
Timp1 Promotes Cell Survival by Activating the PDK1 Signaling Pathway in Melanoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
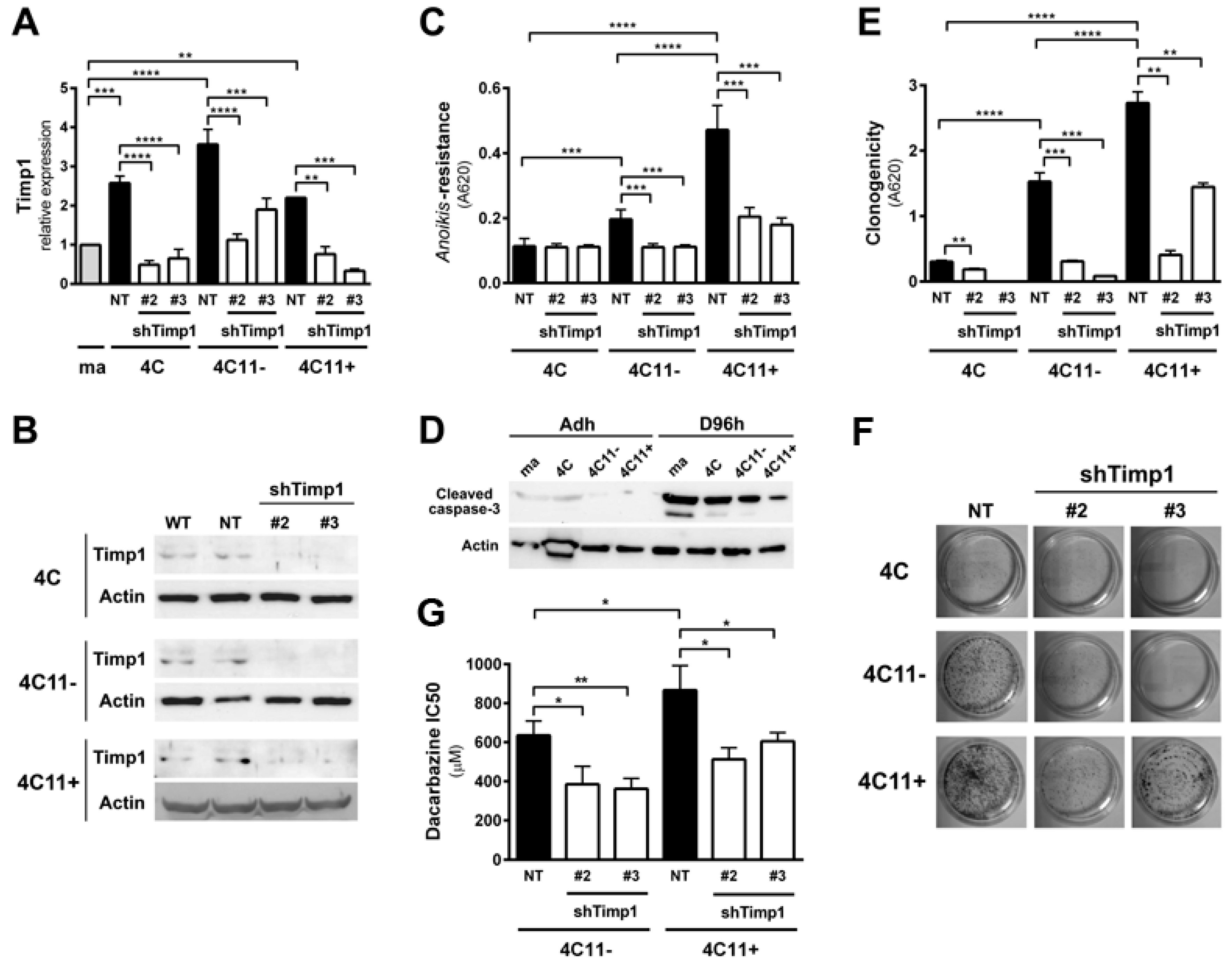
