
Targeting the TAM Receptors in Leukemia

 ,
,
Abstract
:1. Introduction
2. TAM Receptors in Normal Hematopoeisis
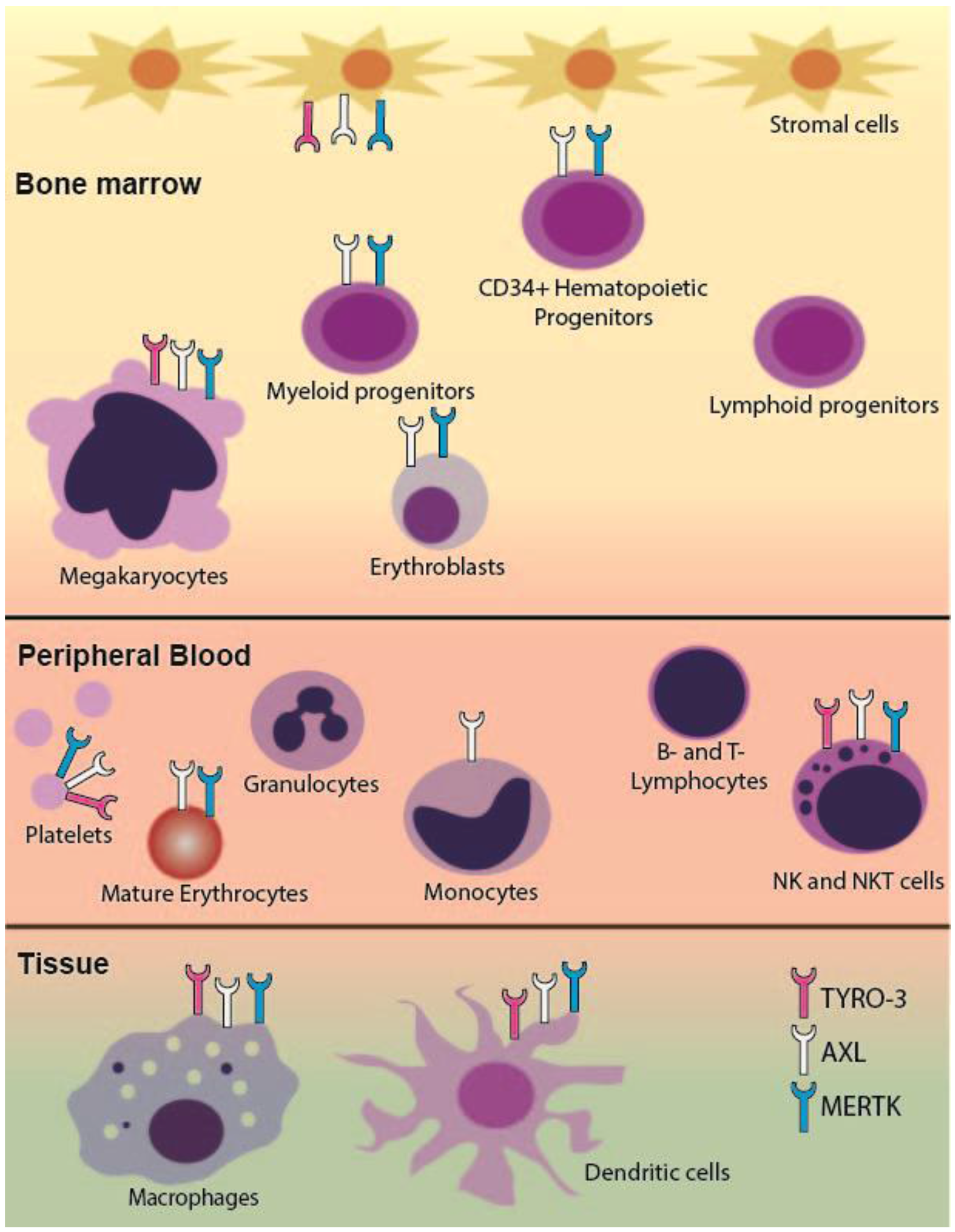
2.1. TAM RTK Expression in the Hematopoietic System
2.2. Regulation of Erythropoiesis
2.3. Role in Megakaryopoiesis
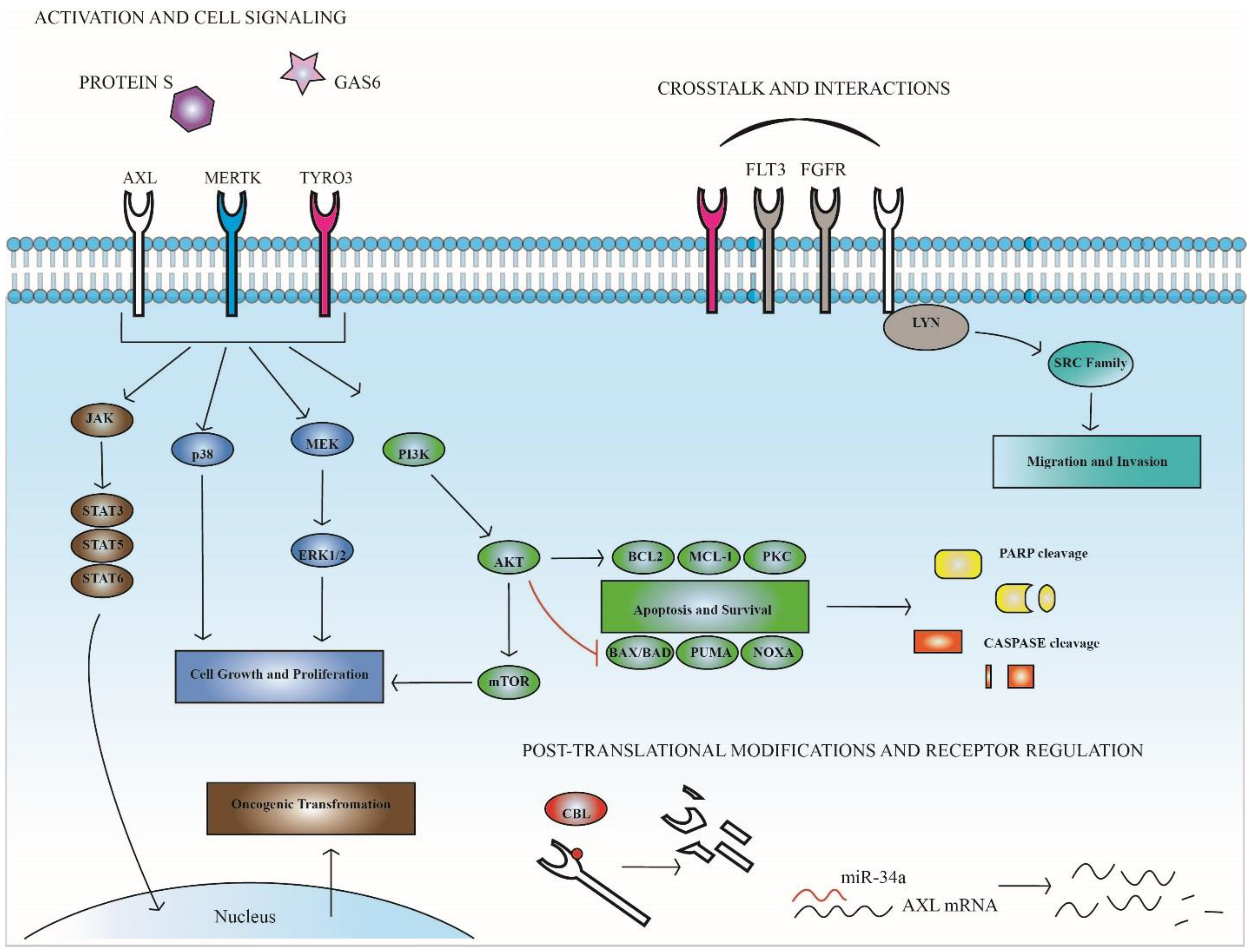
3. TAM Receptors in Hematopoietic Malignancies
3.1. Acute Leukemia
3.1.1. MERTK in Acute Myeloid and Acute Lymphoblastic Leukemia
3.1.2. AXL in Acute Myeloid Leukemia
3.2. Chronic Lymphocytic Leukemia
3.2.1. AXL and TYRO3 in Chronic Lymphocytic Leukemia
3.2.2. Regulation of AXL Expression in Chronic Lymphocytic Leukemia
3.3. Multiple Myeloma
MERTK in Multiple Myeloma
4. TAM Expression and Function in Therapeutic Resistance
4.1. AXL and Resistance in Chronic Myeloid Leukemia
4.2. AXL and Resistance in Acute Myeloid Leukemia
5. Therapeutic Targeting of TAM Receptors
5.1. Tyrosine Kinase Inhibitors
5.1.1. AXL
BGB324 (R428)
TP-0903
ASP2215 (gilteritinib)
5.1.2. MERTK
5.1.3. TYRO3
5.2. Biologic TAM RTK Inhibitors
6. Future Areas of Research
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- U.S. Cancer Statistics Working Group. United States Cancer Statistics: 1999–2013 Incidence and Mortality Web-Based Report; Department of Health and Human Services, Center for Disease Control and Prevention and National Cancer Institute: Atlanta, GA, USA, 2016.
- Varnum, B.C.; Young, C.; Elliott, G.; Garcia, A.; Bartley, T.D.; Fridell, Y.W.; Hunt, R.W.; Trail, G.; Clogston, C.; Toso, R.J.; et al. Axl receptor tyrosine kinase stimulated by the vitamin k-dependent protein encoded by growth-arrest-specific gene 6. Nature 1995, 373, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein s and its relative, gas6, are ligands for the tyro 3/axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagorska, A.; Traves, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 2014. [Google Scholar] [CrossRef] [PubMed]
- Shaver, T.M.; Lehmann, B.D.; Beeler, J.S.; Li, C.I.; Li, Z.; Jin, H.; Stricker, T.P.; Shyr, Y.; Pietenpol, J.A. Diverse, biologically relevant, and targetable gene rearrangements in triple-negative breast cancer and other malignancies. Cancer Res. 2016, 76, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.P.; Kohlschmidt, J.; Maharry, K.; Volinia, S.; Mrozek, K.; Nicolet, D.; Schwind, S.; Becker, H.; Metzeler, K.H.; Mendler, J.H.; et al. Gas6 expression identifies high-risk adult aml patients: Potential implications for therapy. Leukemia 2014, 28, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Brandao, L.; Migdall-Wilson, J.; Eisenman, K.; Graham, D.K. TAM receptors in leukemia: Expression, signaling, and therapeutic implications. Crit. Rev. Oncog. 2011, 16, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, J.H.; van der Poll, T.; van‘t Veer, C. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Gadue, P.; Gong, S.Y.; Garrett, S.; Stein, P.L.; Cohen, P.L. The mer receptor tyrosine kinase: Expression and function suggest a role in innate immunity. Eur. J. Immunol. 2003, 33, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.; Fiebeler, A.; Graham, D.K.; O’Bryan, J.P.; Schmidt, C.A.; Barckow, P.; Serke, S.; Siegert, W.; Snodgrass, H.R.; Huhn, D.; et al. Expression of axl, a transforming receptor tyrosine kinase, in normal and malignant hematopoiesis. Blood 1994, 84, 1931–1941. [Google Scholar] [PubMed]
- Graham, D.K.; Dawson, T.L.; Mullaney, D.L.; Snodgrass, H.R.; Earp, H.S. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994, 5, 647–657. [Google Scholar] [PubMed]
- Lee-Sherick, A.B.; Eisenman, K.M.; Sather, S.; McGranahan, A.; Armistead, P.M.; McGary, C.S.; Hunsucker, S.A.; Schlegel, J.; Martinson, H.; Cannon, C.; et al. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene 2013, 32, 5359–5368. [Google Scholar] [CrossRef] [PubMed]
