
Updates in Therapy for Advanced Melanoma

Abstract
:1. Background
A Historical Perspective
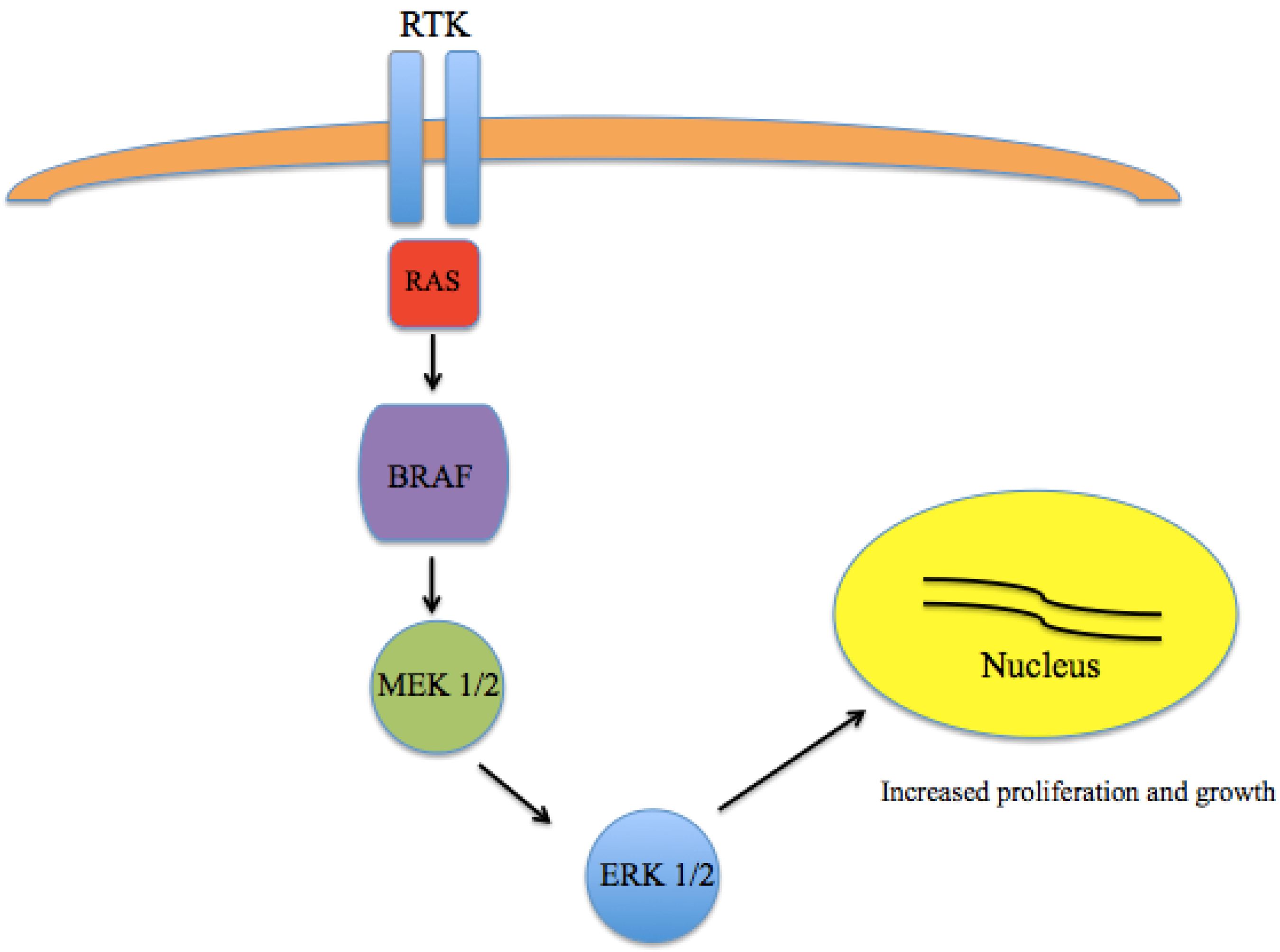
2. Targeted Therapy in Melanoma
2.1. BRAF

2.2. MEK
2.3. KIT
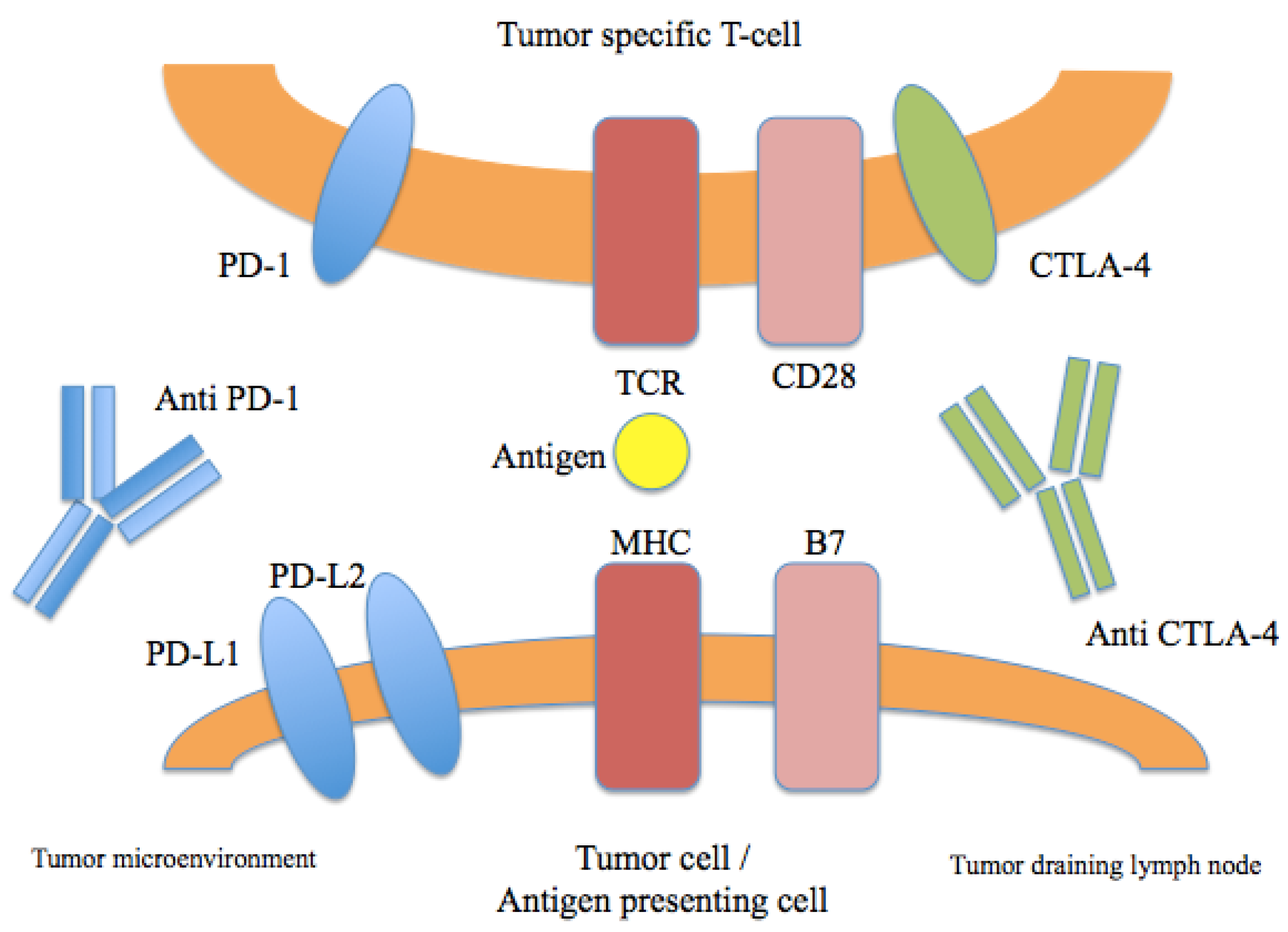
3. Immune Checkpoint Inhibition
3.1. CTLA-4

3.2. PD-1
4. Adoptive Cell Immunotherapy
5. Combination Therapy
5.1. Targeted Therapies

{kind=link}
{kind=link}
| Study | Trial Design | Agents Studied | N | RR (%) | Median PFS (Months) | OS (%) |
|---|---|---|---|---|---|---|
| Dabrafenib + trametinib studies | ||||||
| Robert et al. [5] | Randomized, phase III | Dabrafenib + trametinib | 352 | 64 | 11.4 | 72 (12 months) |
| Vemurafenib | 352 | 51 | 7.3 | 65 (12 months) | ||
| Long et al. [6,74] | Randomized, phase III | Dabrafenib + trametinib | 211 | 67 | 11 | 93 (9 months) |
| Dabrafenib + placebo | 212 | 51 | 8.8 | 85 (9 months) | ||
| Daud et al. [77] | Randomized, phase I—II | Dabrafenib + trametinib (150/2) | 54 | 76 | 9.4 | 51 (24 months) |
| Flaherty et al. [70] | ||||||
| Dabrafenib + trametinib (150/1) | 54 | 50 | NR a | NR | ||
| Dabrafenib (150) | 54 | 54 | 5.8 | 44 (24 months) | ||
| Vemurafenib + cobimetinib studies | ||||||
| Larkin et al. [4,75] | Randomized, phase III | Vemurafenib + cobimetinib (960/60) | 247 | 70 | 12.3 | 81 (9 months) |
| Vemurafenib (960) + placebo | 248 | 50 | 7.2 | 73 (9 months) | ||
| Encorafenib + binimetinib studies | ||||||
