
Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer

Abstract
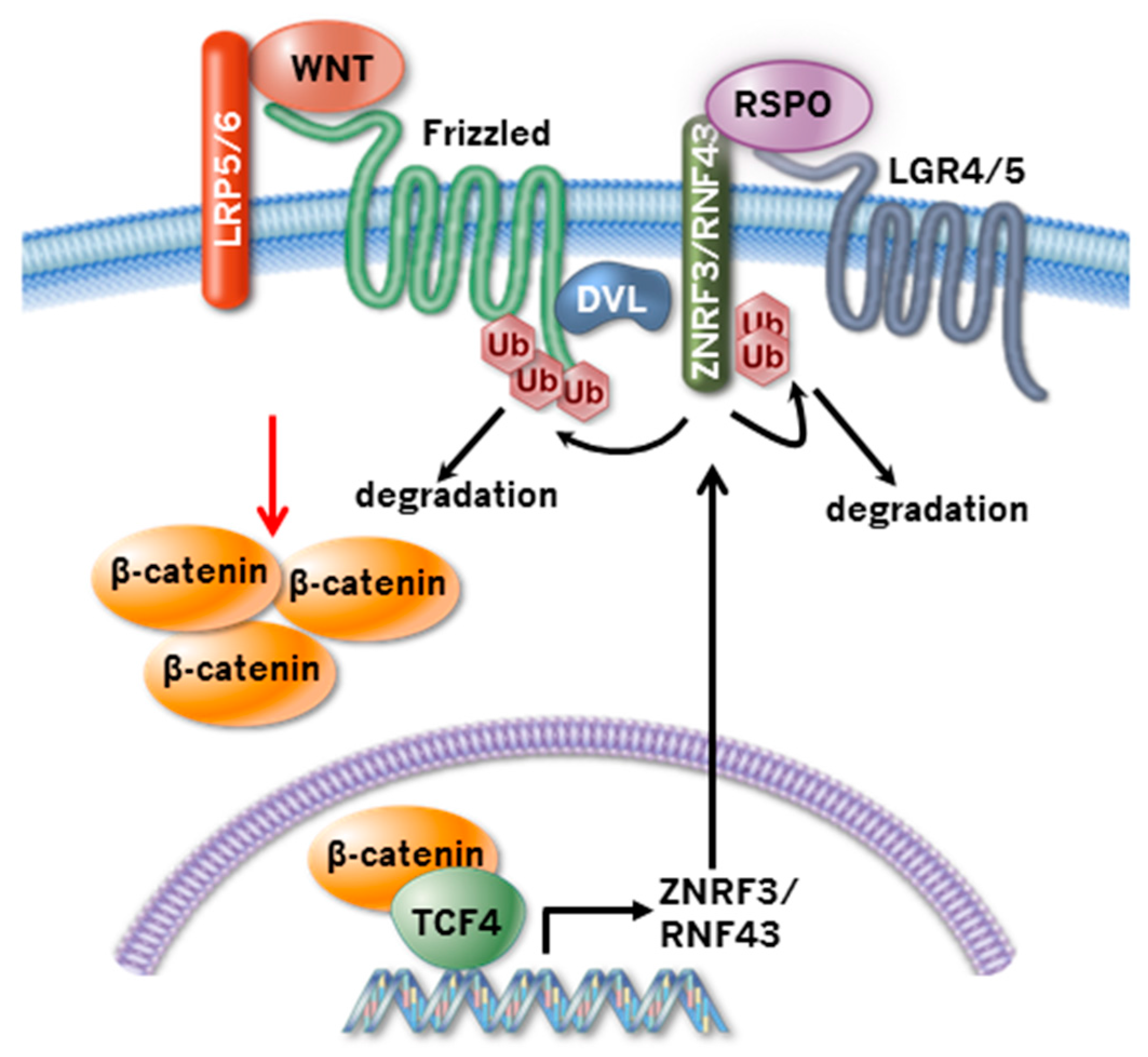
:1. Introduction
2. ZNRF3 and RNF43 Regulate Wnt Receptor Turnover
3. R-spondin-ZNRF3/RNF43 Signaling Module
4. RNF43/ZNRF3 Mutations and RSPO2/3 Translocations in Cancer
4.1. RNF43/ZNRF3 Mutations
4.2. RSPO2/3 Translocations
5. Function of R-spondin-ZNRF3/RNF43 in Tumor Biology
6. RNF43/ZNRF3 Mutations and RSPO2/RSPO3 Translocations as Biomarkers to Identify Wnt-Dependent Tumors
7. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Mukai, A.; Yamamoto-Hino, M.; Awano, W.; Watanabe, W.; Komada, M.; Goto, S. Balanced ubiquitylation and deubiquitylation of Frizzled regulate cellular responsiveness to Wg/Wnt. EMBO J. 2010, 29, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Walker, M.P.; Young, R.; Quick, L.; Orgel, K.A.; Ryan, M.; Gupta, P.; Henrich, I.C.; Ferrer, M.; Marine, S.; et al. USP6 oncogene promotes Wnt signaling by deubiquitylating Frizzleds. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Moffat, L.L.; Robinson, R.E.; Bakoulis, A.; Clark, S.G. The conserved transmembrane RING finger protein PLR-1 downregulates Wnt signaling by reducing Frizzled, Ror and Ryk cell-surface levels in C. elegans. Development 2014, 141, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Charlat, O.; Zamponi, R.; Yang, Y.; Cong, F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol. Cell 2015, 58, 522–533. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.B.; Snel, B.; Clevers, H.C. The R-spondin protein family. Genome Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.R.; Yoon, J.K. The R-spondin family of proteins: Emerging regulators of Wnt signaling. Int. J. Biochem. Cell Biol. 2012, 44, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Kazanskaya, O.; Glinka, A.; del Barco Barrantes, I.; Stannek, P.; Niehrs, C.; Wu, W. R-spondin2 is a secreted activator of Wnt/beta-catenin signaling and is required for xenopus myogenesis. Dev. Cell 2004, 7, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Wagle, M.; Tran, K.; Zhan, X.; Dixon, M.A.; Liu, S.; Gros, D.; Korver, W.; Yonkovich, S.; Tomasevic, N.; et al. R-spondin family members regulate the Wnt pathway by a common mechanism. Mol. Biol. Cell 2008, 19, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Li, S.J.; Yen, T.Y.; Endo, Y.; Klauzinska, M.; Baljinnyam, B.; Macher, B.; Callahan, R.; Rubin, J.S. Loss-of-function point mutations and two-furin domain derivatives provide insights about R-spondin2 structure and function. Cell Signal. 2009, 21, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. LGR5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Ruffner, H.; Sprunger, J.; Charlat, O.; Leighton-Davies, J.; Grosshans, B.; Salathe, A.; Zietzling, S.; Beck, V.; Therier, M.; Isken, A.; et al. R-spondin potentiates Wnt/beta-catenin signaling through orphan receptors LGR4 and LGR5. PLoS ONE 2012, 7, e40976. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Chen, X.; Lin, Z.; Fang, D.; He, X. The structural basis of R-spondin recognition by LGR5 and RNF43. Genes Dev. 2013, 27, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.C.; de Lau, W.; Forneris, F.; Granneman, J.C.; Huch, M.; Clevers, H.; Gros, P. Structure of stem cell growth factor R-spondin 1 in complex with the ectodomain of its receptor LGR5. Cell Rep. 2013, 3, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.C.; de Lau, W.; Madoori, P.K.; Forneris, F.; Granneman, J.C.; Clevers, H.; Gros, P. Structures of Wnt-antagonist ZNRF3 and its complex with R-spondin 1 and implications for signaling. PLoS ONE 2013, 8, e83110. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Huang, B.; Zhang, S.; Yu, X.; Wu, W.; Wang, X. Structural basis for R-spondin recognition by LGR4/5/6 receptors. Genes Dev. 2013, 27, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zamponi, R.; Charlat, O.; Ramones, M.; Swalley, S.; Jiang, X.; Rivera, D.; Tschantz, W.; Lu, B.; Quinn, L.; et al. Interaction with both ZNRF3 and LGR4 is required for the signalling activity of R-spondin. EMBO Rep. 2013, 14, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Xu, Y.; Rajashankar, K.R.; Robev, D.; Nikolov, D.B. Crystal structures of LGR4 and its complex with R-spondin1. Structure 2013, 21, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Zebisch, M.; Xu, Y.; Krastev, C.; MacDonald, B.T.; Chen, M.; Gilbert, R.J.; He, X.; Jones, E.Y. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the wnt agonist R-spondin. Nat. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Moad, H.E.; Pioszak, A.A. Reconstitution of R-spondin:LGR4:ZNRF3 adult stem cell growth factor signaling complexes with recombinant proteins produced in Escherichia coli. Biochemistry 2013, 52, 7295–7304. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Schreiner, C.M.; Wert, S.E.; Mucenski, M.L.; Scott, W.J.; Whitsett, J.A. R-spondin 2 is required for normal laryngeal-tracheal, lung and limb morphogenesis. Development 2008, 135, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Blaydon, D.C.; Ishii, Y.; O’Toole, E.A.; Unsworth, H.C.; Teh, M.T.; Ruschendorf, F.; Sinclair, C.; Hopsu-Havu, V.K.; Tidman, N.; Moss, C.; et al. The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nat. Genet. 2006, 38, 1245–1247. [Google Scholar] [CrossRef] [PubMed]
- Cadieu, E.; Neff, M.W.; Quignon, P.; Walsh, K.; Chase, K.; Parker, H.G.; Vonholdt, B.M.; Rhue, A.; Boyko, A.; Byers, A.; et al. Coat variation in the domestic dog is governed by variants in three genes. Science 2009, 326, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Kazanskaya, O.; Ohkawara, B.; Heroult, M.; Wu, W.; Maltry, N.; Augustin, H.G.; Niehrs, C. The Wnt signaling regulator R-spondin 3 promotes angioblast and vascular development. Development 2008, 135, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Parma, P.; Radi, O.; Vidal, V.; Chaboissier, M.C.; Dellambra, E.; Valentini, S.; Guerra, L.; Schedl, A.; Camerino, G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet. 2006, 38, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Kakitani, M.; Zhao, J.; Oshima, T.; Tang, T.; Binnerts, M.; Liu, Y.; Boyle, B.; Park, E.; Emtage, P.; et al. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science 2005, 309, 1256–1259. [Google Scholar] [CrossRef] [PubMed]
