
Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a Key Regulator of Cell Migration and Cancer Dissemination


Abstract
:1. Introduction
1.1. Cell Migration
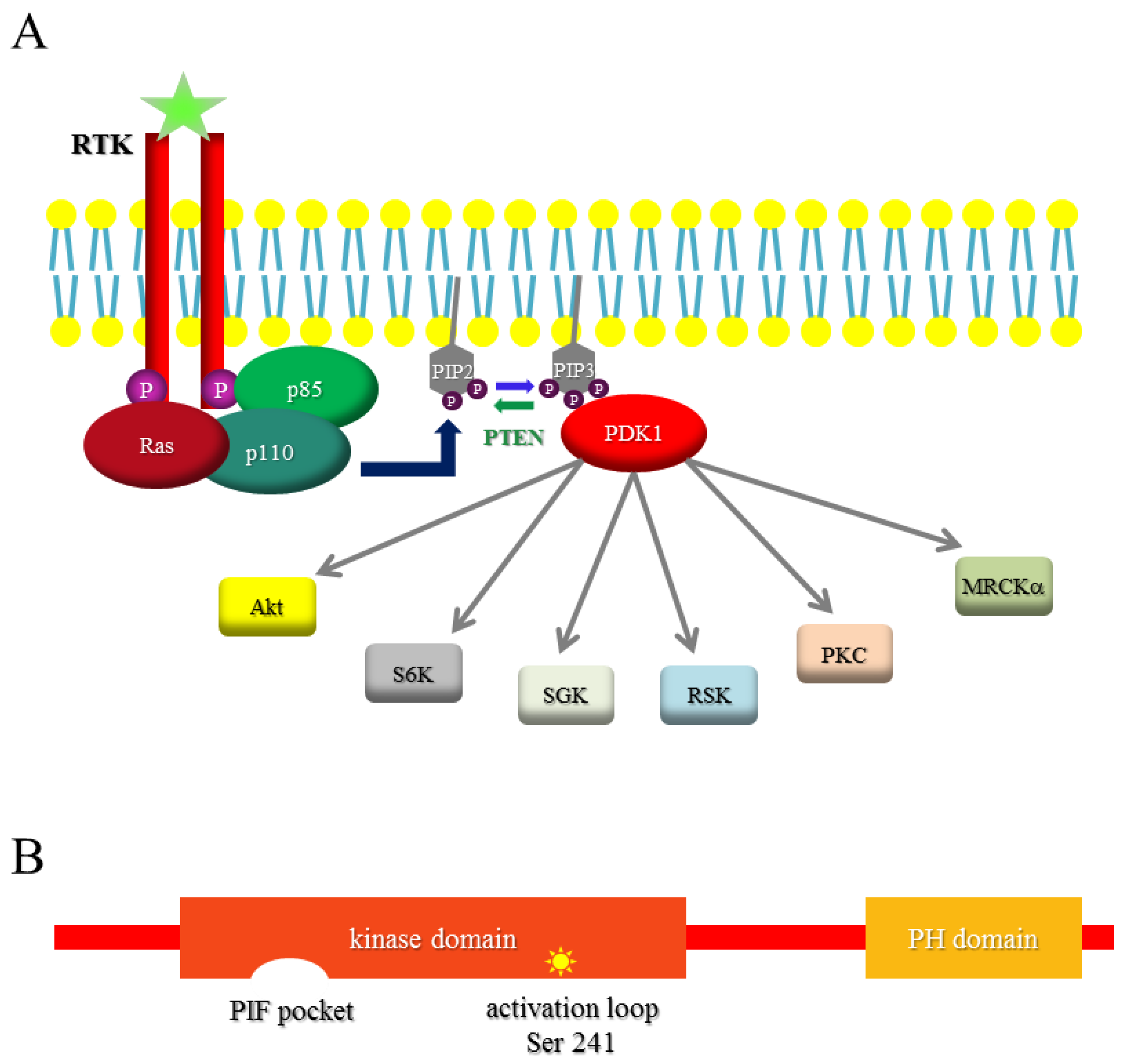
1.2. PI3K
1.3. PDK1
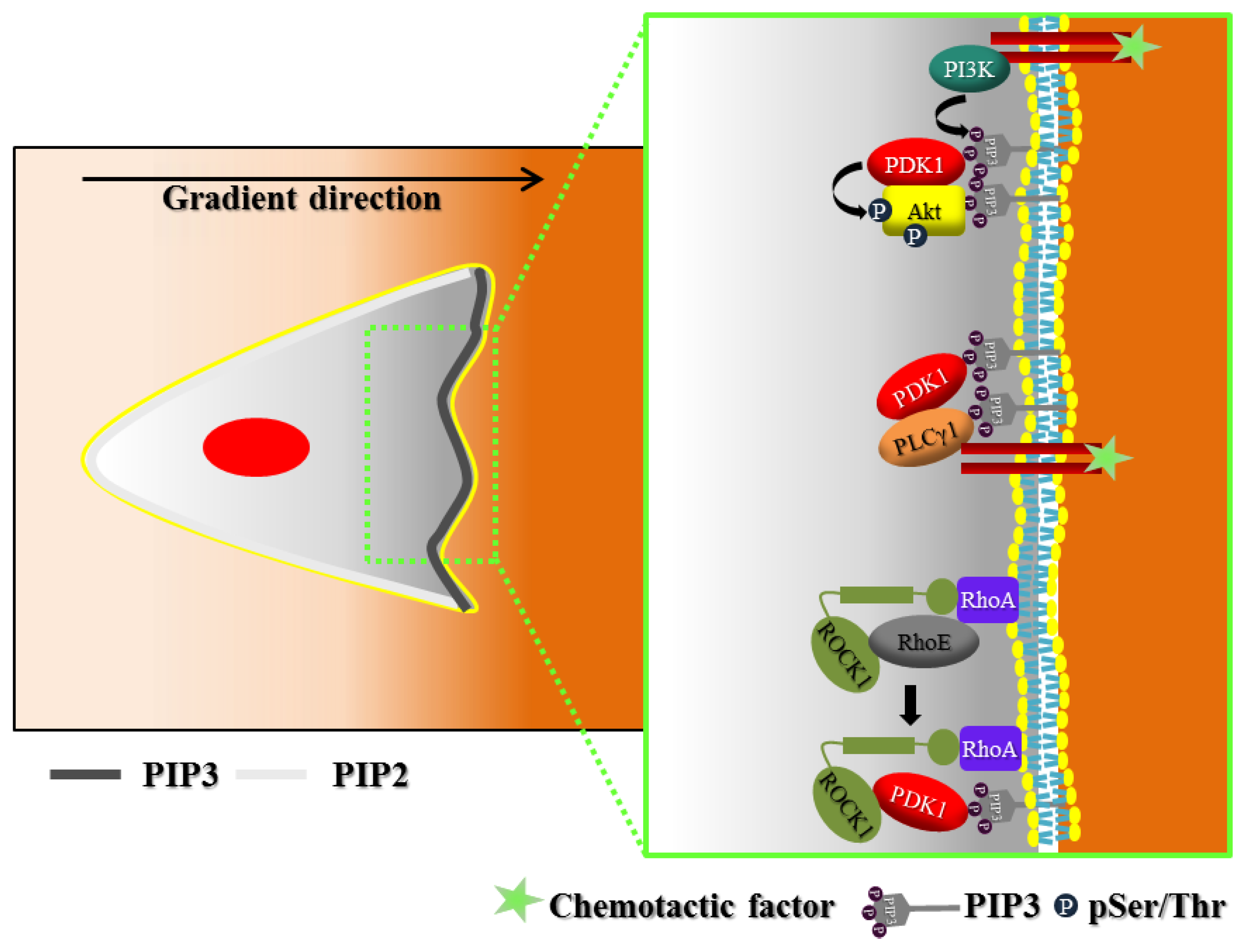
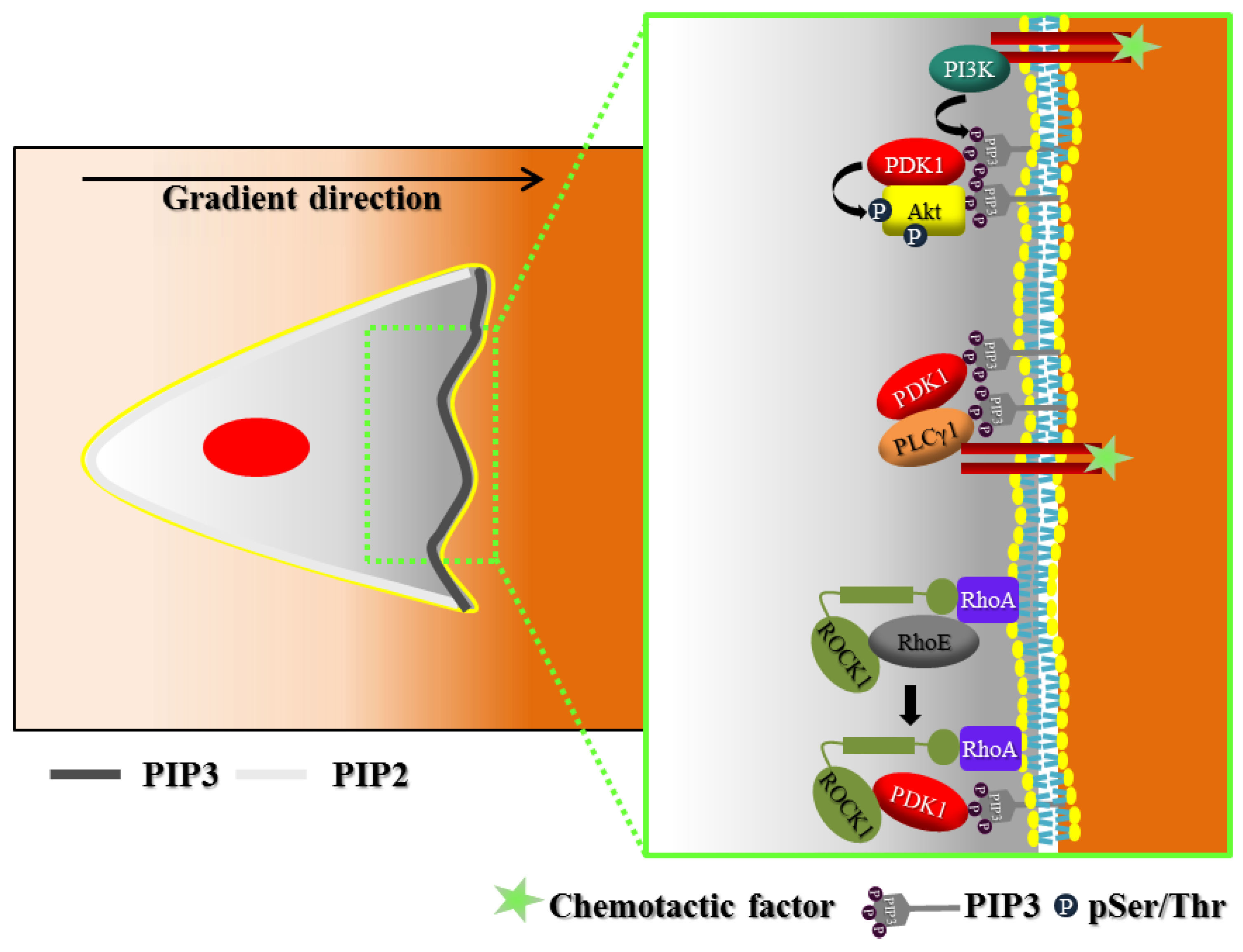
2. Polarization of Signaling
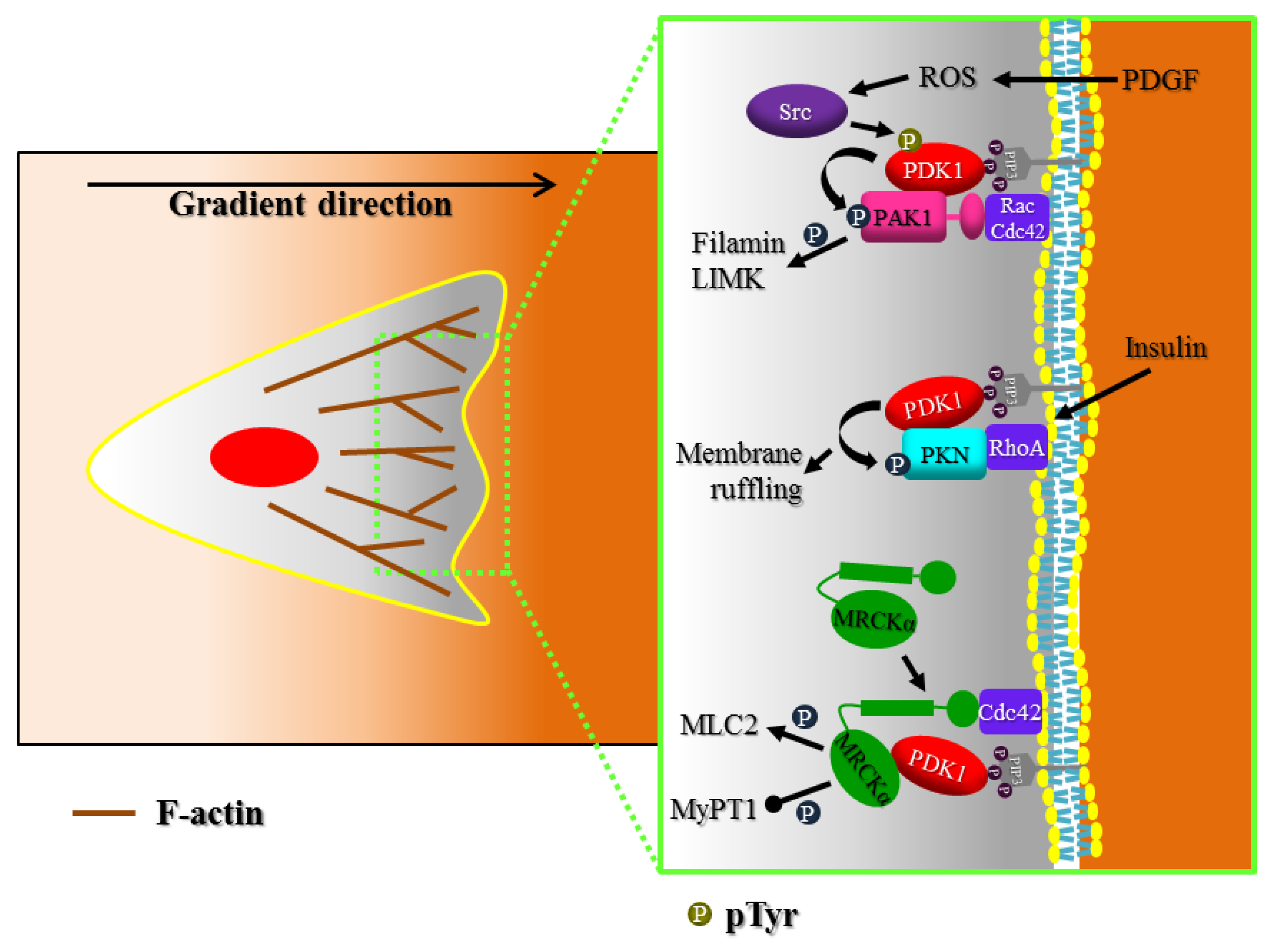
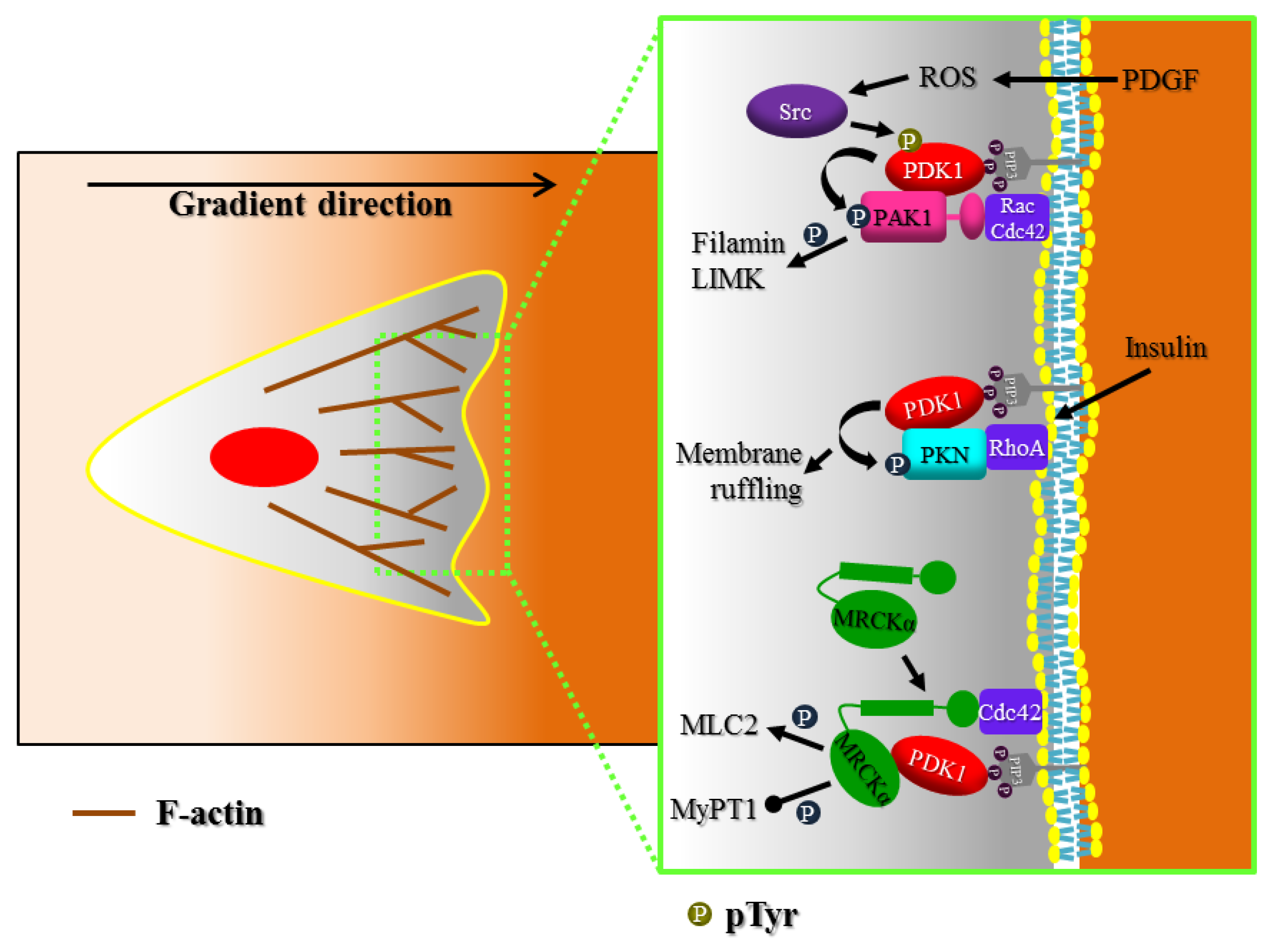
3. Actin Cytoskeleton Regulation
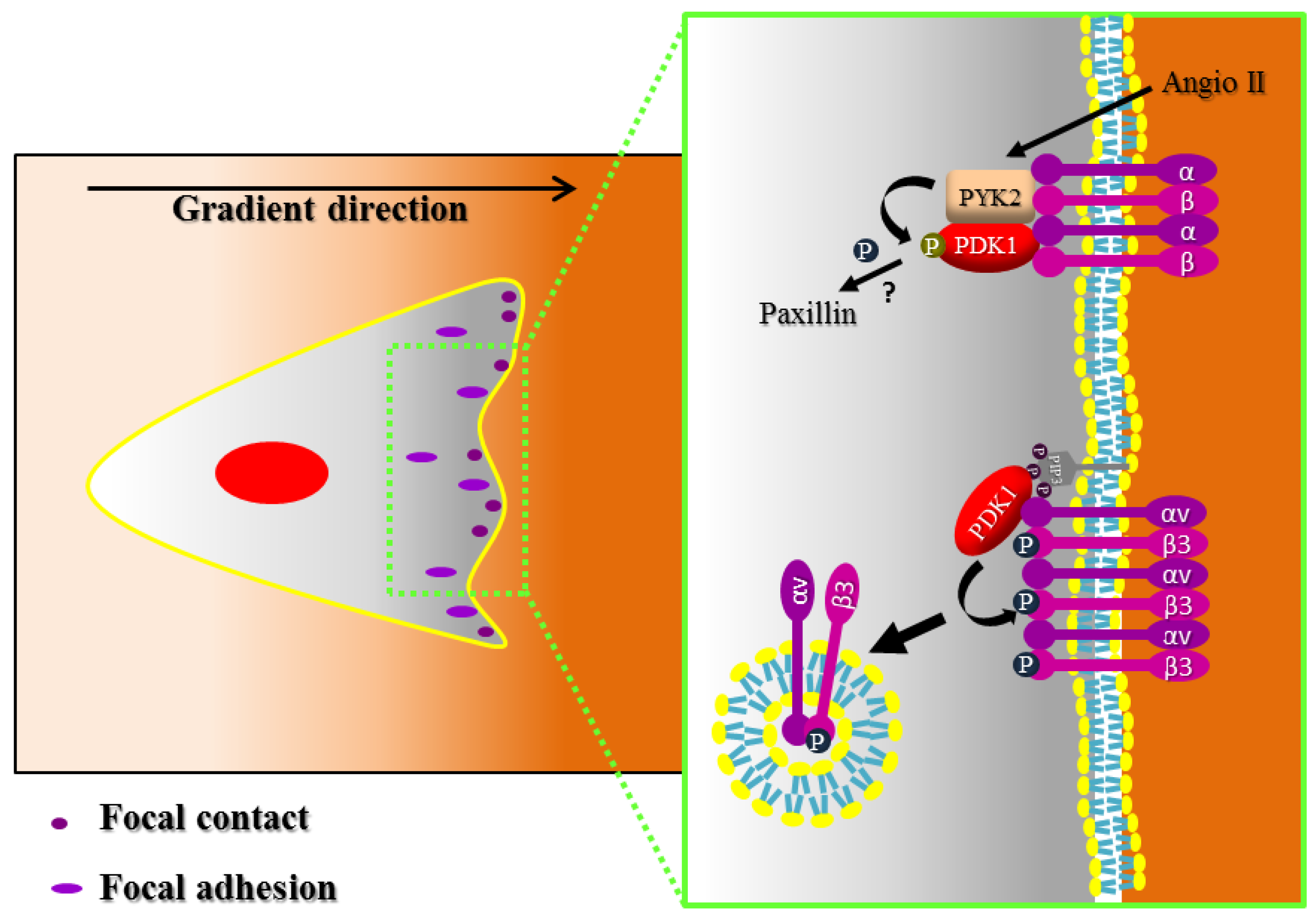
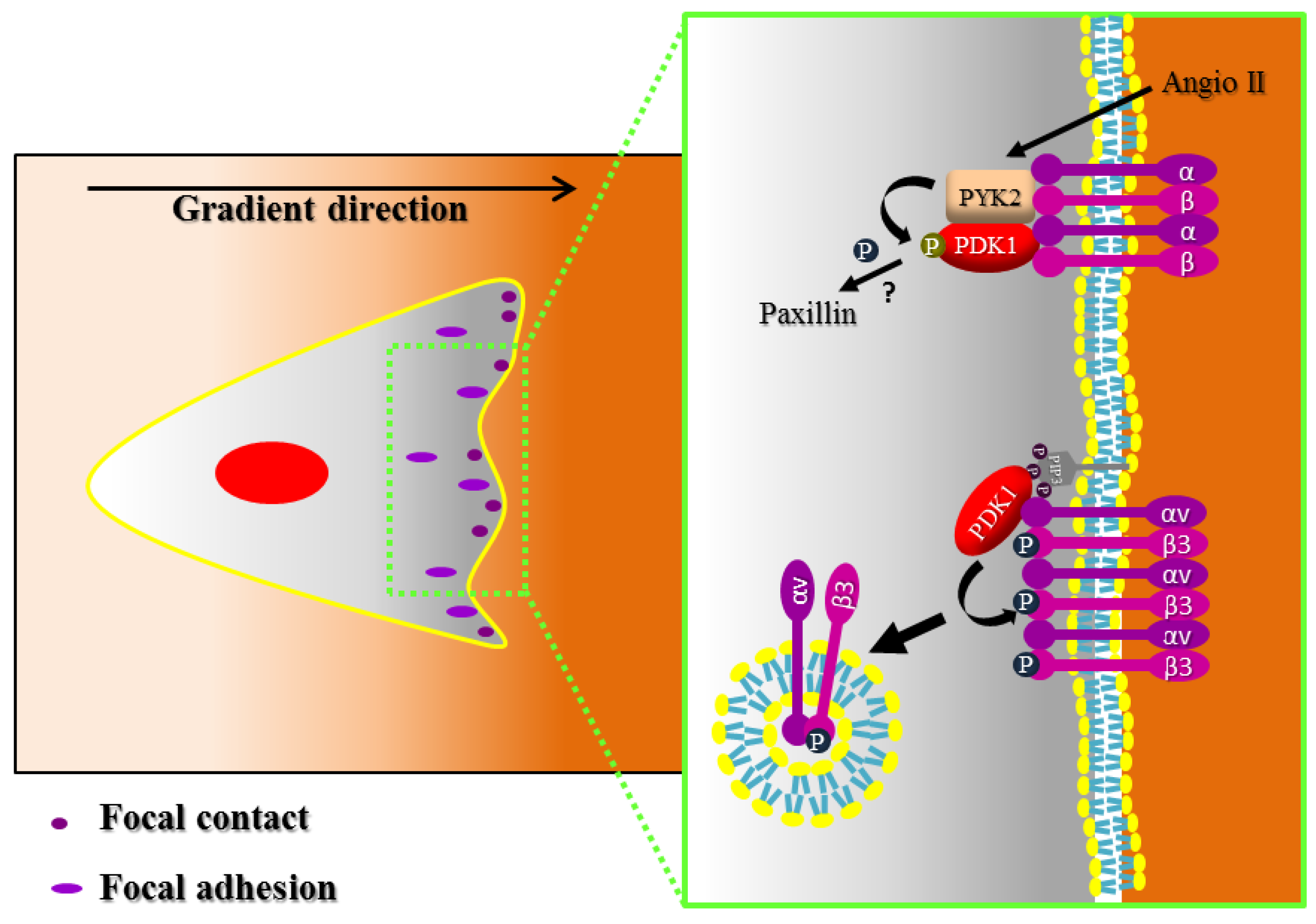
4. Focal Adhesion and Integrin Signaling
5. Tumor Invasiveness and Dissemination
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wyckoff, J.; Condeelis, J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005, 17, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Jones, J.; Segall, J.E. Chemotaxis of metastatic tumor cells: Clues to mechanisms from the dictyostelium paradigm. Cancer Metastasis Rev. 1992, 11, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Borgmann, S.; Brocker, E.B. Amoeboid leukocyte crawling through extracellular matrix: Lessons from the dictyostelium paradigm of cell movement. J. Leukoc. Biol. 2001, 70, 491–509. [Google Scholar] [PubMed]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Cell Biol. 2009, 188, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Cain, R.J.; Ridley, A.J. Phosphoinositide 3-kinases in cell migration. Biol. Cell 2009, 101, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of agc protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/Akt signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Wick, M.J.; Ramos, F.J.; Chen, H.; Quon, M.J.; Dong, L.Q.; Liu, F. Mouse 3-phosphoinositide-dependent protein kinase-1 undergoes dimerization and trans-phosphorylation in the activation loop. J. Biol. Chem. 2003, 278, 42913–42919. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Komander, D.; van Aalten, D.M.; Alessi, D.R. PDK1, the master regulator of agc kinase signal transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Currie, R.A.; Walker, K.S.; Gray, A.; Deak, M.; Casamayor, A.; Downes, C.P.; Cohen, P.; Alessi, D.R.; Lucocq, J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem. J. 1999, 337, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Balendran, A.; Casamayor, A.; Deak, M.; Paterson, A.; Gaffney, P.; Currie, R.; Downes, C.P.; Alessi, D.R. PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr. Biol. 1999, 9, 393–404. [Google Scholar] [CrossRef]
- Biondi, R.M.; Cheung, P.C.; Casamayor, A.; Deak, M.; Currie, R.A.; Alessi, D.R. Identification of a pocket in the pdk1 kinase domain that interacts with pif and the c-terminal residues of pka. EMBO J. 2000, 19, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.J.; Deak, M.; Arthur, J.S.; Armit, L.J.; Alessi, D.R. In vivo role of the pif-binding docking site of pdk1 defined by knock-in mutation. EMBO J. 2003, 22, 4202–4211. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Hill, M.M.; Hess, D.; Brazil, D.P.; Hofsteenge, J.; Hemmings, B.A. Identification of tyrosine phosphorylation sites on 3-phosphoinositide-dependent protein kinase-1 and their role in regulating kinase activity. J. Biol. Chem. 2001, 276, 37459–37471. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.S.; Taniyama, Y.; Rocic, P.; Seshiah, P.N.; Dechert, M.A.; Gerthoffer, W.T.; Griendling, K.K. Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration. Circ. Res. 2004, 94, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Weber, D.S.; Rocic, P.; Hilenski, L.; Akers, M.L.; Park, J.; Hemmings, B.A.; Alexander, R.W.; Griendling, K.K. PYK2- and SRC-dependent tyrosine phosphorylation of PDK1 regulates focal adhesions. Mol. Cell Biol. 2003, 23, 8019–8029. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.A.; Mora, A.; Ashby, P.R.; Williams, M.R.; Murray-Tait, V.; Malone, L.; Prescott, A.R.; Lucocq, J.M.; Alessi, D.R. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002, 21, 3728–3738. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, P.A.; di Blasio, L.; Primo, L. PDK1: A signaling hub for cell migration and tumor invasion. BBA-Rev. Cancer 2015, 1856, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Davies, A.M.; Bertrand, L.; Sharif, I.; Budas, G.R.; Jovanovic, S.; Mouton, V.; Kahn, C.R.; Lucocq, J.M.; Gray, G.A.; et al. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J. 2003, 22, 4666–4676. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Sakamoto, K.; McManus, E.J.; Alessi, D.R. Role of the pdk1-pkb-gsk3 pathway in regulating glycogen synthase and glucose uptake in the heart. FEBS Lett. 2005, 579, 3632–3638. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Jiang, Y.; Yang, Y.; Li, X.; Yang, Z.; Cao, K.; Wang, D.W. Deletion of pdk1 causes cardiac sodium current reduction in mice. PLoS ONE 2015, 10, e0122436. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Akazawa, H.; Tamagawa, M.; Furukawa, K.; Ogawa, W.; Yasuda, N.; Kudo, Y.; Liao, C.H.; Yamamoto, R.; Sato, T.; et al. PDK1 coordinates survival pathways and beta-adrenergic response in the heart. Proc. Natl. Acad. Sci. USA 2009, 106, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Long, M.; Kang, J.A.; Kim, W.S.; Lee, C.R.; Im, S.H.; Strickland, I.; Schulze-Luehrmann, J.; Hayden, M.S.; Ghosh, S. The kinase PDK1 is essential for B-cell receptor mediated survival signaling. PLoS ONE 2013, 8, e55378. [Google Scholar] [CrossRef] [PubMed]
- Venigalla, R.K.; McGuire, V.A.; Clarke, R.; Patterson-Kane, J.C.; Najafov, A.; Toth, R.; McCarthy, P.C.; Simeons, F.; Stojanovski, L.; Arthur, J.S. PDK1 regulates VDJ recombination, cell-cycle exit and survival during b-cell development. EMBO J 2013, 32, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Zaru, R.; Matthews, S.P.; Edgar, A.J.; Prescott, A.R.; Gomez-Nicola, D.; Hanauer, A.; Watts, C. The pdk1-rsk signaling pathway controls langerhans cell proliferation and patterning. J. Immunol. 2015, 195, 4264–4272. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Schulze-Luehrman, J.; Hayden, M.S.; Hashimoto, N.; Ogawa, W.; Kasuga, M.; Ghosh, S. The kinase PDK1 integrates t cell antigen receptor and CD28 coreceptor signaling to induce NF-κB and activate T cells. Nat. Immunol. 2009, 10, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.K.; Kelly, A.P.; Clarke, R.; Sinclair, L.V.; Deak, M.; Alessi, D.R.; Cantrell, D.A. Temporal differences in the dependency on phosphoinositide-dependent kinase 1 distinguish the development of invariant Vα 14 NKT cells and conventional T cells. J. Immunol. 2010, 185, 5973–5982. [Google Scholar] [CrossRef] [PubMed]