| Sullivan et al. [76] | Randomized, phase II | Encorafenib + binimetinib (600/45) | 38 | 72 | 11.3 | NR |
| (all doses combined) | ||||||
| Encorafenib + binimetinib (400/45) | 4 | 78 | NR | |||
5.2. Immunotherapy Combinations
| Study | Trial Design | Agents Studied | N | RR (%) | PFS (Months) | OS (%) |
|---|---|---|---|---|---|---|
| Wolchok et al. [83] | Phase I | Nivolumab + ipilimumab | 314 | 57.6 | 11.5 | 75 (2 years) |
| Sznol et al. [84] | multiple cohort | |||||
| Nivolumab | 316 | 43.7 | 6.9 | NC a | ||
| Ipilimumab | 315 | 19 | 2.9 | NC | ||
| Hodi et al. [85], | Randomized, phase II | Ipilimumab + nivolumab | 72 * | 60 | 8.9 | NR b |
| Postow et al. [81] | ||||||
| Ipilimumab + placebo | 37 * | 11 | 4.7 | NR | ||
| Larkin et al. [82] | Randomized, phase III | Ipilimumab + nivolumab | 314 | 57.6 | 11.5 | NR |
| Ipilimumab monotherapy | 315 | 19 | 2.9 | NR | ||
| Nivolumab monotherapy | 316 | 43.7 | 6.9 | NR |
5.3. Future Opportunities for Combination Therapy
| Combination | Study Population | Status | Study Design |
|---|---|---|---|
| Nivolumab + ipilimumab (NCT02320058) | Patients with melanoma brain metastases | Recruiting | Single arm phase II |
| Pembrolizumab + pegylated IFN alfa-2b and pembrolizumab + ipilimumab (NCT02089685) | Advanced/unresectable or metastatic melanoma or renal cell carcinoma | Recruiting | Single arm phase I Randomized expansion cohorts |