- Kinzel, B.; Pikiolek, M.; Orsini, V.; Sprunger, J.; Isken, A.; Zietzling, S.; Desplanches, M.; Dubost, V.; Breustedt, D.; Valdez, R.; et al. Functional roles of LGR4 and LGR5 in embryonic gut, kidney and skin development in mice. Dev. Biol. 2014, 390, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Storm, E.E.; Durinck, S.; de Sousa e Melo, F.; Tremayne, J.; Kljavin, N.; Tan, C.; Ye, X.; Chiu, C.; Pham, T.; Hongo, J.A.; et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 2016, 529, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single LGR5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. Wnt signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jiao, Y.; Dal Molin, M.; Maitra, A.; de Wilde, R.F.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Wolfgang, C.L.; Canto, M.I.; et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 21188–21193. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Rowley, S.M.; Christie, M.; Allan, P.E.; Bowtell, D.D.; Australian Ovarian Cancer Study, G.; Gorringe, K.L.; Campbell, I.G. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. J. Pathol. 2013, 229, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Yonescu, R.; Offerhaus, G.J.; Klimstra, D.S.; Maitra, A.; Eshleman, J.R.; Herman, J.G.; Poh, W.; Pelosof, L.; Wolfgang, C.L.; et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J. Pathol. 2014, 232, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Assie, G.; Letouze, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; Rene-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Chan-On, W.; Nairismagi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Kahyo, T.; Kato, H.; Igarashi, H.; Matsuura, S.; Nakamura, S.; Kurachi, K.; Nakamura, T.; Ogawa, H.; Funai, K.; et al. RSPO fusion transcripts in colorectal cancer in japanese population. Mol. Biol. Rep. 2014, 41, 5375–5384. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Yamashita, S.; Tanabe, T.; Hashimoto, T.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Shinmura, K.; Saito, Y.; Hiraoka, N.; et al. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J. Pathol. 2016, 239, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.L.; Rahrmann, E.P.; Moriarity, B.S.; Choi, K.; Conboy, C.B.; Greeley, A.D.; Halfond, A.L.; Anderson, L.K.; Wahl, B.R.; Keng, V.W.; et al. Canonical Wnt/beta-catenin signaling drives human schwann cell transformation, progression, and tumor maintenance. Cancer Discov. 2013, 3, 674–689. [Google Scholar] [CrossRef] [PubMed]
