
DNA Repair Pathway Alterations in Bladder Cancer

Abstract
:1. Introduction
1.1. Bladder Cancer Is a Global Health Problem
1.2. DNA Repair Pathway Alterations Are Biomarkers and Therapeutic Targets
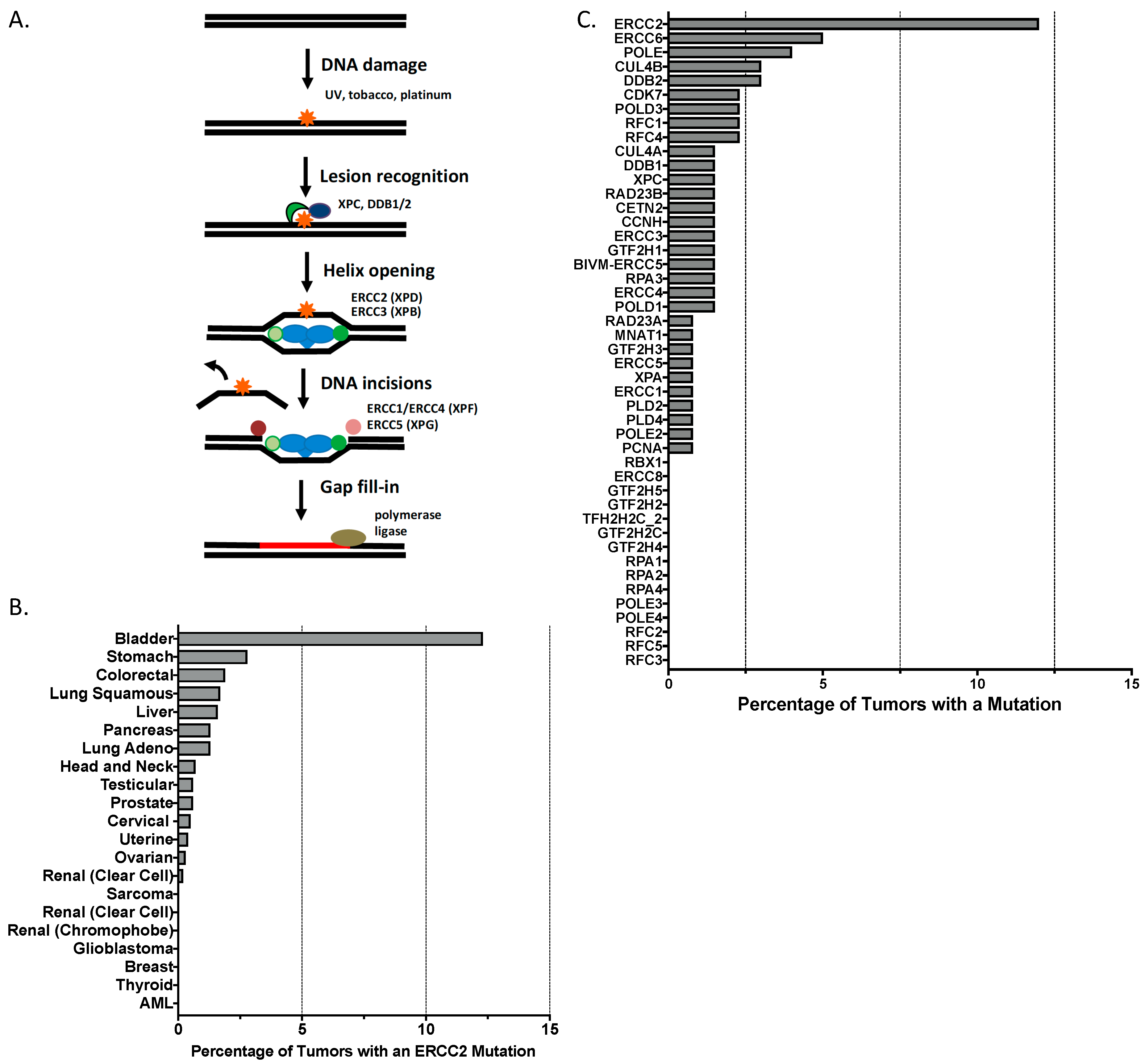
2. The Genomic Landscape of Bladder Cancer Reveals Alterations in DNA Repair Pathways
2.1. Bladder Tumors Exhibit a Complex Somatic Mutational Landscape
2.2. Mutational Signatures Reflect Underlying DNA Damage and Repair Processes
2.3. Gene Expression Profiles Define Distinct MIBC Subtypes
3. DNA Repair Pathways Are Predictive Biomarkers in Bladder Cancer
3.1. Low ERCC1 Expression Is Associated with Improved Outcomes in Cisplatin-Treated Patients
3.2. High MRE11A Expression Is Associated with Improved Outcomes in Radiation-Treated Patients
3.3. Basal-like Tumors Benefit Most from Chemotherapy
3.4. Somatic ERCC2 Mutations Are Associated with Improved Outcomes among Cisplatin-Treated Patients
3.5. Mutations in DNA Repair Genes beyond ERCC2 Are also Associated with Improved Outcomes
3.6. Germline DNA Repair Polymorphisms Are Associated with Bladder Cancer Risk and Treatment Response
4. Ongoing Efforts to Optimize Bladder Cancer Treatment
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, C.; Bosetti, C.; Negri, E.; Malvezzi, M.; La Vecchia, C. Mechanisms of disease: The epidemiology of bladder cancer. Nat. Clin. Pract. Urol. 2006, 3, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmadie, H.A.; Iyer, G.; Lee, B.H.; Scott, S.N.; Mehra, R.; Bagrodia, A.; Jordan, E.J.; Gao, S.P.; Ramirez, R.; Cha, E.K.; et al. Frequent somatic CDH1 loss-of-function mutations in plasmacytoid variant bladder cancer. Nat. Genet. 2016, 48, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.G.; Palapattu, G.S.; Shariat, S.F.; Karakiewicz, P.I.; Bastian, P.J.; Lotan, Y.; Gupta, A.; Vazina, A.; Gilad, A.; Sagalowsky, A.I.; et al. Clinical outcomes following radical cystectomy for primary nontransitional cell carcinoma of the bladder compared to transitional cell carcinoma of the bladder. J. Urol. 2006, 175, 2048–2053. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J. DNA-Damaging Anticancer Drugs—A Perspective for DNA Repair-Oriented Therapy. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keapl represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Hayden, A.; Douglas, J.; Sommerlad, M.; Andrews, L.; Gould, K.; Hussain, S.; Thomas, G.J.; Packham, G.; Crabb, S.J. The Nrf2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol. Oncol. 2014, 32, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Morganella, S.; Alexandrov, L.B.; Glodzik, D.; Zou, X.; Davies, H.; Staaf, J.; Sieuwerts, A.M.; Brinkman, A.B.; Martin, S.; Ramakrishna, M.; et al. The topography of mutational processes in breast cancer genomes. Nat. Commun. 2016, 7, 11383. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Tiao, G.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.D.; et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Czerniak, B.; Ochoa, A.; Su, X.; Siefker-Radtke, A.; Dinney, C.; McConkey, D.J. Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nat. Rev. Urol. 2014, 11, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Damrauer, J.S.; Hoadley, K.A.; Chism, D.D.; Fan, C.; Tiganelli, C.J.; Wobker, S.E.; Yeh, J.J.; Milowsky, M.I.; Iyer, G.; Parker, J.S.; et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc. Natl. Acad. Sci. USA 2014, 111, 3110–3115. [Google Scholar] [CrossRef] [PubMed]