- Hinton, H.J.; Alessi, D.R.; Cantrell, D.A. The serine kinase phosphoinositide-dependent kinase 1 (PDK1) regulates T cell development. Nat. Immunol. 2004, 5, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.P.; Finlay, D.K.; Hinton, H.J.; Clarke, R.G.; Fiorini, E.; Radtke, F.; Cantrell, D.A. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J. 2007, 26, 3441–3450. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Owens, D.M.; Ghosh, S.; Farber, D.L. Conditional PDK1 ablation promotes epidermal and t-cell-mediated dysfunctions leading to inflammatory skin disease. J. Investig. Dermatol. 2015, 135, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Dainichi, T.; Hayden, M.S.; Park, S.G.; Oh, H.; Seeley, J.J.; Grinberg-Bleyer, Y.; Beck, K.M.; Miyachi, Y.; Kabashima, K.; Hashimoto, T.; et al. Pdk1 is a regulator of epidermal differentiation that activates and organizes asymmetric cell division. Cell Rep. 2016, 15, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Watatani, K.; Hirabayashi, Y.; Itoh, Y.; Gotoh, Y. PDK1 regulates the generation of oligodendrocyte precursor cells at an early stage of mouse telencephalic development. Genes Cells 2012, 17, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Kido, Y.; Uchida, T.; Asahara, S.; Shigeyama, Y.; Matsuda, T.; Takeda, A.; Tsuchihashi, D.; Nishizawa, A.; Ogawa, W.; et al. Ablation of pdk1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat. Genet. 2006, 38, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Westmoreland, J.J.; Wang, Q.; Bouzaffour, M.; Baker, S.J.; Sosa-Pineda, B. PDK1 activity controls proliferation, survival, and growth of developing pancreatic cells. Dev. Biol. 2009, 334, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Di, R.; Tao, F.; Chang, Z.; Lu, S.; Fan, W.; Shan, C.; Li, X.; Yang, Z. PDK1 regulates vascular remodeling and promotes epithelial-mesenchymal transition in cardiac development. Mol. Cell Biol. 2010, 30, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Parent, C.A.; Devreotes, P.N. A cell’s sense of direction. Science 1999, 284, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, A.; Iglesias, P.A. Models of eukaryotic gradient sensing: Application to chemotaxis of amoebae and neutrophils. Biophys. J. 2002, 82, 50–63. [Google Scholar] [CrossRef]
- Kutscher, B.; Devreotes, P.; Iglesias, P.A. Local excitation, global inhibition mechanism for gradient sensing: An interactive applet. Sci. STKE 2004. [Google Scholar] [CrossRef] [PubMed]
- Skupsky, R.; Losert, W.; Nossal, R.J. Distinguishing modes of eukaryotic gradient sensing. Biophys. J. 2005, 89, 2806–2823. [Google Scholar] [CrossRef] [PubMed]
- Firtel, R.A.; Chung, C.Y. The molecular genetics of chemotaxis: Sensing and responding to chemoattractant gradients. Bioessays 2000, 22, 603–615. [Google Scholar] [CrossRef]
- Bourne, H.R.; Weiner, O. A chemical compass. Nature 2002. [Google Scholar] [CrossRef] [PubMed]
- Servant, G.; Weiner, O.D.; Neptune, E.R.; Sedat, J.W.; Bourne, H.R. Dynamics of a chemoattractant receptor in living neutrophils during chemotaxis. Mol. Biol. Cell 1999, 10, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Zhang, N.; Murphy, D.B.; Devreotes, P.N. Dynamic distribution of chemoattractant receptors in living cells during chemotaxis and persistent stimulation. J. Cell Biol. 1997, 139, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Zhang, N.; Long, Y.; Parent, C.A.; Devreotes, P.N. Localization of the g protein betagamma complex in living cells during chemotaxis. Science 2000, 287, 1034–1036. [Google Scholar] [CrossRef] [PubMed]