| Ipilimumab ± T-VEC (NCT01740297) | Advanced/unresectable melanoma, with injectable tumor | Recruiting | Phase Ib, II |
| Pembrolizumab + T-VEC (NCT02263508) | Advanced/unresectable melanoma, with injectable tumor | Active but not recruiting | Phase Ib/III |
| Ipilimumab + nivolumab and dabrafenib + trametinib (NCT02224781) | Advanced/unresectable melanoma, BRAF mutated | Recruiting | Randomized phase III, comparing sequence |
| Ipilimumab ± dabrafenib ± trametinib (NCT01940809) | Unresectable or metastatic malignant melanoma, BRAF mutated | Recruiting | Phase I |
| Pembrolizumab + trametinib and dabrafenib (NCT02130466) | Advanced (unresectable Stage III) or metastatic (Stage IV) melanoma | Recruiting | Phase II/III |
| MPDL3280A + vemurafenib or vemurafenib + cobimetinib (NCT01656642) | Metastatic melanoma, with BRAFV600 mutation | Recruiting | Phase II |
| MEDI4736 + dabrafenib and trametinib or with trametinib alone (NCT02027961) | Stage IIIc (unresectable) or Stage IV (metastatic) melanoma | Recruiting | Phase II/III |
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.; Krapcho, M.; Garshell, J.; Miller, D.; Altekruse, S.; Kosary, C.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. SEER Cancer Statistics Review, 1975–2012; National Cancer Institute: Bethesda, MD, USA, April 2015. Available online: http://seer.cancer.gov/csr/1975_2012/ (accessed on 11 September 2015).