- Bernsmeier, C.; Pop, O.T.; Singanayagam, A.; Triantafyllou, E.; Patel, V.C.; Weston, C.J.; Curbishley, S.; Sadiq, F.; Vergis, N.; Khamri, W.; et al. Patients with acute-on-chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology 2015. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, B.A.; Zizzo, G.; Ulas, M.; Linan, M.K.; Schreiter, J.; Cohen, P.L. Increased expression of Mer tyrosine kinase in circulating dendritic cells and monocytes of lupus patients: Correlations with plasma interferon activity and steroid therapy. Arthritis Res. Ther. 2014. [Google Scholar] [CrossRef] [PubMed]
- Guignant, C.; Venet, F.; Planel, S.; Demaret, J.; Gouel-Cheron, A.; Nougier, C.; Friggeri, A.; Allaouchiche, B.; Lepape, A.; Monneret, G. Increased MerTK expression in circulating innate immune cells of patients with septic shock. Intensive Care Med. 2013, 39, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, S.; Wang, H.; Wu, H.; Lu, Q.; Han, D. TAM receptors and the regulation of erythropoiesis in mice. Haematologica 2009, 94, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Burnier, L.; Lambrechts, D.; Fish, R.J.; Tjwa, M.; Plaisance, S.; Sugamele, R.; DeMol, M.; Martinez-Soria, E.; Maxwell, P.H.; et al. Role of Gas6 in erythropoiesis and anemia in mice. J. Clin. Investig. 2008, 118, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Lee-Sherick, A.B.; DeRyckere, D.; Cohen, R.A.; Jacobsen, K.M.; McGranahan, A.; Brandao, L.N.; Winges, A.; Sawczyn, K.K.; Liang, X.; et al. Mer receptor tyrosine kinase is a therapeutic target in pre-B-cell acute lymphoblastic leukemia. Blood 2013, 122, 1599–1609. [Google Scholar] [CrossRef]
- Satomura, K.; Derubeis, A.R.; Fedarko, N.S.; Ibaraki-O’Connor, K.; Kuznetsov, S.A.; Rowe, D.W.; Young, M.F.; Gehron Robey, P. Receptor tyrosine kinase expression in human bone marrow stromal cells. J. Cell. Physiol. 1998, 177, 426–438. [Google Scholar] [CrossRef]
- Anam, K.; Davis, T.A. Comparative analysis of gene transcripts for cell signaling receptors in bone marrow-derived hematopoietic stem/progenitor cell and mesenchymal stromal cell populations. Stem Cell Res. Ther. 2013. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.P.; Chen, C.W.; Chen, J.S.; Mao, H.C.; Chou, C.L. Circulating growth arrest-specific protein 6 levels are associated with erythropoietin resistance in hemodialysis patients. Springerplus 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, S.; Chen, Y.; Wang, H.; Wu, H.; Tang, H.; Xiong, W.; Ma, J.; Ge, Y.; Lu, Q.; et al. The role of Tyro 3 subfamily receptors in the regulation of hemostasis and megakaryocytopoiesis. Haematologica 2007, 92, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; de Frutos, P.; Aparicio, C.; Melis, E.; Savi, P.; Lupu, F.; Arnout, J.; Dewerchin, M.; Hoylaerts, M.; Herbert, J.; et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat. Med. 2001, 7, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, Q.; Darrow, A.L.; Wang, Y.; Derian, C.K.; Yang, J.; de Garavilla, L.; Andrade-Gordon, P.; Damiano, B.P. Mer receptor tyrosine kinase signaling participates in platelet function. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Cosemans, J.M.; van Kruchten, R.; Olieslagers, S.; Schurgers, L.J.; Verheyen, F.K.; Munnix, I.C.; Waltenberger, J.; Angelillo-Scherrer, A.; Hoylaerts, M.F.; Carmeliet, P.; et al. Potentiating role of Gas6 and Tyro3, Axl and Mer (TAM) receptors in human and murine platelet activation and thrombus stabilization. J. Thromb. Haemost. 2010, 8, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.M.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Karol, S.E.; Coustan-Smith, E.; Cao, X.; Shurtleff, S.A.; Raimondi, S.C.; Choi, J.K.; Ribeiro, R.C.; Dahl, G.V.; Bowman, W.P.; Taub, J.W.; et al. Prognostic factors in children with acute myeloid leukaemia and excellent response to remission induction therapy. Br. J. Haematol. 