- Karkera, J.; Martinez, G.; Bell, K.; Portale, J.; Gaffney, D.; Loenzi, M.V.; Platero, S. Identification of R-spondin fusions in NSCLC. In Proceedings of the AACR-IASLC Joint Conference on Molecular Origins of Lung Cancer, San Diego, CA, USA, 6–9 January 2014.
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [PubMed]
- Callahan, R.; Mudunur, U.; Bargo, S.; Raafat, A.; McCurdy, D.; Boulanger, C.; Lowther, W.; Stephens, R.; Luke, B.T.; Stewart, C.; et al. Genes affected by mouse mammary tumor virus (MMTV) proviral insertions in mouse mammary tumors are deregulated or mutated in primary human mammary tumors. Oncotarget 2012, 3, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Lowther, W.; Wiley, K.; Smith, G.H.; Callahan, R. A new common integration site, Int7, for the mouse mammary tumor virus in mouse mammary tumors identifies a gene whose product has furin-like and thrombospondin-like sequences. J. Virol. 2005, 79, 10093–10096. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, V.; Kimm, M.A.; Boer, M.; Wessels, L.; Theelen, W.; Jonkers, J.; Hilkens, J. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat. Genet. 2007, 39, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.K.; Allaei, R.; Silverstein, K.A.; Staggs, R.A.; Sarver, A.L.; Bergemann, T.L.; Gupta, M.; O’Sullivan, M.G.; Matise, I.; Dupuy, A.J.; et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009, 323, 1747–1750. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; van Es, J.H.; van den Born, M.; Clevers, H. Porcupine inhibitor suppresses paracrine Wnt-driven growth of RNF43;ZNRF3-mutant neoplasia. Proc. Natl. Acad. Sci. USA 2015, 112, 7548–7550. [Google Scholar] [CrossRef] [PubMed]
- Klauzinska, M.; Baljinnyam, B.; Raafat, A.; Rodriguez-Canales, J.; Strizzi, L.; Greer, Y.E.; Rubin, J.S.; Callahan, R. RSPO2/Int7 regulates invasiveness and tumorigenic properties of mammary epithelial cells. J. Cell Physiol. 2012, 227, 1960–1971. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Yi, J.; Carmon, K.S.; Crumbley, C.A.; Xiong, W.; Thomas, A.; Fan, X.; Guo, S.; An, Z.; Chang, J.T.; et al. Aberrant RSPO3-LGR4 signaling in KEAP1-deficient lung adenocarcinomas promotes tumor aggressiveness. Oncogene 2015, 34, 4692–4701. [Google Scholar] [CrossRef] [PubMed]
- Loregger, A.; Grandl, M.; Mejias-Luque, R.; Allgauer, M.; Degenhart, K.; Haselmann, V.; Oikonomou, C.; Hatzis, P.; Janssen, K.P.; Nitsche, U.; et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated beta-catenin by sequestering TCF4 to the nuclear membrane. Sci. Signal. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Qiu, S.; Lu, L.; Zou, J.; Li, W.F.; Wang, O.; Zhao, H.; Wang, H.; Tang, J.; Chen, L.; et al. RSPO2-LGR5 signaling has tumour-suppressive activity in colorectal cancer. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Ettenberg, S.A.; Charlat, O.; Daley, M.P.; Liu, S.; Vincent, K.J.; Stuart, D.D.; Schuller, A.G.; Yuan, J.; Ospina, B.; Green, J.; et al. Inhibition of tumorigenesis driven by different Wnt proteins requires blockade of distinct ligand-binding regions by LRP6 antibodies. Proc. Natl. Acad. Sci. USA 2010, 107, 15473–15478. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Bourhis, E.; Chiu, C.; Stawicki, S.; DeAlmeida, V.I.; Liu, B.Y.; Phamluong, K.; Cao, T.C.; Carano, R.A.; Ernst, J.A.; et al. Wnt isoform-specific interactions with coreceptor specify inhibition or potentiation of signaling by LRP6 antibodies. PLoS ONE 2010, 5, e12682. [Google Scholar] [CrossRef] [PubMed]