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Long, G.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, JJ.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.; O'Day, S.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Scadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Weber, J.; D’Angelo, S.; Minor, D.; Hodi, F.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Middleton, M.; Grob, J.; Aaronson, N.; Fierlbeck, G.; Tilgen, W.; Seiter, S.; Gore, M.; Aamdal, S.; Cebon, J.; Coates, A.; et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J. Clin. Oncol. 2000, 18, 158–166. [Google Scholar] [PubMed]
- Danson, S.; Middleton, M. Temozolomide: A novel oral alkylating agent. Expert Rev. Anticancer Ther. 2001, 1, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.; Lotze, M.; Dutcher, J.; Fisher, R.; Rosenberg, S. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [PubMed]
- Atkins, M.; Kunkel, L.; Sznol, M.; Rosenberg, S. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J. Sci. Am. 2000, 6, S11–S14. [Google Scholar] [PubMed]
- Pollock, P.; Meltzer, P. A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell 2002, 2, 5–7. [Google Scholar] [CrossRef]
- Brose, M.; Volpe, P.; Feldman, M.; Kumar, M.; Rishi, I.; Gerrero, R.; Einhorn, E.; Herlyn, M.; Minna, J.; Nicholson, A.; et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002, 62, 6997–7000. [Google Scholar] [PubMed]
- Pollock, P.; Harper, U.; Hansen, K. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- bioMérieux. THxID BRAF. 2013. Available online: http://www.accessdata.fda.gov/cdrh_docs/pdf12/P120014c.pdf (accessed on 11 September 2015). [Google Scholar]
- Roche Molecular Systems. I cobas® 4800 BRAF V600 Mutation Test—P110020. 2011. Available online: http://www.accessdata.fda.gov/cdrh_docs/pdf11/P110020c.pdf (accessed on 11 September 2015). [Google Scholar]
- Tetzlaff, M.; Pattanaprichakul, P.; Wargo, J.; Fox, P.S.; Patel, K.P.; Estrella, J.S.; Broaddus, R.R.; Williams, M.D.; Davies, M.A.; Routbort, M.J.; et al. Utility of BRAF V600E Immunohistochemistry Expression Pattern as a Surrogate of BRAF Mutation Status in 154 Patients with Advanced Melanoma. Hum. Pathol. 2015, 46, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Pearlstein, M.; Zedek, D.; Ollila, D.W.; Treece, A.; Gulley, M.L.; Groben, P.A.; Thomas, N.E. Validation of the VE1 immunostain for the BRAF V600E mutation in melanoma. J. Cutan. Pathol. 2014, 41, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Long, G.; Wilmott, J.; Capper, D.; Preusser, M.; Zhang, Y.; Thompson, J.; Kefford, R.F.; von Deimling, A.; Scolyer, R.A. Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am. J. Surg. Pathol. 2013, 37, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O'Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.; Chapman, P.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.; Demidov, L.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.; Demidov, L.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. An update on BREAK-3, a phase III, randomized trial: Dabrafenib (DAB) versus dacarbazine (DTIC) in patients with BRAF V600E-positive mutation metastatic melanoma (MM). In Proceedings of the 49th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, USA, 31 May–4 June 2013; Volume 31.