2015, 168, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Redaniel, M.T.; Pulte, D.; Jeffreys, M. Survival disparities by age and country of diagnosis for patients with acute leukemia. Leuk. Lymphoma 2015, 56, 2787–2792. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Hsieh, S.; Shinohara, E.T.; DeWees, T.; Frangoul, H.; Perkins, S.M. Effects of race/ethnicity and socioeconomic status on outcome in childhood acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2016, 38, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Alvarnas, J.C.; Brown, P.A.; Aoun, P.; Ballen, K.K.; Bellam, N.; Blum, W.; Boyer, M.W.; Carraway, H.E.; Coccia, P.F.; Coutre, S.E.; et al. Acute lymphoblastic leukemia. J. Natl. Compr. Cancer Netw. 2012, 10, 858–914. [Google Scholar]
- Brandwein, J.M.; Gupta, V.; Wells, R.A.; Schuh, A.C.; Schimmer, A.D.; Lipton, J.H.; Messner, H.A.; Yi, Q.L.; Chun, K.; Kamel-Reid, S.; et al. Treatment of elderly patients with acute lymphoblastic leukemia--evidence for a benefit of imatinib in BCR-ABL positive patients. Leuk. Res. 2005, 29, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Meshinchi, S. Pediatric acute myeloid leukemia: Biology and therapeutic implications of genomic variants. Pediatr. Clin. N. Am. 2015, 62, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.; Hudson, M.M.; Strickland, D.K.; Phipps, S.; Srivastava, D.K.; Ribeiro, R.C.; Rubnitz, J.E.; Sandlund, J.T.; Kun, L.E.; Bowman, L.C.; et al. Late effects of treatment in survivors of childhood acute myeloid leukemia. J. Clin. Oncol. 2000, 18, 3273–3279. [Google Scholar] [PubMed]
- Kaspers, G.J. Pediatric acute myeloid leukemia. Expert Rev. Anticancer Ther. 2012, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Mulrooney, D.A.; Dover, D.C.; Li, S.; Yasui, Y.; Ness, K.K.; Mertens, A.C.; Neglia, J.P.; Sklar, C.A.; Robison, L.L.; Davies, S.M. Twenty years of follow-up among survivors of childhood and young adult acute myeloid leukemia: A report from the Childhood Cancer Survivor Study. Cancer 2008, 112, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Small, D. FLT3 inhibitors: A paradigm for the development of targeted therapeutics for paediatric cancer. Eur. J. Cancer 2004, 40, 707–721, discussion 722–724. [Google Scholar] [CrossRef] [PubMed]
- Meshinchi, S.; Woods, W.G.; Stirewalt, D.L.; Sweetser, D.A.; Buckley, J.D.; Tjoa, T.K.; Bernstein, I.D.; Radich, J.P. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood 2001, 97, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Horibe, K.; Takahashi, Y.; Matsumoto, K.; Fukuda, M.; Inaba, J.; Kato, K.; Kojima, S.; Matsuyama, T. Prognostic value of internal tandem duplication of the FLT3 gene in childhood acute myelogenous leukemia. Med. Pediatr. Oncol. 1999, 33, 525–529. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.; Sanford, B.L.; Geyer, S.; Bloomfield, C.D.; Dohner, K.; Thiede, C.; Marcucci, G.; Lo-Coco, F.; Klisovic, R.B.; et al. The Multi-Kinase Inhibitor Midostaurin (M) Prolongs Survival Compared with Placebo (P) in Combination with Daunorubicin (D)/Cytarabine (C) Induction (ind), High-Dose C Consolidation (consol), and As Maintenance (maint) Therapy in Newly Diagnosed Acute Myeloid Leukemia (AML) Patients (pts) Age 18–60 with FLT3 Mutations (muts): An International Prospective Randomized (rand) P-Controlled Double-Blind Trial (CALGB 10603/RATIFY [Alliance]). Blood 2015, 126, 6. [Google Scholar]
- Levis, M.J.; Perl, A.E.; Dombret, H.; Döhner, H.; Steffen, B.; Rousselot, P.; Martinelli, G.; Estey, E.H.; Burnett, A.K.; Gammon, G.; et al. Final Results of a Phase 2 Open-Label, Monotherapy Efficacy and Safety Study of Quizartinib (AC220) in Patients with FLT3-ITD Positive or Negative Relapsed/Refractory Acute Myeloid Leukemia After Second-Line Chemotherapy or Hematopoietic Stem Cell Transplantation. Blood 2015, 120, 673. [Google Scholar]