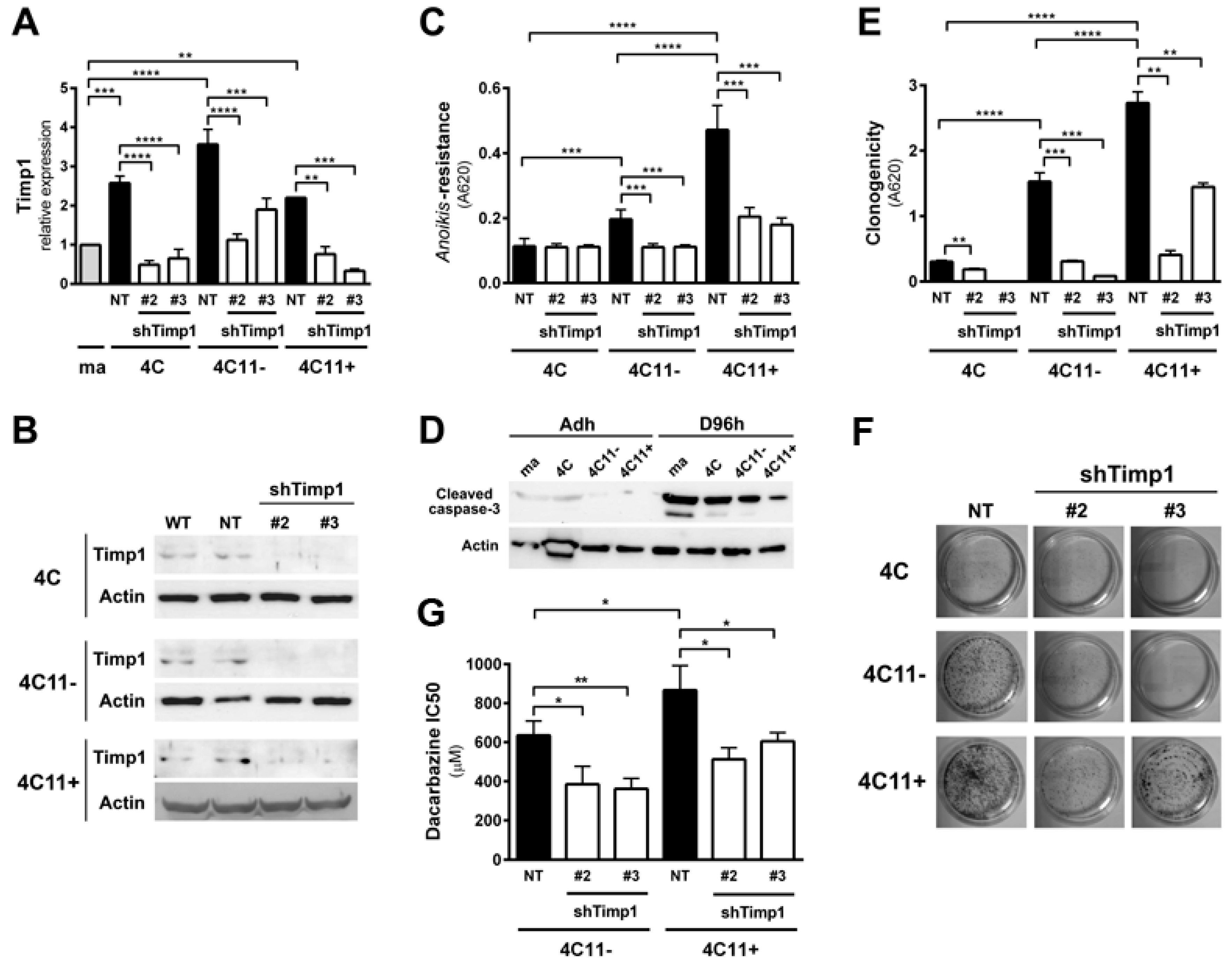
2.1. Timp1 Silencing Results in Decreased Cell Survival in Vitro along