- Hauschild, A.; Grobb, J.; Demidov, L.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.; Miller, W.; Martin-Algarra, S.; et al. An update on overall survival (OS) and follow-on therapies in BREAK-3, a phase III, randomized trial: dabrafenib (D) vs. dacarbazine (DTIC) in patients (pts) with BRAF V600E mutation-positive metastatic melanoma (MM). Ann. Oncol. 2014, 25, iv378. [Google Scholar] [CrossRef]
- Chapman, P. Mechanisms of resistance to RAF inhibition in melanomas harboring a BRAF mutation. In Proceedings of the 2013 ASCO Annual Meeting, Chicago, IL, USA, 31 May–3 June 2013.
- Rizos, H.; Menzies, A.; Pupo, G.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Kim, K. MEK inhibition in the treatment of advanced melanoma. Curr. Oncol. Rep. 2013, 15, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.; Schadendorf, D.; Berking, C.; Agarwala, S.; van Herpen, C.; Queirolo, P.; Blank, C.; Hauschild, A.; Beck, J.; Dummer, R. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Carvajal, R.; Sosman, J.; Quevedo, J.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.; Busam, K.; Pinkel, D.; Bastian, B. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Beadling, C.; Jacobsen-Dunlop, E.; Hodi, F.; Le, C.; Warrick, A.; Patterson, J.; Town, A. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.; Antonescu, C.; Wolchok, J.; Chapman, P.; Roman, R.A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.; Corless, C.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.; Lawrence, D.; Weber, J.; Gajewski, T.F.; Gonzalez, R.; Lutzky, J.; O’Day, S.J.; Hamid, O.; Wolchok, J.D.; Chapman, P.B.; et al. Phase II Study of Nilotinib in Melanoma Harboring KIT Alterations Following Progression to Prior KIT Inhibition. Clin. Cancer Res. 2015, 21, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Grob, J.; Aamdal, S.; Bondarenko, I.; Robert, C.; Thomas, L.; Garbe, C.; Chiarion-Sileni, V.; Testori, A.; Chen, T.; et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J. Clin. Oncol. 2015, 33, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.; Robert, C.; Weber, J.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival from phase II and phase III trials of ipilimumab. J. Clinical. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.; Hodi, F.; Brahmer, J.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.; Sznol, M.; McDermott, D.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti–PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.; Ribas, A.; Robert, C.; Hodi, F.S.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W.; et al. Long-term efficacy of pembrolizumab (pembro; MK-3475) in a pooled analysis of 655 patients (pts) with advanced melanoma (MEL) enrolled in KEYNOTE-001. In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015; Volume 33.
- Berger, R.; Rotem-Yehudar, R.; Slama, G.; Landes, S.; Kneller, A.; Leiba, M.; Koren-Michowitz, M.; Shimoni, A.; Nagler, A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 3044–3051. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.; Kudchadkar, R.; Sznol, M.; McDermott, D.F.; Lotem, M.; Schachter, J.; Wolchok, J.D.; Urba, W.J.; Kuzel, T.; Schuchter, L.M.; et al. Phase 2, multicenter, safety and efficacy study of pidilizumab in patients with metastatic melanoma. In Proceedings of the 2014 ASCO Annual Meeting, Chicago, IL, 29 May–2 June 2014; Volume 32.