- Kung, L.L.a.H.-J. Mitogenic signals and transforming potential of Nyk, a newly identified nueral cell adhesion molecule-related receptor tyrosine kinase. Mol. Cell. Biol. 1995, 15, 6582–6592. [Google Scholar]
- Brandao, L.N.; Winges, A.; Christoph, S.; Sather, S.; Migdall-Wilson, J.; Schlegel, J.; McGranahan, A.; Gao, D.; Liang, X.; Deryckere, D.; et al. Inhibition of MerTK increases chemosensitivity and decreases oncogenic potential in T-cell acute lymphoblastic leukemia. Blood Cancer J. 2013, 3, e101. [Google Scholar] [CrossRef]
- Keating, A.K.; Salzberg, D.B.; Sather, S.; Liang, X.; Nickoloff, S.; Anwar, A.; Deryckere, D.; Hill, K.; Joung, D.; Sawczyn, K.K.; et al. Lymphoblastic leukemia/lymphoma in mice overexpressing the Mer (MerTK) receptor tyrosine kinase. Oncogene 2006, 25, 6092–6100. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Lee-Sherick, A.B.; Zhang, W.; Menachof, K.K.; Hill, A.A.; Rinella, S.; Kirkpatrick, G.; Page, L.S.; Stashko, M.A.; Jordan, C.T.; Wei, Q.; et al. Efficacy of a Mer and Flt3 tyrosine kinase small molecule inhibitor, UNC1666, in acute myeloid leukemia. Oncotarget 2015, 6, 6722–6736. [Google Scholar] [CrossRef]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef]
- Rochlitz, C.; Lohri, A.; Bacchi, M.; Schmidt, M.; Nagel, S.; Fopp, M.; Fey, M.F.; Herrmann, R.; Neubauer, A. Axl expression is associated with adverse prognosis and with expression of Bcl-1 and CD34 in de novo acute myeloid leukemia (AML): Results from a multicenter trial of the Swiss Group for Clinical Cancer Research (SAKK). Leukemia 1999, 13, 1352–1358. [Google Scholar] [CrossRef]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef]
- Park, I.K.; Mishra, A.; Chandler, J.; Whitman, S.P.; Marcucci, G.; Caligiuri, M.A. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: Implications for Axl as a potential therapeutic target. Blood 2013, 121, 2064–2073. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.C.; Lay, J.D.; Huang, J.S.; Cheng, A.L.; Tang, J.L.; Lin, M.T.; Lai, G.M.; Chuang, S.E. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008, 268, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Pleyer, L.; Egle, A.; Hartmann, T.N.; Greil, R. Molecular and cellular mechanisms of CLL: Novel therapeutic approaches. Nat. Rev. Clin. Oncol. 2009, 6, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Secreto, C.; Boysen, J.; Sassoon, T.; Shanafelt, T.D.; Mukhopadhyay, D.; Kay, N.E. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: Implications for therapy. Blood 2011, 117, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Boysen, J.; Nelson, M.; Secreto, C.; Warner, S.L.; Bearss, D.J.; Lesnick, C.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. Targeted axl inhibition primes chronic lymphocytic leukemia B cells to apoptosis and shows synergistic/additive effects in combination with BTK inhibitors. Clin. Cancer Res. 2015, 21, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Contri, A.; Brunati, A.M.; Trentin, L.; Cabrelle, A.; Miorin, M.; Cesaro, L.; Pinna, L.A.; Zambello, R.; Semenzato, G.; Donella-Deana, A. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J. Clin. Investig. 2005, 115, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Boysen, J.; Nelson, M.; Warner, S.L.; Bearss, D.; Kay, N.E.; Ghosh, A.K. Axl activates fibroblast growth factor receptor pathway to potentiate survival signals in B-cell chronic lymphocytic leukemia cells. Leukemia 2016, 30, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Basilico, C.; Moscatelli, D. The FGF family of growth factors and oncogenes. Adv. Cancer Res. 1992, 59, 115–165. [Google Scholar] [PubMed]