- Aine, M.; Eriksson, P.; Liedberg, F.; Sjodahl, G.; Hoglund, M. Biological determinants of bladder cancer gene expression subtypes. Sci. Rep. 2015, 5, 10957. [Google Scholar] [CrossRef] [PubMed]
- Rebouissou, S.; Bernard-Pierrot, I.; de Reynies, A.; Lepage, M.L.; Krucker, C.; Chapeaublanc, E.; Herault, A.; Kamoun, A.; Caillault, A.; Letouze, E.; et al. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Grossman, H.B.; Natale, R.B.; Tangen, C.M.; Speights, V.O.; Vogelzang, N.J.; Trump, D.L.; deVere White, R.W.; Sarosdy, M.F.; Wood, D.P., Jr.; Raghavan, D.; et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N. Engl. J. Med. 2003, 349, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Advanced Bladder Cancer Meta-analysis. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data advanced bladder cancer (ABC) meta-analysis collaboration. Eur. Urol. 2005, 48, 202–205. [Google Scholar]
- Mak, R.H.; Hunt, D.; Shipley, W.U.; Efstathiou, J.A.; Tester, W.J.; Hagan, M.P.; Kaufman, D.S.; Heney, N.M.; Zietman, A.L. Long-term outcomes in patients with muscle-invasive bladder cancer after selective bladder-preserving combined-modality therapy: a pooled analysis of Radiation Therapy Oncology Group protocols 8802, 8903, 9506, 9706, 9906, and 0233. J. Clin. Oncol. 2014, 32, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, N.J.; Shipley, W.U.; Clayman, R.H.; Niemierko, A.; Drumm, M.; Heney, N.M.; Michaelson, M.D.; Lee, R.J.; Saylor, P.J.; Wszolek, M.F.; et al. Long-term Outcomes After Bladder-preserving Tri-modality Therapy for Patients with Muscle-invasive Bladder Cancer: An Updated Analysis of the Massachusetts General Hospital Experience. Eur. Urol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; Paz-Ares, L.; Cuello, M.; Cecere, F.L.; Albiol, S.; Guillem, V.; Gallardo, E.; Carles, J.; Mendez, P.; de la Cruz, J.J.; et al. Gene expression of ERCC1 as a novel prognostic marker in advanced bladder cancer patients receiving cisplatin-based chemotherapy. Ann. Oncol. 2007, 18, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Mullane, S.A.; Werner, L.; Guancial, E.A.; Lis, R.T.; Stack, E.C.; Loda, M.; Kantoff, P.W.; Choueiri, T.K.; Rosenberg, J.; Bellmunt, J. Expression Levels of DNA Damage Repair Proteins Are Associated With Overall Survival in Platinum-Treated Advanced Urothelial Carcinoma. Clin. Genitourin. Cancer 2016, 14, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Klatte, T.; Seitz, C.; Rink, M.; Roupret, M.; Xylinas, E.; Karakiewicz, P.; Susani, M.; Shariat, S.F. ERCC1 as a Prognostic and Predictive Biomarker for Urothelial Carcinoma of the Bladder following Radical Cystectomy. J. Urol. 2015, 194, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Sakano, S.; Ogawa, S.; Yamamoto, Y.; Nishijima, J.; Miyachika, Y.; Matsumoto, H.; Hara, T.; Matsuyama, H. ERCC1 and XRCC1 expression predicts survival in bladder cancer patients receiving combined trimodality therapy. Mol. Clin. Oncol. 2013, 1, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Dabholkar, M.; Vionnet, J.; Bostick-Bruton, F.; Yu, J.J.; Reed, E. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J. Clin. Invest. 1994, 94, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Friboulet, L.; Olaussen, K.A.; Pignon, J.P.; Shepherd, F.A.; Tsao, M.S.; Graziano, S.; Kratzke, R.; Douillard, J.Y.; Seymour, L.; Pirker, R.; et al. ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N. Engl. J. Med. 2013, 368, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Metzger, R.; Leichman, C.G.; Danenberg, K.D.; Danenberg, P.V.; Lenz, H.J.; Hayashi, K.; Groshen, S.; Salonga, D.; Cohen, H.; Laine, L.; et al. ERCC1 mRNA levels complement thymidylate synthase mRNA levels in predicting response and survival for gastric cancer patients receiving combination cisplatin and fluorouracil chemotherapy. J. Clin. Oncol. 1998, 16, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Nelson, L.D.; Teo, M.T.; Chilka, S.; Bhattarai, S.; Johnston, C.F.; Elliott, F.; Lowery, J.; Taylor, C.F.; Churchman, M.; et al. MRE11 expression is predictive of cause-specific survival following radical radiotherapy for muscle-invasive bladder cancer. Cancer Res. 2010, 70, 7017–7026. [Google Scholar] [CrossRef] [PubMed]
- Laurberg, J.R.; Brems-Eskildsen, A.S.; Nordentoft, I.; Fristrup, N.; Schepeler, T.; Ulhoi, B.P.; Agerbaek, M.; Hartmann, A.; Bertz, S.; Wittlinger, M.; et al. Expression of TIP60 (tat-interactive protein) and MRE11 (meiotic recombination 11 homolog) predict treatment-specific outcome of localised invasive bladder cancer. BJU Int. 2012, 110, E1228–E1236. [Google Scholar] [CrossRef] [PubMed]
- Magliocco, A.M.; Moughan, J.; Simko, J.; Efstathiou, J.A.; Gray, P.J.; Hagan, M.P.; Kaufman, D.S.; Tester, W.J.; Zietman, A.L.; McCarthy, S.M.; et al. The impact of MRE11 in nuclear to cytoplasmic ratio on outcomes in muscle invasive bladder cancer an analysis of NRG/RTOG 8802, 8903, 9506, 9706, 9906, and 0233. In Proceedings of the 2017 Genitourinary Cancer Symposium, Orlando, FL, USA, 16–18 February 2017. [Google Scholar]
- Van Allen, E.M.; Mouw, K.W.; Kim, P.; Iyer, G.; Wagle, N.; Al-Ahmadie, H.; Zhu, C.; Ostrovnaya, I.; Kryukov, G.V.; O’Connor, K.W.; et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014, 4, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Plimack, E.R.; Hoffman-Censits, J.; Garraway, L.A.; Bellmunt, J.; Van Allen, E.; Rosenberg, J.E. Clinical Validation of Chemotherapy Response Biomarker ERCC2 in Muscle-Invasive Urothelial Bladder Carcinoma. JAMA Oncol. 2016, 2, 1094–1096. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.B.; Scott, S.N.; Zabor, E.C.; Cha, E.K.; Hreiki, J.; Sfakianos, J.P.; Ramirez, R.; Bagrodia, A.; Rosenberg, J.E.; Bajorin, D.F.; et al. Genomic characterization of response to chemoradiation in urothelial bladder cancer. Cancer 2016, 122, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Plimack, E.R.; Dunbrack, R.L.; Brennan, T.A.; Andrake, M.D.; Zhou, Y.; Serebriiskii, I.G.; Slifker, M.; Alpaugh, K.; Dulaimi, E.; Palma, N.; et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer. Eur. Urol. 2015, 68, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Kiyotani, K.; Tamura, K.; Antic, T.; Jang, M.; Montoya, M.; Campanile, A.; Yew, P.Y.; Ganshert, C.; Fujioka, T.; et al. Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clin. Cancer Res. 2014, 20, 6605–6617. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Kerr, M.; Teo, M.T.; Jevons, S.J.; Koritzinsky, M.; Wouters, B.G.; Bhattarai, S.; Kiltie, A.E. Post-transcriptional regulation of MRE11 expression in muscle-invasive bladder tumours. Oncotarget 2014, 5, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J.; Choi, W.; Ochoa, A.; Siefker-Radtke, A.; Czerniak, B.; Dinney, C.P. Therapeutic opportunities in the intrinsic subtypes of muscle-invasive bladder cancer. Hematol. Oncol. Clin. North Am. 2015, 29, 377–394. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J.; Choi, W.; Shen, Y.; Lee, I.L.; Porten, S.; Matin, S.F.; Kamat, A.M.; Corn, P.; Millikan, R.E.; Dinney, C.; et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-naive Urothelial Cancer is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur. Urol. 2016, 69, 855–862. [Google Scholar] [PubMed]
- Porter, M.P.; Kerrigan, M.C.; Donato, B.M.; Ramsey, S.D. Patterns of use of systemic chemotherapy for Medicare beneficiaries with urothelial bladder cancer. Urol. Oncol. 2011, 29, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Fuss, J.O.; Tainer, J.A. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair 2011, 10, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, B.; Kuper, J.; Kisker, C. Role of XPD in cellular functions: To TFIIH and beyond. DNA Repair 2016, 44, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.Y.; Bambury, R.; Zabor, E.C.; Jordan, E.J.; Al-Ahmadie, H.A.; Boyd, M.; Bouvier, N.; Mullane, S.; Cha, E.K.; Roper, N.; et al. DNA Damage Response and Repair Gene Alterations Are Associated With Improved Survival In Patients With Platinum-Treated Advanced Urothelial Carcinoma. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- van der Post, R.S.; Kiemeney, L.A.; Ligtenberg, M.J.; Witjes, J.A.; Hulsbergen-van de Kaa, C.A.; Bodmer, D.; Schaap, L.; Kets, C.M.; van Krieken, J.H.; Hoogerbrugge, N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J. Med. Genet. 2010, 47, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Skeldon, S.C.; Semotiuk, K.; Aronson, M.; Holter, S.; Gallinger, S.; Pollett, A.; Kuk, C.; van Rhijn, B.; Bostrom, P.; Cohen, Z.; et al. Patients with Lynch syndrome mismatch repair gene mutations are at higher risk for not only upper tract urothelial cancer but also bladder cancer. Eur. Urol. 2013, 63, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.W.; Gu, J.; Wu, X. Germline prognostic markers for urinary bladder cancer: obstacles and opportunities. Urol. Oncol. 2012, 30, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.J.; Vijai, J.; Hamilton, R.J.; Ostrovnaya, I.; Iyer, G.; Garcia-Grossman, I.R.; Kim, P.H.; Przybylo, J.A.; Alanee, S.; Riches, J.C.; et al. Germline single nucleotide polymorphisms associated with response of urothelial carcinoma to platinum-based therapy: the role of the host. Ann. Oncol. 2013, 24, 2414–2421. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, P.H.; Alanee, S.; Stratton, K.L.; Garcia-Grossman, I.R.; Cao, H.; Ostrovnaya, I.; Plimack, E.R.; Manschreck, C.; Ganshert, C.; Smith, N.D.; et al. Clinical Evaluation of Cisplatin Sensitivity of Germline Polymorphisms in Neoadjuvant Chemotherapy for Urothelial Cancer. Clin. Genitourin. Cancer 2016, 14, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.T.; Dyrskjot, L.; Nsengimana, J.; Buchwald, C.; Snowden, H.; Morgan, J.; Jensen, J.B.; Knowles, M.A.; Taylor, G.; Barrett, J.H.; et al. Next-generation sequencing identifies germline MRE11A variants as markers of radiotherapy outcomes in muscle-invasive bladder cancer. Ann. Oncol. 