.D.; Hallahan, A.R. ABC transporter activity linked to radiation resistance and molecular subtype in pediatric medulloblastoma. Exp. Hematol. Oncol. 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, C.; Nakahara, Y.; Ueda, S.; Hawkins, C.; Taylor, M.D.; Smith, C.A.; Rutka, J.T. Expression of MAGE and GAGE genes in medulloblastoma and modulation of resistance to chemotherapy. Laboratory investigation. J. Neurosurg. Pediatr. 2008, 1, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, C.; Eberhart, D.E.; Curran, T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001, 61, 513–516. [Google Scholar] [PubMed]
- Malek, R.; Matta, J.; Taylor, N.; Perry, M.E.; Mendrysa, S.M. The p53 inhibitor MDM2 facilitates Sonic Hedgehog-mediated tumorigenesis and influences cerebellar foliation. PLoS ONE 2011, 6, e17884. [Google Scholar] [CrossRef] [PubMed]
- Furman, M.A.; Shulman, K. Cyclic AMP and adenyl cyclase in brain tumors. J. Neurosurg. 1977, 46, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Warrington, N.M.; Gianino, S.M.; Jackson, E.; Goldhoff, P.; Garbow, J.R.; Piwnica-Worms, D.; Gutmann, D.H.; Rubin, J.B. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res. 2010, 70, 5717–5727. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, L.; Chen, Y.; Remke, M.; Shih, D.; Lu, F.; Wang, H.; Deng, Y.; Yu, Y.; Xia, Y.; et al. The G protein alpha subunit Galphas is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat. Med. 2014, 20, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Forget, A.; Bihannic, L.; Cigna, S.M.; Lefevre, C.; Remke, M.; Barnat, M.; Dodier, S.; Shirvani, H.; Mercier, A.; Mensah, A.; et al. Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Dev. Cell 2014, 29, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Mille, F.; Tamayo-Orrego, L.; Levesque, M.; Remke, M.; Korshunov, A.; Cardin, J.; Bouchard, N.; Izzi, L.; Kool, M.; Northcott, P.A.; et al. The Shh receptor Boc promotes progression of early medulloblastoma to advanced tumors. Dev. Cell 2014, 31, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Aref, D.; Moffatt, C.J.; Agnihotri, S.; Ramaswamy, V.; Dubuc, A.M.; Northcott, P.A.; Taylor, M.D.; Perry, A.; Olson, J.M.; Eberhart, C.G.; et al. Canonical TGF-beta pathway activity is a predictor of SHH-driven medulloblastoma survival and delineates putative precursors in cerebellar development. Brain Pathol. 2013, 23, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Gate, D.; Danielpour, M.; Rodriguez, J., Jr.; Kim, G.B.; Levy, R.; Bannykh, S.; Breunig, J.J.; Kaech, S.M.; Flavell, R.A.; Town, T. T-cell TGF-beta signaling abrogation restricts medulloblastoma progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3458–E3466. [Google Scholar] [CrossRef] [PubMed]
- Rios, I.; Alvarez-Rodriguez, R.; Marti, E.; Pons, S. Bmp2 antagonizes sonic hedgehog-mediated proliferation of cerebellar granule neurones through Smad5 signalling. Development 2004, 131, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Rubin, J.B.; Gibson, H.D.; DeHaan, E.N.; Alvarez-Hernandez, X.; Segal, R.A.; Luster, A.D. SDF-1 alpha induces chemotaxis and enhances Sonic hedgehog-induced proliferation of cerebellar granule cells. Development 2001, 128, 1971–1981. [Google Scholar] [PubMed]
- Sengupta, R.; Dubuc, A.; Ward, S.; Yang, L.; Northcott, P.; Woerner, B.M.; Kroll, K.; Luo, J.; Taylor, M.D.; Wechsler-Reya, R.J.; et al. CXCR4 activation defines a new subgroup of Sonic hedgehog-driven medulloblastoma. Cancer Res. 2012, 72, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.P.; Emmenegger, B.A.; Grasfeder, L.L.; Oliver, T.G.; Wechsler-Reya, R.J. Fibroblast growth factor blocks Sonic hedgehog signaling in neuronal precursors and tumor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 2973–2978. [Google Scholar] [CrossRef] [PubMed]
- Emmenegger, B.A.; Hwang, E.I.; Moore, C.; Markant, S.L.; Brun, S.N.; Dutton, J.W.; Read, T.A.; Fogarty, M.P.; Singh, A.R.; Durden, D.L.; et al. Distinct roles for fibroblast growth factor signaling in cerebellar development and medulloblastoma. Oncogene 2013, 32, 4181–4188. [Google Scholar] [CrossRef] [PubMed]