- Kay, N.E.; Bone, N.D.; Tschumper, R.C.; Howell, K.H.; Geyer, S.M.; Dewald, G.W.; Hanson, C.A.; Jelinek, D.F. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia 2002, 16, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Menzel, T.; Rahman, Z.; Calleja, E.; White, K.; Wilson, E.L.; Wieder, R.; Gabrilove, J. Elevated intracellular level of basic fibroblast growth factor correlates with stage of chronic lymphocytic leukemia and is associated with resistance to fludarabine. Blood 1996, 87, 1056–1063. [Google Scholar] [PubMed]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Luthar, N.; Toulany, M.; Gill, P.S.; Salgia, R.; Kimple, R.J.; et al. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014, 74, 5152–5164. [Google Scholar] [CrossRef] [PubMed]
- Boysen, J.; Sinha, S.; Price-Troska, T.; Warner, S.L.; Bearss, D.J.; Viswanatha, D.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: Implications for therapy in p53-defective CLL patients. Leukemia 2014, 28, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Zenz, T.; Mohr, J.; Eldering, E.; Kater, A.P.; Buhler, A.; Kienle, D.; Winkler, D.; Durig, J.; van Oers, M.H.; Mertens, D.; et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 2009, 113, 3801–3808. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, M.K.; van Lom, K.; Tielemans, D.; Elstrodt, F.; Langerak, A.W.; van’t Veer, M.B.; Jongen-Lavrencic, M. 17p13/TP53 deletion in B-CLL patients is associated with microRNA-34a downregulation. Leukemia 2009, 23, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Zenz, T.; Habe, S.; Denzel, T.; Mohr, J.; Winkler, D.; Buhler, A.; Sarno, A.; Groner, S.; Mertens, D.; Busch, R.; et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): Dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood 2009, 114, 2589–2597. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J. Clin. Investig. 2012, 122, 3456–3463. [Google Scholar] [CrossRef]
- Kumar, S.K.; Lee, J.H.; Lahuerta, J.J.; Morgan, G.; Richardson, P.G.; Crowley, J.; Haessler, J.; Feather, J.; Hoering, A.; Moreau, P.; et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Leukemia 2012, 26, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Al-Hujaily, E.M.; Oldham, R.A.; Hari, P.; Medin, J.A. Development of Novel Immunotherapies for Multiple Myeloma. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef]
- Kocoglu, M.; Badros, A. The role of immunotherapy in multiple myeloma. Pharmaceuticals 2016. [Google Scholar] [CrossRef]
- Noonan, K.A.; Huff, C.A.; Davis, J.; Lemas, M.V.; Fiorino, S.; Bitzan, J.; Ferguson, A.; Emerling, A.; Luznik, L.; Matsui, W.; et al. Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma. Sci. Transl. Med. 2015. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Waizenegger, J.S.; Ben-Batalla, I.; Weinhold, N.; Meissner, T.; Wroblewski, M.; Janning, M.; Riecken, K.; Binder, M.; Atanackovic, D.; Taipaleenmaeki, H.; et al. Role of Growth arrest-specific gene 6-Mer axis in multiple myeloma. Leukemia 2015, 29, 696–704. [Google Scholar] [CrossRef]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef]
- Park, I.K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef]
- Macleod, K.; Mullen, P.; Sewell, J.; Rabiasz, G.; Lawrie, S.; Miller, E.; Smyth, J.F.; Langdon, S.P. Altered ErbB receptor signaling and gene expression in cisplatin-resistant ovarian cancer. Cancer Res. 2005, 65, 6789–6800. [Google Scholar] [CrossRef]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Hurlburt, W.; Greer, A.; Reeves, K.A.; Hillerman, S.; Chang, H.; Fargnoli, J.; Graf Finckenstein, F.; Gottardis, M.M.; Carboni, J.M. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010, 70, 7221–7231. [Google Scholar] [CrossRef] [PubMed]