- Parent, C.A.; Blacklock, B.J.; Froehlich, W.M.; Murphy, D.B.; Devreotes, P.N. G protein signaling events are activated at the leading edge of chemotactic cells. Cell 1998, 95, 81–91. [Google Scholar] [CrossRef]
- Meili, R.; Ellsworth, C.; Lee, S.; Reddy, T.B.; Ma, H.; Firtel, R.A. Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to camp in dictyostelium. EMBO J. 1999, 18, 2092–2105. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Araki, M. Tumor suppressor pten: Modulator of cell signaling, growth, migration and apoptosis. J. Cell Sci. 2001, 114, 2375–2382. [Google Scholar] [PubMed]
- Iijima, M.; Devreotes, P. Tumor suppressor pten mediates sensing of chemoattractant gradients. Cell 2002, 109, 599–610. [Google Scholar] [CrossRef]
- Funamoto, S.; Meili, R.; Lee, S.; Parry, L.; Firtel, R.A. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and pten mediates chemotaxis. Cell 2002, 109, 611–623. [Google Scholar] [CrossRef]
- Primo, L.; di Blasio, L.; Roca, C.; Droetto, S.; Piva, R.; Schaffhausen, B.; Bussolino, F. Essential role of pdk1 in regulating endothelial cell migration. J. Cell Biol. 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.N.; Wu, M.; Wan, W.Z.; Sun, R.H.; Yang, D.; Sun, X.J.; Ma, D.L.; Ying, G.G.; Zhang, N. Down-regulation of 3-phosphoinositide-dependent protein kinase-1 levels inhibits migration and experimental metastasis of human breast cancer cells. Mol. Cancer Res. 2009, 7, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Waugh, C.; Sinclair, L.; Finlay, D.; Bayascas, J.R.; Cantrell, D. Phosphoinositide (3,4,5)-triphosphate binding to phosphoinositide-dependent kinase 1 regulates a protein kinase B/Akt signaling threshold that dictates T-cell migration, not proliferation. Mol. Cell Biol. 2009, 29, 5952–5962. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Higuchi, M.; Oishi, K.; Kishi, Y.; Okazaki, T.; Sakai, H.; Miyata, T.; Nakajima, K.; Gotoh, Y. PDK1-AKT pathway regulates radial neuronal migration and microtubules in the developing mouse neocortex. Proc. Natl. Acad. Sci. USA 2016, 113, E2955–E2964. [Google Scholar] [CrossRef] [PubMed]
- Pinner, S.; Sahai, E. PDK1 regulates cancer cell motility by antagonising inhibition of rock1 by rhoe. Nat. Cell Biol. 2008, 10, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, P.A.; di Blasio, L.; Puliafito, A.; Seano, G.; Sessa, R.; Chianale, F.; Leung, T.; Bussolino, F.; Primo, L. PDK1-mediated activation of MRCKα regulates directional cell migration and lamellipodia retraction. J. Cell Biol. 2014, 206, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by rho-associated kinase (rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by rho and rho-associated kinase (rho-kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Chikh, A.; Wheeler, A.P.; Maffucci, T.; Falasca, M. A novel regulatory mechanism links plcgamma1 to PDK1. J. Cell Sci. 2012, 125, 3153–3163. [Google Scholar] [CrossRef] [PubMed]