- Le, P.N.; McDermott, J.D.; Jimeno, A. Targeting the Wnt pathway in human cancers: Therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Moon, J.; Dodge, M.E.; Pan, X.; Zhang, L.; Hanson, J.M.; Tuladhar, R.; Ma, Z.; Shi, H.; Williams, N.S.; et al. The development of highly potent inhibitors for porcupine. J. Med. Chem. 2013, 56, 2700–2704. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Blagodatski, A.; Poteryaev, D.; Katanaev, V.L. Targeting the Wnt pathways for therapies. Mol. Cell. Ther. 2014. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Chartier, C.; Raval, J.; Axelrod, F.; Bond, C.; Cain, J.; Dee-Hoskins, C.; Ma, S.; Fischer, M.M.; Shah, J.; Wei, J.; et al. Therapeutic targeting of tumor-derived R-spondin attenuates beta-catenin signaling and tumorigenesis in multiple cancer types. Cancer Res. 2016, 76, 713–723. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cancers | Mutation Frequency | Sample Size | Ref. | Comments |
|---|---|---|---|---|
| Intraductal papillary mucinous neoplasm (IPMN) and mucinous cystic neoplasm (MCN) | 75% 37.5% | 8 IPMN 8 MCN | [37] | These tumors can progress to pancreatic ductal adenocarcinoma. |
| Cholangiocarcinoma (CCA)
O. viverrini associated Non-O. viverrini associated | 9.3% 3.5% | 54 86 | [45,46] | |
| Mucinous ovarian carcinomas | 21% | 29 | [39] | 2/22 (9%) in mucinous ovarian borderline tumors. |
| Gastric cancer
Microsatellite-stable (MSS) microsatellite instability (MSI) | 4.8% 54.6% | 100 | [42] | Recurrent G659fs mutations in MSI subtype |
| Colorectal adenocarcinomas
NHS and HPFS dataset TCGA dataset | 18.9% 17.6% | 185 224 | [38] | Recurrent G659fs mutations and R117fs mutations. |
| Endometrial carcinomas | 18.1% | 248 | [38] | Recurrent G659fs mutations |
| Pancreatic carcinomas with acinar differentiation | 4% | 23 | [40] | |
| Pancreatic cancer | 6% | 109 | [41] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, H.-X.; Jiang, X.; Cong, F. Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers 2016, 8, 54. https://doi.org/10.3390/cancers8060054
Hao H-X, Jiang X, Cong F. Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers. 2016; 8(6):54. https://doi.org/10.3390/cancers8060054
Chicago/Turabian StyleHao, Huai-Xiang, Xiaomo Jiang, and Feng Cong. 2016. "Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer" Cancers 8, no. 6: 54. https://doi.org/10.3390/cancers8060054
APA StyleHao, H.-X., Jiang, X., & Cong, F. (2016). Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers, 8(6), 54. https://doi.org/10.3390/cancers8060054