- Marum, J.E.; Branford, S. Current developments in molecular monitoring in chronic myeloid leukemia. Ther. Adv. Hematol. 2016, 7, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; O’Brien, S.; Jabbour, E.; Garcia-Manero, G.; Quintas-Cardama, A.; Shan, J.; Rios, M.B.; Ravandi, F.; Faderl, S.; Kadia, T.; et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: A single-institution historical experience. Blood 2012, 119, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Hehlmann, R.; Muller, M.C.; Lauseker, M.; Hanfstein, B.; Fabarius, A.; Schreiber, A.; Proetel, U.; Pletsch, N.; Pfirrmann, M.; Haferlach, C.; et al. Deep molecular response is reached by the majority of patients treated with imatinib, predicts survival, and is achieved more quickly by optimized high-dose imatinib: Results from the randomized CML-study IV. J. Clin. Oncol. 2014, 32, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Kalmanti, L.; Saussele, S.; Lauseker, M.; Muller, M.C.; Dietz, C.T.; Heinrich, L.; Hanfstein, B.; Proetel, U.; Fabarius, A.; Krause, S.W.; et al. Safety and efficacy of imatinib in CML over a period of 10 years: Data from the randomized CML-study IV. Leukemia 2015, 29, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Eide, C.A.; O’Hare, T. Chronic myeloid leukemia: Advances in understanding disease biology and mechanisms of resistance to tyrosine kinase inhibitors. Curr. Hematol. Malig. Rep. 2015, 10, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Jacquel, A.; Belhacene, N.; Robert, G.; Cluzeau, T.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget 2011, 2, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Grosso, S.; Puissant, A.; Dufies, M.; Colosetti, P.; Jacquel, A.; Lebrigand, K.; Barbry, P.; Deckert, M.; Cassuto, J.P.; Mari, B.; et al. Gene expression profiling of imatinib and PD166326-resistant CML cell lines identifies Fyn as a gene associated with resistance to BCR-ABL inhibitors. Mol. Cancer Ther. 2009, 8, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Gioia, R.; Tregoat, C.; Dumas, P.Y.; Lagarde, V.; Prouzet-Mauleon, V.; Desplat, V.; Sirvent, A.; Praloran, V.; Lippert, E.; Villacreces, A.; et al. CBL controls a tyrosine kinase network involving AXL, SYK and LYN in nilotinib-resistant chronic myeloid leukaemia. J. Pathol. 2015, 237, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Baladi, T.; Abet, V.; Piguel, S. State-of-the-art of small molecule inhibitors of the TAM family: The point of view of the chemist. Eur. J. Med. Chem. 2015, 105, 220–237. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. First Axl inhibitor enters clinical trials. Nat. Biotechnol. 2013, 31, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: www.clinicaltrials.gov (accessed on 16 August 2016).
- Wnuk-Lipinska, K.; Tiron, C.; Gausdal, G.; Sandal, T.; Frink, R.; Hinz, S.; Hellesøy, M.; Ahmed, L.; Haugen, H.; Liang, X.; et al. BGB324, a selective small molecule Axl kinase inhibitor to overcome EMT-associated drug resistance in carcinomas: Therapeutic rationale and early clinical studies. Cancer Res. 2014. [Google Scholar] [CrossRef]
- Mollard, A.; Warner, S.L.; Call, L.T.; Wade, M.L.; Bearss, J.J.; Verma, A.; Sharma, S.; Vankayalapati, H.; Bearss, D.J. Design, synthesis and biological evaluation of a series of novel axl kinase inhibitors. ACS Med. Chem. Lett. 2011, 2, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Keating, M.J.; Wierda, W.G.; Gandhi, V. Preclinical combination of TP-0903, an AXL inhibitor and B-PAC-1, a procaspase-activating compound with ibrutinib in chronic lymphocytic leukemia. Leuk. Lymphoma 2016, 57, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Tolero Pharmaceuticals. Available online: http://toleropharmaceuticals.com/TP-0903.html (accessed on 15 October 2016).