with Melanoma Progression
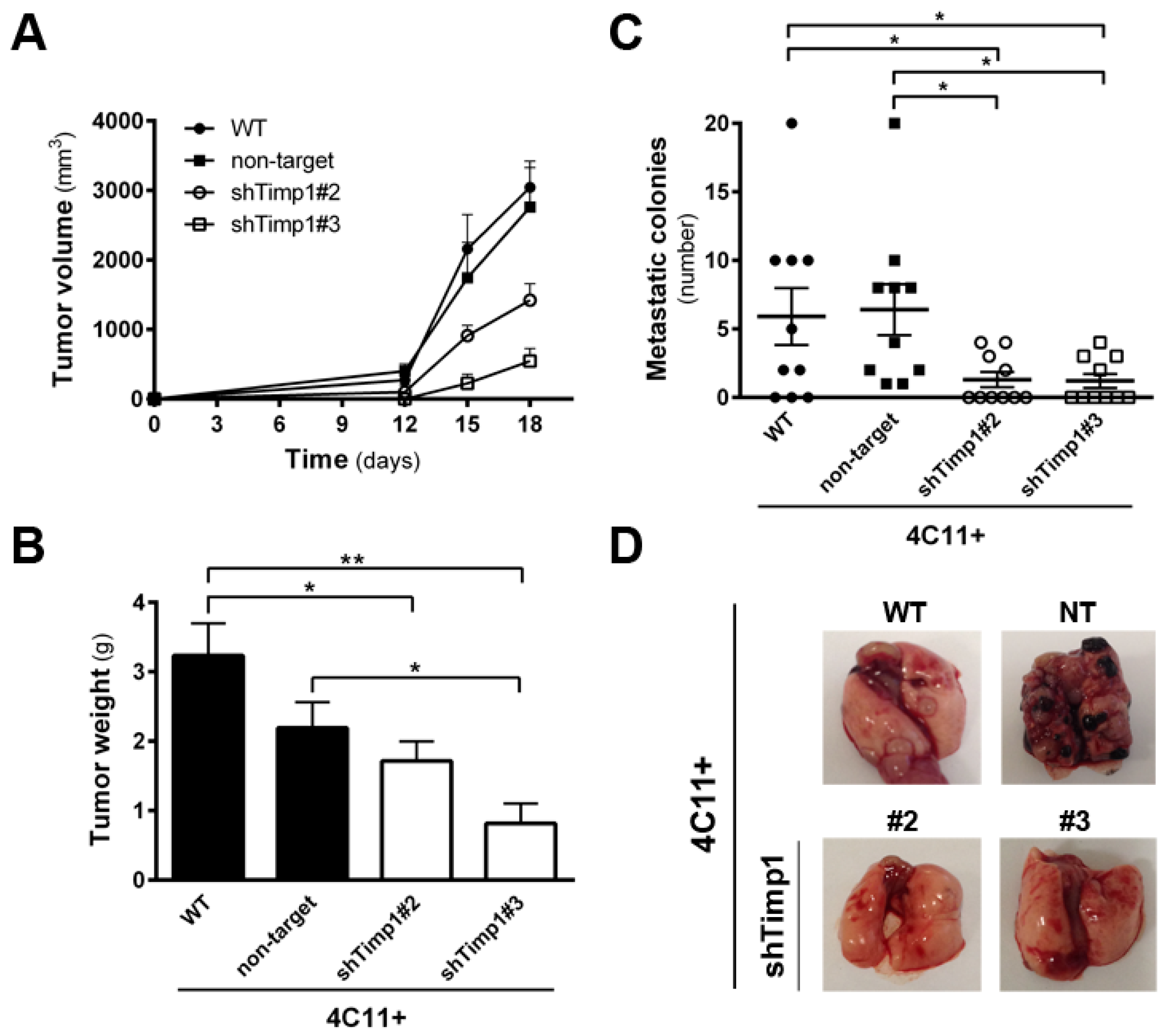
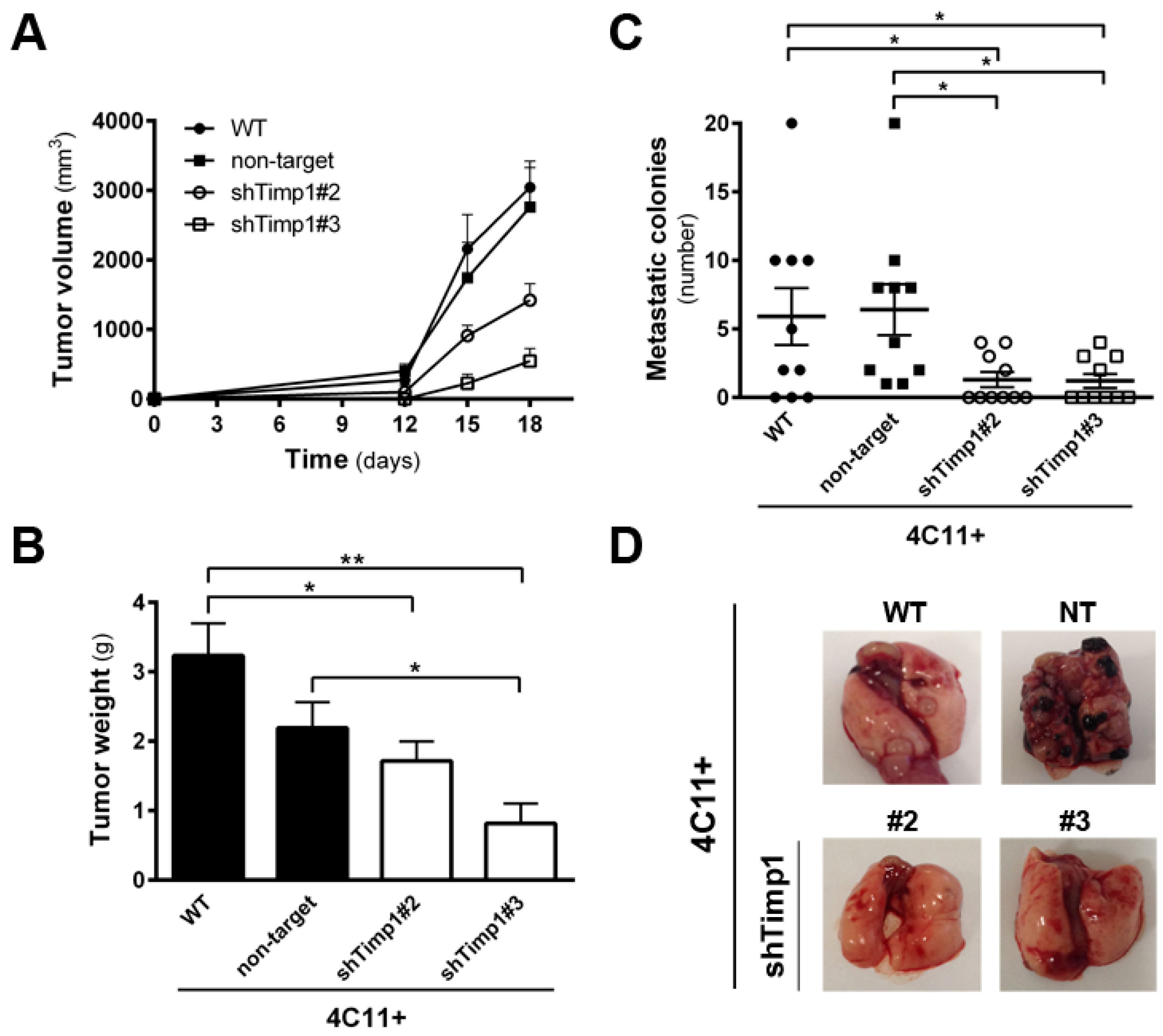