- Sah, N.K.; Khan, Z.; Khan, G.J.; Bisen, P.S. Structural, functional and therapeutic biology of survivin. Cancer Lett. 2006, 244, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Brun, S.N.; Markant, S.L.; Esparza, L.A.; Garcia, G.; Terry, D.; Huang, J.M.; Pavlyukov, M.S.; Li, X.N.; Grant, G.A.; Crawford, J.R.; et al. Survivin as a therapeutic target in Sonic hedgehog-driven medulloblastoma. Oncogene 2015, 34, 3770–3779. [Google Scholar] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.A.A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Pasca di Magliano, M.; Sekine, S.; Ermilov, A.; Ferris, J.; Dlugosz, A.A.; Hebrok, M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006, 20, 3161–3173. [Google Scholar] [CrossRef] [PubMed]
- Whisenant, T.C.; Ho, D.T.; Benz, R.W.; Rogers, J.S.; Kaake, R.M.; Gordon, E.A.; Huang, L.; Baldi, P.; Bardwell, L. Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.H.; Wang, H.C.; Zhao, D.M.; Ji, X.X.; Song, M.; Yang, X.J.; Cui, W. Cooperatively transcriptional and epigenetic regulation of sonic hedgehog overexpression drives malignant potential of breast cancer. Cancer Sci. 2015, 106, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, W.; Yang, Q.; Zhou, S. Nuclear localization of GLI1 and elevated expression of FOXC2 in breast cancer is associated with the basal-like phenotype. Histol. Histopathol. 2012, 27, 475–484. [Google Scholar] [PubMed]
- Memmi, E.M.; Sanarico, A.G.; Giacobbe, A.; Peschiaroli, A.; Frezza, V.; Cicalese, A.; Pisati, F.; Tosoni, D.; Zhou, H.; Tonon, G.; et al. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013, 58, 995–1010. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Li, J.; Gao, T.; Xie, J.; Evers, B.M. Protein kinase Cdelta negatively regulates hedgehog signaling by inhibition of Gli1 activity. J. Biol. Chem. 2009, 284, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, X.; Zheng, X.; Yao, Y.; Liu, Q. PKM2 regulates Gli1 expression in hepatocellular carcinoma. Oncol. Lett. 2014, 8, 1973–1979. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, X.; Zheng, X.; Yao, Y.; Wang, M.; Liu, Q. The transcriptional activity of Gli1 is negatively regulated by AMPK through Hedgehog partial agonism in hepatocellular carcinoma. Int. J. Mol. Med. 2014, 34, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Chi, S.; Zhang, X.; Xie, J. Regulation of Gli1 localization by the cAMP/protein kinase A signaling axis through a site near the nuclear localization signal. J. Biol. Chem. 2006, 281, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Asaoka, Y.; Kanai, F.; Ichimura, T.; Tateishi, K.; Tanaka, Y.; Ohta, M.; Seto, M.; Tada, M.; Ijichi, H.; Ikenoue, T.; et al. Identification of a suppressive mechanism for Hedgehog signaling through a novel interaction of Gli with 14-3-3. J. Biol. Chem. 2010, 285, 4185–4194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Volny, N.; Lee, S.; Qiao, J.; Chung, D.H. Gli1 transcriptional activity is negatively regulated by AKT2 in neuroblastoma. Oncotarget 2013, 4, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Luo, J.; Mosley, Y.Y.; Hedrick, V.E.; Paul, L.N.; Chang, J.; Zhang, G.; Wang, Y.K.; Banko, M.R.; Brunet, A.; et al. AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep. 2015, 12, 599–609. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.Y.; Yang, J.-Y. Targeting the Hedgehog Pathway in Pediatric Medulloblastoma. Cancers 2015, 7, 2110-2123. https://doi.org/10.3390/cancers7040880
Huang SY, Yang J-Y. Targeting the Hedgehog Pathway in Pediatric Medulloblastoma. Cancers. 2015; 7(4):2110-2123. https://doi.org/10.3390/cancers7040880
Chicago/Turabian StyleHuang, Sherri Y., and Jer-Yen Yang. 2015. "Targeting the Hedgehog Pathway in Pediatric Medulloblastoma" Cancers 7, no. 4: 2110-2123. https://doi.org/10.3390/cancers7040880
APA StyleHuang, S. Y., & Yang, J.-Y. (2015). Targeting the Hedgehog Pathway in Pediatric Medulloblastoma. Cancers, 7(4), 2110-2123. https://doi.org/10.3390/cancers7040880