- Mori, M.; Kaneko, N.; Ueno, Y.; Tanaka, R.; Cho, K.; Saito, R.; Kondoh, Y.; Shimada, I.; Kuromitsu, S. ASP2215, a novel FLT3/AXL inhibitor: Preclinical evaluation in acute myeloid leukemia (AML). J. Clin. Oncol. 2014, 32, 7070. [Google Scholar]
- Smith, C.C.; Levis, M.J.; Litzow, M.R.; Perl, A.E.; Altman, J.K.; Gill, S.C.; Kadokura, T.; Yuen, G.J.; Fisniku, O.; Liu, C.; et al. Pharmacokinetic profile and pharmacodynamic effects of ASP2215, a selective, potent inhibitor of FLT3/AXL, in patients with relapsed or refractory acute myeloid leukemia: Results from a first-in-human phase 1/2 study. Blood 2015, 126, 4836. [Google Scholar]
- Levis, M.J.; Perl, A.E.; Altman, J.K.; Cortes, J.E.; Ritchie, E.K.; Larson, R.A.; Smith, C.C.; Wang, E.S.; Strickland, S.A.; Baer, M.R.; et al. Results of a first-in-human, phase I/II trial of ASP2215, a selective, potent inhibitor of FLT3/Axl in patients with relapsed or refractory (R/R) acute myeloid leukemia (L/L). J. Clin. Oncol. 2015, 33, 7003. [Google Scholar]
- Altman, J.K.; Perl, A.E.; Cortes, J.E.; Levis, M.J.; Smith, C.C.; Litzow, M.R.; Baer, M.R.; Claxton, D.F.; Erba, H.P.; Gill, S.C.; et al. Antileukemic activity and tolerability of ASP2215 80 mg and greater in FLT3 mutation-positive subjects with relapsed or refractory acute myeloid leukemia: Results from a phase 1/2, open-label, dose-escalation/dose-response study. Blood 2015, 126, 321. [Google Scholar]
- Liu, J.; Yang, C.; Simpson, C.; Deryckere, D.; van Deusen, A.; Miley, M.J.; Kireev, D.; Norris-Drouin, J.; Sather, S.; Hunter, D.; et al. Discovery of novel small molecule mer kinase inhibitors for the treatment of pediatric acute lymphoblastic leukemia. ACS Med. Chem. Lett. 2012, 3, 129–134. [Google Scholar] [CrossRef]
- Christoph, S.; Deryckere, D.; Schlegel, J.; Frazer, J.K.; Batchelor, L.A.; Trakhimets, A.Y.; Sather, S.; Hunter, D.M.; Cummings, C.T.; Liu, J.; et al. UNC569, a novel small-molecule mer inhibitor with efficacy against acute lymphoblastic leukemia in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 2367–2377. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, W.; Stashko, M.A.; Deryckere, D.; Cummings, C.T.; Hunter, D.; Yang, C.; Jayakody, C.N.; Cheng, N.; Simpson, C.; et al. UNC1062, a new and potent Mer inhibitor. Eur. J. Med. Chem. 2013, 65, 83–93. [Google Scholar] [CrossRef]
- Zhang, W.; DeRyckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J. Med. Chem. 2014, 57, 7031–7041. [Google Scholar] [CrossRef]
- DeRyckere, D.; Lee Sherick, A.B.; Huey, M.G.; Hill, A.A.; Tyner, J.W.; Jacobsen, K.M.; Page, L.S.; Kirkpatrick, G.D.; Eryildiz, F.; Montgomery, S.A.; et al. UNC2025, a MerTK small molecule inhibitor, is therapeutically effective alone and in combination with methotrexate in leukemia models. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef]
- Powell, N.A.; Kohrt, J.T.; Filipski, K.J.; Kaufman, M.; Sheehan, D.; Edmunds, J.E.; Delaney, A.; Wang, Y.; Bourbonais, F.; Lee, D.Y.; et al. Novel and selective spiroindoline-based inhibitors of Sky kinase. Bioorg. Med. Chem. Lett. 2012, 22, 190–193. [Google Scholar] [CrossRef]
- Powell, N.A.; Hoffman, J.K.; Ciske, F.L.; Kaufman, M.D.; Kohrt, J.T.; Quin, J., 3rd; Sheehan, D.J.; Delaney, A.; Baxi, S.M.; Catana, C.; et al. Highly selective 2,4-diaminopyrimidine-5-carboxamide inhibitors of Sky kinase. Bioorg. Med. Chem. Lett. 2013, 23, 1046–1050. [Google Scholar] [CrossRef]
- Powell, N.A.; Hoffman, J.K.; Ciske, F.L.; Kohrt, J.T.; Baxi, S.M.; Peng, Y.W.; Zhong, M.; Catana, C.; Ohren, J.; Perrin, L.A.; et al. Optimization of highly selective 2,4-diaminopyrimidine-5-carboxamide inhibitors of Sky kinase. Bioorg. Med. Chem. Lett. 2013, 23, 1051–1055. [Google Scholar] [CrossRef]
- Cerchia, L.; Esposito, C.L.; Camorani, S.; Rienzo, A.; Stasio, L.; Insabato, L.; Affuso, A.; de Franciscis, V. Targeting Axl with an high-affinity inhibitory aptamer. Mol. Ther. 2012, 20, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.J.; Gardner, J.; Vendel, M.C.; Ailor, E.; Szak, S.; Huang, F.; Doern, A.; Tan, X.; Yang, W.; Grueneberg, D.A.; et al. Evaluation of Tyro3 expression, Gas6-mediated Akt phosphorylation, and the impact of anti-Tyro3 antibodies in melanoma cell lines. Biochemistry 2013, 52, 3102–3118. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.T.; Linger, R.M.; Cohen, R.A.; Sather, S.; Kirkpatrick, G.D.; Davies, K.D.; DeRyckere, D.; Earp, H.S.; Graham, D.K. Mer590, a novel monoclonal antibody targeting MER receptor tyrosine kinase, decreases colony formation and increases chemosensitivity in non-small cell lung cancer. Oncotarget 2014, 5, 10434–10445. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.E.; Le, J.P.; Sather, S.; Pernu, B.M.; Graham, D.K.; Pierce, A.M.; Keating, A.K. Mer receptor tyrosine kinase inhibition impedes glioblastoma multiforme migration and alters cellular morphology. Oncogene 2012, 31, 4171–4181. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gong, M.; Li, X.; Zhou, Y.; Gao, W.; Tulpule, A.; Chaudhary, P.M.; Jung, J.; Gill, P.S. Induction, regulation, and biologic function of Axl receptor tyrosine kinase in Kaposi sarcoma. Blood 2010, 116, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.J.; Pei, L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28, 3442–3455. [Google Scholar] [CrossRef] [PubMed]