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.; Gulley, J.L.; Tsang, K.Y.; Schlom, J. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol. Res. 2015, 3, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lee-Gabel, L.; Nadeau, M.; Ferencz, T.; Soefje, S. Clinical evaluation of compounds targeting PD-1/PD-L1 pathway for cancer immunotherapy. J. Oncol. Pharm. Pract. 2015, 21, 451–467. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Tykodi, S.; Chow, L.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.; Kluger, H.; Sullivan, R.J.; Flaherty, K.T.; Soria, J.C.; Xiao, Y.; Chappey, C.; Kowanetz, M.; Ballinger, M.; Sosman, J.A. Clinical activity of the PD-L1 inhibitor MPDL3280A in patients with metastatic melanoma: updated phase I data. Pigment Cell Melanoma Res. 2014, 27, 1169. [Google Scholar]
- Cha, E.; Wallin, J.; Kowanetz, M. PD-L1 inhibition with MPDL3280A for solid tumors. Semin. Oncol. 2015, 42, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Lutzky, J.; Antonia, S.; Blake-Haskins, A.; Li, X.; Robbins, P.B.; Shalabi, A.M.; Vasselli, J.; Ibrahim, R.A.; Khleif, S.; Segal, N.H. A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors. In Proceedings of the 2014 ASCO Annual Meeting, Chicago, IL, 29 May–2 June 2014; Volume 32.
- Johnson, L.; Morgan, R.; Dudley, M.; Rosenberg, S. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.; Dudley, M.; Rosenberg, S. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.; Yang, J.; Sherry, R.; Dudley, M. Durable complete responses in heavily pretreated patients with metastatic melanoma using T cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Forget, M.; Chacon, J.; Radvanyi, L. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.; Shapira-Frommer, R.; Treves, A.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N.; et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2010, 23, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Phan, G.; Rosenberg, S. Adoptive cell transfer for patients with metastatic melanoma: the potential and promise of cancer immunotherapy. Cancer Control 2013, 20, 289–297. [Google Scholar] [PubMed]
- Lu, Y.; Yao, X.; Crystal, J.; Robbins, P. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, Y.G.; Pyo, M.; Lee, H.K.; Hong, J.T.; Kim, Y.; Han, S.B. Adoptive cell therapy of melanoma with cytokine-induced killer cells. Immune Netw. 2015, 15, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.; Chinnasamy, N.; Rosenberg, S. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.; Kassim, S.; Tran, T.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.; Infante, J.; Daud, A.; Gonzalez, R.; Kefford, R.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Flaherty, K. BRAF in melanoma: current strategies and future directions. Clin. Cancer Res. 2013, 19, 4326–4334. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, O.; ElHalawani, H.; Ahmed, H. Doublet BRAF/MEK inhibition versus single-agent BRAF inhibition in the management of BRAF-mutant advanced melanoma, biological rationale and meta-analysis of published data. Clin. Transl. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Arnone, M.R.; Bleam, M.R.; Moss, K.G.; Yang, J.; Fedorowicz, K.E.; Smitheman, K.N.; Erhardt, J.A.; Hughes-Earle, A.; Kane-Carson, L.S.; et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS ONE 2013, 8, e67583. [Google Scholar] [CrossRef] [PubMed]
- Long, G.; Stroyakovsky, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.; et al. COMBI-d: A randomized, double-blinded, phase III study comparing the combination of dabrafenib and trametinib to dabrafenib and trametinib placebo as first-line therapy in patients (pts) with unresectable or metastatic BRAFV600E/K mutation-positive cutaneous melanoma. In Proceedings of the 2014 ASCO Annual Meeting, Chicago, IL, 29 May–2 June 2014; Volume 32.
- Larkin, J.; Yan, Y.; McArthur, G.; Ascierto, P.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Update of progression-free survival (PFS) and correlative biomarker analysis from coBRIM: Phase III study of cobimetinib (cobi) plus vemurafenib (vem) in advanced BRAF-mutated melanoma. In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Sullivan, R.; Weber, J.; Patel, S.; Dummer, R.; Miller, W.; Cosgrove, D.; Carlino, M.; Tan, D.; Lebbe, C.; Cipani, T.; et al. A phase Ib/II study of BRAF inhibitor (BRAFi) encorafenib (ENCO) plus MEK inhibitor (MEKi) binimetinib (BINI) in cutaneous melanoma patients naive to BRAFi treatment. In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015; Volume 33.
- Daud, A.; Weber, J.; Sosman, J.A.; Kim, K.; Gonzalez, R.; Hamid, O.; Infante, J.; Cebon, J.; Schuchter, L.; Long, G. Updated overall survival (OS) results for BRF113220, a phase I–II study of dabrafenib alone versus combined dabrafenib and trametinib in patients with BRAF V600 metastatic melanoma (MM). In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015; Volume 33.