- Gresset, A.; Hicks, S.N.; Harden, T.K.; Sondek, J. Mechanism of phosphorylation-induced activation of phospholipase C-gamma isozymes. J. Biol. Chem. 2010, 285, 35836–35847. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Logan, S.K.; Lehto, V.P.; Baccante, G.; Lemmon, M.A.; Schlessinger, J. Activation of phospholipase c gamma by PI 3-kinase-induced ph domain-mediated membrane targeting. EMBO J. 1998, 17, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.D.; Mullins, R.D. Cellular control of actin nucleation. Annu. Rev. Cell Dev. Biol. 2002, 18, 247–288. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulou, M.; Hall, A. Cell migration: Rho gtpases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Takenawa, T.; Miki, H. Wasp and wave family proteins: Key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 2001, 114, 1801–1809. [Google Scholar] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Dos Remedios, C.G.; Chhabra, D.; Kekic, M.; Dedova, I.V.; Tsubakihara, M.; Berry, D.A.; Nosworthy, N.J. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol. Rev. 2003, 83, 433–473. [Google Scholar] [CrossRef] [PubMed]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef] [PubMed]
- King, C.C.; Gardiner, E.M.; Zenke, F.T.; Bohl, B.P.; Newton, A.C.; Hemmings, B.A.; Bokoch, G.M. P21-activated kinase (PAK1) is phosphorylated and activated by 3- phosphoinositide-dependent kinase-1 (PDK1). J. Biol. Chem. 2000, 275, 41201–41209. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Gururaj, A.E.; Barnes, C.J. P21-activated kinases in cancer. Nat. Rev. Cancer 2006, 6, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.Q.; Landa, L.R.; Wick, M.J.; Zhu, L.; Mukai, H.; Ono, Y.; Liu, F. Phosphorylation of protein kinase n by phosphoinositide-dependent protein kinase-1 mediates insulin signals to the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2000, 97, 5089–5094. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Geiger, B.; Bershadsky, A.; Pankov, R.; Yamada, K.M. Transmembrane crosstalk between the extracellular matrix—Cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2001, 2, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Kiosses, W.B.; Shattil, S.J.; Pampori, N.; Schwartz, M.A. Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat. Cell Biol. 2001, 3, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Chrzanowska-Wodnicka, M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996, 12, 463–518. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.J.; Parsons, J.T.; Horwitz, A.F. Adhesion assembly, disassembly and turnover in migrating cells—Over and over and over again. Nat. Cell Biol. 2002, 4, E97–E100. [Google Scholar] [CrossRef] [PubMed]
- Regen, C.M.; Horwitz, A.F. Dynamics of βb 1 integrin-mediated adhesive contacts in motile fibroblasts. J. Cell Biol. 1992, 119, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Smilenov, L.B.; Mikhailov, A.; Pelham, R.J.; Marcantonio, E.E.; Gundersen, G.G. Focal adhesion motility revealed in stationary fibroblasts. Science 1999, 286, 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- Broussard, J.A.; Webb, D.J.; Kaverina, I. Asymmetric focal adhesion disassembly in motile cells. Curr. Opin. Cell Biol. 2008, 20, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Ezratty, E.J.; Partridge, M.A.; Gundersen, G.G. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 2005, 7, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.T.; Kunz, J. Focal adhesion disassembly requires clathrin-dependent endocytosis of integrins. FEBS Lett. 2009, 583, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Ezratty, E.J.; Bertaux, C.; Marcantonio, E.E.; Gundersen, G.G. Clathrin mediates integrin endocytosis for focal adhesion disassembly in migrating cells. J. Cell Biol. 2009, 187, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, L.; Gagliardi, P.A.; Puliafito, A.; Sessa, R.; Seano, G.; Bussolino, F.; Primo, L. PDK1 regulates focal adhesion disassembly by modulating endocytosis of alpha v β3 integrin. J. Cell Sci. 2015, 128, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Cell-matrix adhesion in vascular development. J. Thromb. Haemost 2007, 5, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Kirk, R.I.; Sanderson, M.R.; Lerea, K.M. Threonine phosphorylation of the β3 integrin cytoplasmic tail, at a site recognized by PDK1 and AKT/PKB in vitro, regulates shc binding. J. Biol. Chem. 2000, 275, 30901–30906. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Yuan, H.; Yin, Y.; Zeng, X.; Bai, R.; Glazer, R.I. 3-phosphoinositide-dependent protein kinase-1 (PDK1) promotes invasion and activation of matrix metalloproteinases. BMC Cancer 2006. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Yoshida, S.; Muroi, E.; Yoshida, N.; Kawamura, M.; Kouchi, Z.; Nakamura, Y.; Sakai, R.; Fukami, K. Phosphoinositide 3-kinase signaling pathway mediated by p110α regulates invadopodia formation. J. Cell Biol. 2011, 193, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.S.; Whigham, A.S.; Yarbrough, W.G.; Weaver, A.M. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007, 67, 4227–4235. [Google Scholar] [CrossRef] [PubMed]
- Coopman, P.J.; Do, M.T.; Thompson, E.W.; Mueller, S.C. Phagocytosis of cross-linked gelatin matrix by human breast carcinoma cells correlates with their invasive capacity. Clin. Cancer Res. 1998, 4, 507–515. [Google Scholar] [PubMed]
- Yamaguchi, H.; Pixley, F.; Condeelis, J. Invadopodia and podosomes in tumor invasion. Eur. J. Cell Biol. 2006, 85, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Segall, J.E. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 2003, 3, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Ruller, C.; Feng, Y.; Lazova, R.; Kluger, H.; Li, J.L.; De, S.K.; Rickert, R.; Pellecchia, M.; Bosenberg, M.; et al. Genetic inactivation or pharmacological inhibition of PDK1 delays development and inhibits metastasis of braf(v600e)::Pten(−/−) melanoma. Oncogene 2014, 33, 4330–4339. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective requirement of PI3K/PDK1 signaling for kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lei, J.; Fang, Z.L.; Xiong, J.P. Mir-128b is down-regulated in gastric cancer and negatively regulates tumour cell viability by targeting PDK1/AKT/NF-κB axis. J. Biosci. 2016, 41, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. Akt-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, P.A.; di Blasio, L.; Orso, F.; Seano, G.; Sessa, R.; Taverna, D.; Bussolino, F.; Primo, L. 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia 2012, 14, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Ericson, K.; Gan, C.; Cheong, I.; Rago, C.