- Leconet, W.; Larbouret, C.; Chardes, T.; Thomas, G.; Neiveyans, M.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Pugniere, M.; Bernex, F.; et al. Preclinical validation of AXL receptor as a target for antibody-based pancreatic cancer immunotherapy. Oncogene 2014, 33, 5405–5414. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef] [PubMed]
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 2007, 109, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S., 2nd; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Moody, G.; Belmontes, B.; Masterman, S.; Wang, W.; King, C.; Murawsky, C.; Tsuruda, T.; Liu, S.; Radinsky, R.; Beltran, P.J. Antibody-mediated neutralization of autocrine Gas6 inhibits the growth of pancreatic ductal adenocarcinoma tumors in vivo. Int. J. Cancer 2016, 139, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Batlevi, C.L.; Matsuki, E.; Brentjens, R.J.; Younes, A. Novel immunotherapies in lymphoid malignancies. Nat. Rev. Clin. Oncol. 2016, 13, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Manabe, A.; Coustan-Smith, E.; Behm, F.G.; Raimondi, S.C.; Campana, D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood 1992, 79, 2370–2377. [Google Scholar]
- Iwamoto, S.; Mihara, K.; Downing, J.R.; Pui, C.H.; Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J. Clin. Investig. 2007, 117, 1049–1057. [Google Scholar] [CrossRef]
- Sison, E.A.; Rau, R.E.; McIntyre, E.; Li, L.; Small, D.; Brown, P. MLL-rearranged acute lymphoblastic leukaemia stem cell interactions with bone marrow stroma promote survival and therapeutic resistance that can be overcome with CXCR4 antagonism. Br. J. Haematol. 2013, 160, 785–797. [Google Scholar] [CrossRef]
- Dormady, S.P.; Zhang, X.M.; Basch, R.S. Hematopoietic progenitor cells grow on 3T3 fibroblast monolayers that overexpress growth arrest-specific gene-6 (GAS6). Proc. Natl. Acad. Sci. USA 2000, 97, 12260–12265. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Taichman, R.S. GAS6/Mer axis regulates the homing and survival of the E2A/PBX1-positive B-cell precursor acute lymphoblastic leukemia in the bone marrow niche. Exp. Hematol. 2010, 38, 132–140. [Google Scholar] [CrossRef]
- Sison, E.A.; Magoon, D.; Li, L.; Annesley, C.E.; Rau, R.E.; Small, D.; Brown, P. Plerixafor as a chemosensitizing agent in pediatric acute lymphoblastic leukemia: Efficacy and potential mechanisms of resistance to CXCR4 inhibition. Oncotarget 2014, 5, 8947–8958. [Google Scholar] [CrossRef]
- Crosier, P.S.; Hall, L.R.; Vitas, M.R.; Lewis, P.M.; Crosier, K.E. Identification of a novel receptor tyrosine kinase expressed in acute myeloid leukemic blasts. Leuk. Lymphoma 1995, 18, 443–449. [Google Scholar] [CrossRef]
- De Vos, J.; Couderc, G.; Tarte, K.; Jourdan, M.; Requirand, G.; Delteil, M.C.; Rossi, J.F.; Mechti, N.; Klein, B. Identifying intercellular signaling genes expressed in malignant plasma cells by using complementary DNA arrays. Blood 2001, 98, 771–780. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound | IC50 Values | Other Targeted Kinases | |||||
|---|---|---|---|---|---|---|---|
| TYRO3 | AXL | MERTK | |||||
| Enzymatic | Cell-Based | Enzymatic | Cell-Based | Enzymatic | Cell-Based | ||
| BGB324 | 200 nM | >1400 nM | 14 nM | 14 nM | 220 nM | 700 nM | ABL, RET, TIE2, FLT3 |
| TP-0903 | <200 nM | 27 nM | 222 nM | <200 nM | AURKA, AURKB, JAK2, ALK, ABL1 | ||
| ASP2215 | 0.7 nM | 2.9 nM | FLT3, LTK, ALK | ||||
| UNC2025 | 17 nM | 301 nM | 14 nM | 122 nM | 0.74 nM | 2.7 nM | FLT3, TRKA, KIT |
| MRX-2843 | 17 nM | 15 nM | 1.3 nM | FLT3, TRKA | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huey, M.G.; Minson, K.A.; Earp, H.S.; DeRyckere, D.; Graham, D.K. Targeting the TAM Receptors in Leukemia. Cancers 2016, 8, 101. https://doi.org/10.3390/cancers8110101
Huey MG, Minson KA, Earp HS, DeRyckere D, Graham DK. Targeting the TAM Receptors in Leukemia. Cancers. 2016; 8(11):101. https://doi.org/10.3390/cancers8110101
Chicago/Turabian StyleHuey, Madeline G., Katherine A. Minson, H. Shelton Earp, Deborah DeRyckere, and Douglas K. Graham. 2016. "Targeting the TAM Receptors in Leukemia" Cancers 8, no. 11: 101. https://doi.org/10.3390/cancers8110101
APA StyleHuey, M. G., Minson, K. A., Earp, H. S., DeRyckere, D., & Graham, D. K. (2016). Targeting the TAM Receptors in Leukemia. Cancers, 8(11), 101. https://doi.org/10.3390/cancers8110101