- Curran, M.; Montalvo, M.; Yagita, H.; Allison, J. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Verma, R.; Sznol, M.; Boddupalli, C.S.; Gettinger, S.N.; Kluger, H.; Callahan, M.; Wolchok, J.D.; Halaban, R.; Dhodapkar, M.V.; et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J. Immunol. 2015, 194, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.; Kluger, H.; Callahan, M.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.; Chesney, J.; Pavlick, A.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.; Meyer, N.; Giguere, J.; Agarwala, S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.; Cowey, C.; Lao, C.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.; Cowey, C.; Lao, C.; Schadendorf, D.; Ferrucci, P.; Smylie, M.; et al. Efficacy and safety results from a phase III trial of nivolumab (NIVO) alone or combined with ipilimumab (IPI) versus IPI alone in treatment-naive patients (pts) with advanced melanoma (MEL) (CheckMate 067). In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015; Volume 33.
- Sznol, M.; Kluger, H.; Callahan, M.; Postow, M.; Gordon, R.; Segal, N.; Rizvi, N.; Lesokhin, A.; Atkins, M.; Kirkwood, J.; et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL). In Proceedings of the 2014 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2014; Volume 32.
- Hodi, F.; Postow, M.; Chesney, J.; Pavlick, A.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.; Meyer, N.; Giguere, J.; et al. Clinical response, progression-free survival (PFS), and safety in patients (pts) with advanced melanoma (MEL) receiving nivolumab (NIVO) combined with ipilimumab (IPI) vs IPI monotherapy in CheckMate 069 study. In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015; Volume 33.
- Bajor, D.; Mick, R.; Riese, M.; Richman, L.; Xu, X.; Torigian, D.A.; Stelekati, E.; Sweeney, M.; Sullivan, B.; Schuchter, L.M.; et al. Combination of agonistic CD40 monoclonal antibody CP-870,893 and anti-CTLA-4 antibody tremelimumab in patients with metastatic melanoma. In Proceedings of the 106th Annual Meeting of the American Association for Cancer Research, Philadelphia, PA, USA, 18–22 April 2015.
- Andtbacka, R.; Kaufman, H.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.; Andtbacka, R.; Minor, D.; Hamid, O.; Li, A.; Chou, J.; Kaufman, H. Survival, safety, and response patterns in a phase 1b multicenter trial of talimogene laherparepvec (T-VEC) and ipilimumab (ipi) in previously untreated, unresected stage IIIB-IV melanoma. In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015; Volume 33.
- Boni, A.; Cogdill, A.; Dang, P.; Udayakumar, D.; Wargo, J. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.; Piris, A.; Cogdill, A.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hodi, F.; Callahan, M.K.; Konto, C.; Wolchok, J.D. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 2013, 368, 1365–1366. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Lawson, D.; Salama, A.; Koon, H.B.; Guthrie, T.H.; Thomas, S.S.; O’Day, S.; Shaheen, M.F.; Zhang, B.; Francis, S.; et al. A single-arm, open-label, phase II study to evaluate the safety of vemurafenib (VEM) followed by ipilimumab (IPI) in BRAF V600-mutated metastatic melanoma (MM). In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015; Volume 33.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, B.P.; Salama, A.K.S. Updates in Therapy for Advanced Melanoma. Cancers 2016, 8, 17. https://doi.org/10.3390/cancers8010017
Singh BP, Salama AKS. Updates in Therapy for Advanced Melanoma. Cancers. 2016; 8(1):17. https://doi.org/10.3390/cancers8010017
Chicago/Turabian StyleSingh, Bhavana P., and April K. S. Salama. 2016. "Updates in Therapy for Advanced Melanoma" Cancers 8, no. 1: 17. https://doi.org/10.3390/cancers8010017
APA StyleSingh, B. P., & Salama, A. K. S. (2016). Updates in Therapy for Advanced Melanoma. Cancers, 8(1), 17. https://doi.org/10.3390/cancers8010017