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Huso, D.L.; Vogelstein, B.; Papadopoulos, N. Genetic inactivation of AKT1, AKT2, and PDPK1 in human colorectal cancer cells clarifies their roles in tumor growth regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Calleja, V.; Ferro, R.; Fantin, A.; Riley, A.M.; Potter, B.V.; Brennan, C.H.; Maffucci, T.; Larijani, B.; Falasca, M. A small molecule inhibitor of PDK1/plcgamma1 interaction blocks breast and melanoma cancer cell invasion. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, C.; Falasca, M. Targeting pdk1 in cancer. Curr. Med. Chem. 2011, 18, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.J.; Hsieh, F.C.; Song, H.; Lin, J. Elevated phosphorylation and activation of PDK-1/Akt pathway in human breast cancer. Br. J. Cancer 2005, 93, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Su, T.; Saal, L.H.; Koujak, S.; Hopkins, B.D.; Barkley, C.R.; Wu, J.; Nandula, S.; Dutta, B.; Xie, Y.; et al. 3-phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009, 69, 6299–6306. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, O.C.; Lai, E.W.; Vissapragada, S.; Cromelin, C.; Avetian, M.; Salinas, P.; Ramos, H.; Kallakury, B.; Casimiro, M.; Lisanti, M.P.; et al. A reduction in pten tumor suppressor activity promotes ErbB-2-induced mouse prostate adenocarcinoma formation through the activation of signaling cascades downstream of PDK1. Am. J. Pathol. 2009, 174, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Lee, P.L.; Li, Z.; Jiang, X.; Lim, Y.C.; Hooi, S.C.; Yu, Q. B55β-associated PP2A complex controls PDK1-directed MYC signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell 2010, 18, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.; Wee, Z.N.; et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mtor-targeted therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Chang, T.H.; Huang, Y.F.; Chen, C.C.; Chou, C.Y. COL11A1 confers chemoresistance on ovarian cancer cells through the activation of Akt/c/EBPβ pathway and PDK1 stabilization. Oncotarget 2015, 6, 23748–23763. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, Z.; Liu, T.; Han, L.; Wang, C.; Yang, B.; Zheng, F. Upregulation of PDK1 associates with poor prognosis in esophageal squamous cell carcinoma with facilitating tumorigenicity in vitro. Med. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lian, S.; Shao, Y.; Liu, H.; He, J.; Lu, W.; Zhang, Y.; Jiang, Y.; Zhu, J. PDK1 induces JunB, EMT, cell migration and invasion in human gallbladder cancer. Oncotarget 2015, 6, 29076–29086. [Google Scholar] [PubMed]
- Zabkiewicz, J.; Pearn, L.; Hills, R.K.; Morgan, R.G.; Tonks, A.; Burnett, A.K.; Darley, R.L. The PDK1 master kinase is over-expressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica 2014, 99, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Lau, E.; Zhang, T.; Feng, Y.; Sereduk, C.; Yin, H.; De, S.K.; Meeth, K.; Platt, J.T.; Langdon, C.G.; et al. PDK1 and SGK3 contribute to the growth of BRAF-mutant melanomas and are potential therapeutic targets. Cancer Res. 2015, 75, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Bayascas, J.R.; Leslie, N.R.; Parsons, R.; Fleming, S.; Alessi, D.R. Hypomorphic mutation of PDK1 suppresses tumorigenesis in pten(+/−) mice. Curr. Biol. 2005, 15, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Clarke, P.A.; Raynaud, F.I.; van Montfort, R.L. Drugging the PI3 kinome: From chemical tools to drugs in the clinic. Cancer Res. 2010, 70, 2146–2157. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue (Promoter) | Phenotype | Viable/Lethal | References |