2014, 25, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Ghazani, A.A.; Oliver, N.M.; St Pierre, J.P.; Garofalo, A.; Rainville, I.R.; Hiller, E.; Treacy, D.J.; Rojas-Rudilla, V.; Wood, S.; Bair, E.; et al. Assigning clinical meaning to somatic and germ-line whole-exome sequencing data in a prospective cancer precision medicine study. Genet. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’Amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Isono, M.; Hoffmann, M.J.; Pinkerneil, M.; Sato, A.; Michaelis, M.; Cinatl, J., Jr.; Niegisch, G.; Schulz, W.A. Checkpoint kinase inhibitor AZD7762 strongly sensitises urothelial carcinoma cells to gemcitabine. J. Exp. Clin. Cancer Res. 2017, 36, 1. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.; Xu, H.G.; Chen, J.; Xu, Z.X.; Levitt, J.M.; Stanley, J.A.; Yang, E.S.; Lerner, S.P.; Sonpavde, G. Activity of CEP-9722, a poly (ADP-ribose) polymerase inhibitor, in urothelial carcinoma correlates inversely with homologous recombination repair response to DNA damage. Anticancer Drugs 2014, 25, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.; Yang, J.C.; Lu, M.C.; Lee, C.L.; Peng, C.Y.; Hsu, W.Y.; Dai, Y.H.; Chang, F.R.; Zhang, D.Y.; Wu, W.J.; et al. ATR-Chk1 signaling inhibition as a therapeutic strategy to enhance cisplatin chemosensitivity in urothelial bladder cancer. Oncotarget 2016, 7, 1947–1959. [Google Scholar] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Pathway | Cohort/Study Details | References |
|---|---|---|---|
| MRE11A | DSB repair | Low MRE11 staining associated with worse survival following RT for MIBC; no association between MRE11 levels and survival in cystectomy patients | [40,41,42] |
| ERCC1 | NER | Low ERCC1 mRNA levels associated with improved survival in advanced/metastatic BC patients treated with cisplatin-based chemotherapy | [32] |
| High nuclear ERCC1 staining associated with worse survival in metastatic BC patients treated with cisplatin-based chemotherapy | [33] | ||
| ERCC2 | NER | Somatic ERCC2 missense mutations associated with improved pathologic response and survival in MIBC patients receiving neoadjuvant cisplatin-based chemotherapy followed by cystectomy | [43,44] |
| Somatic ERCC2 missense mutations associated with decreased metastatic recurrence rate in MIBC patients receiving chemoradiotherapy | [45] | ||
| >1 gene | DSB repair, others | Alteration(s) in ATM, RB1, or FANCC associated with improved pathologic response in MIBC patients receiving neoadjuvant cisplatin-based chemotherapy followed by cystectomy | [46] |
| Alteration(s) in ATM, ERCC2, FANCD2, PALB2, BRCA1, or BRCA2 associated with increased RFS in MIBC patients treated with cystectomy and peri-operative chemotherapy | [47] | ||
| Alteration(s) in ≥1 of 20 DDR genes associated with trend towards decreased recurrence in MIBC patients treated with chemoradiotherapy | [45] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouw, K.W. DNA Repair Pathway Alterations in Bladder Cancer. Cancers 2017, 9, 28. https://doi.org/10.3390/cancers9040028
Mouw KW. DNA Repair Pathway Alterations in Bladder Cancer. Cancers. 2017; 9(4):28. https://doi.org/10.3390/cancers9040028
Chicago/Turabian StyleMouw, Kent W. 2017. "DNA Repair Pathway Alterations in Bladder Cancer" Cancers 9, no. 4: 28. https://doi.org/10.3390/cancers9040028
APA StyleMouw, K. W. (2017). DNA Repair Pathway Alterations in Bladder Cancer. Cancers, 9(4), 28. https://doi.org/10.3390/cancers9040028