|---|---|---|---|
| Whole body | Lack of somites, forebrain and neural crest-derived tissue; vasculature not functional | Lethal E9.5 | [23] |
| Cardiac muscles (MCK-Cre) | Heart failure; no activation of Akt and S6K. No activation of glycogen synthase after insulin stimulation; glucose uptake defects | Death between 5 and 11 weeks of age | [25,26] |
| Myocardium (αMHC-Cre) | Slow heart rate, decreased sodium current density | Death at 11 weeks of age | [27] |
| Myocardium (tamoxifen-inducible αMHC-Cre) | Cardiac dysfunction 1 week after Tamox; impaired responsiveness of βAR; increased apoptosis | Death at 5–15 weeks after tamoxifen | [28] |
| B cells (CD19-Cre) | Defective B cell development; increased apoptosis | Viable | [29] |
| Hematopoietic cells (Vav-Cre) | B cell development arrest; increased myeloid cell recruitment in lung and liver. Lack of Langerhans cells | Viable | [30,31] |
| T cells (CD4-Cre) | T cells activation and proliferation defects | Viable | [32,33] |
| Thymocytes (Lck-Cre) | No maturation of T cells | Viable | [34,35] |
| CD4 T cells/keratinocytes (OX40-Cre) | Inflammatory skin diseases | Viable | [36] |
| Keratinocytes (K14-Cre) | Thin and shiny epidermis; hypoplasia of vibrissae; deficient barrier function; asymmetric cell division defects | Death within several hours after birth | [37] |
| Neural precursors cells (Nestin-Cre) | Reduction in number of oligodendrocytes precursors cells during telencephalic development | Viable | [38] |
| Pancreas β cells (Rat insulin 2-Cre) | Alterate glucose homeostasis (diabetes); increased level of blood glucose and decreased level of insulin | Males die at 12.24 weeks of age | [39] |
| Pancreas progenitors (PDX1-Cre) | Pancreas hypoplasia; hyperglycemia; reduced number of endocrine and exocrine cells during development | Viable | [40] |
| Vascular endothelial cells (Tie2-Cre) | Growth retardation; hemorrhages; heart with abnormal morphology; defective vessels in yolk sac and in placenta; defective epithelial-mesenchymal transition | Lethal E11.5 | [41] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Blasio, L.; Gagliardi, P.A.; Puliafito, A.; Primo, L. Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a Key Regulator of Cell Migration and Cancer Dissemination. Cancers 2017, 9, 25. https://doi.org/10.3390/cancers9030025
Di Blasio L, Gagliardi PA, Puliafito A, Primo L. Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a Key Regulator of Cell Migration and Cancer Dissemination. Cancers. 2017; 9(3):25. https://doi.org/10.3390/cancers9030025
Chicago/Turabian StyleDi Blasio, Laura, Paolo A. Gagliardi, Alberto Puliafito, and Luca Primo. 2017. "Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a Key Regulator of Cell Migration and Cancer Dissemination" Cancers 9, no. 3: 25. https://doi.org/10.3390/cancers9030025
APA StyleDi Blasio, L., Gagliardi, P. A., Puliafito, A., & Primo, L. (2017). Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a Key Regulator of Cell Migration and Cancer Dissemination. Cancers, 9(3), 25. https://doi.org/10.3390/cancers9030025