2.2. Timp1 Silencing Reverses the Aggressive Phenotype of Metastatic Melanoma Cells In Vivo
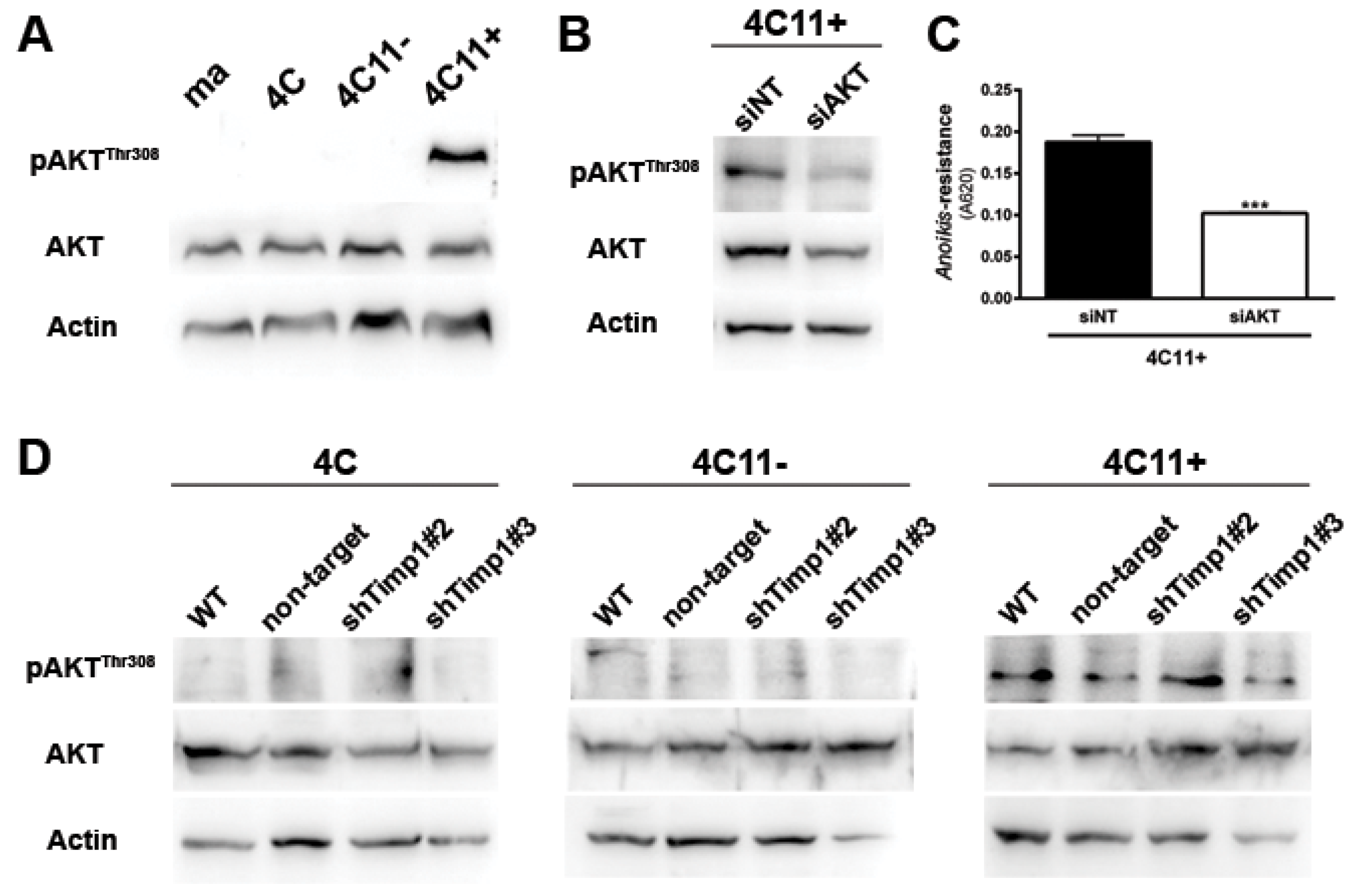
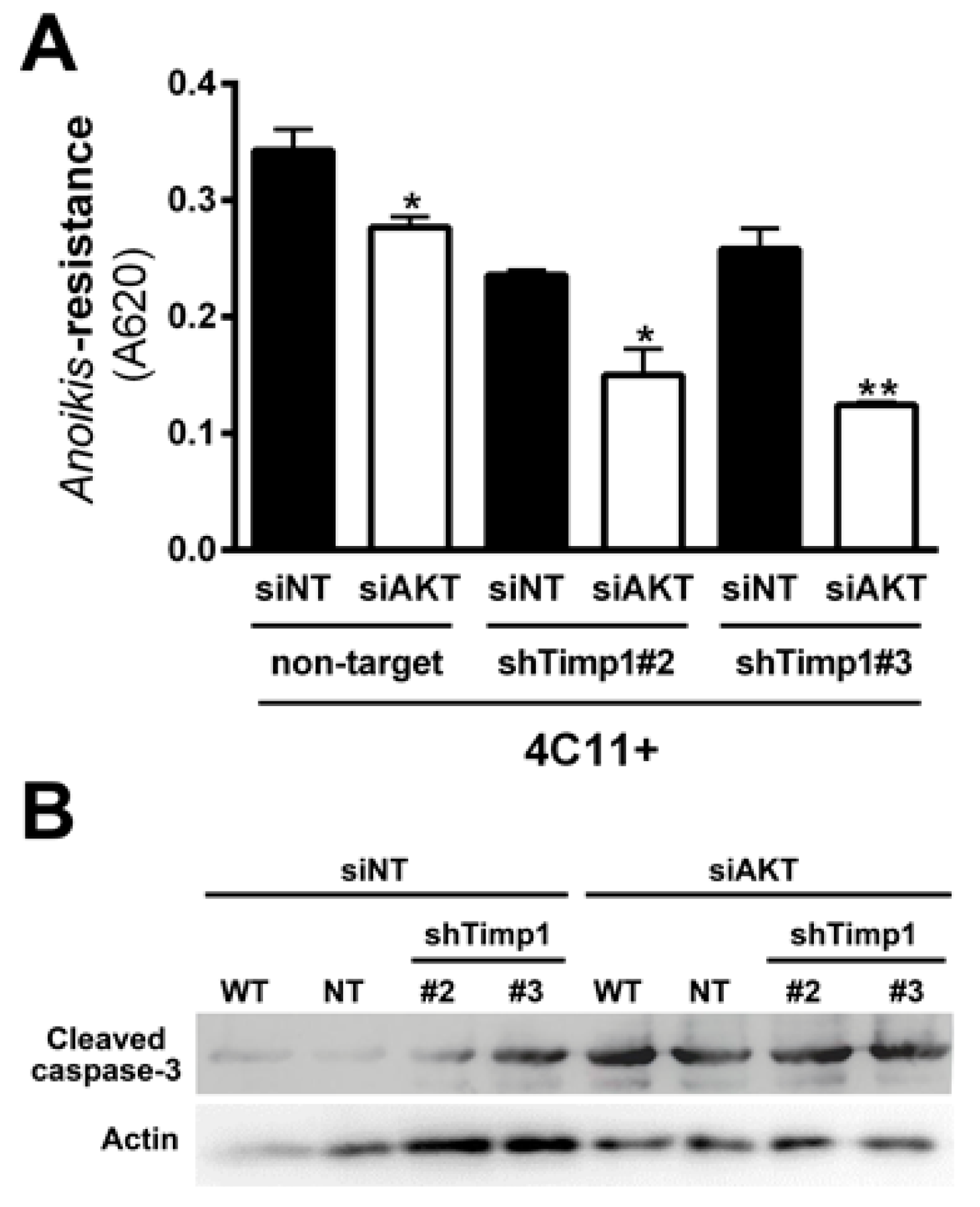
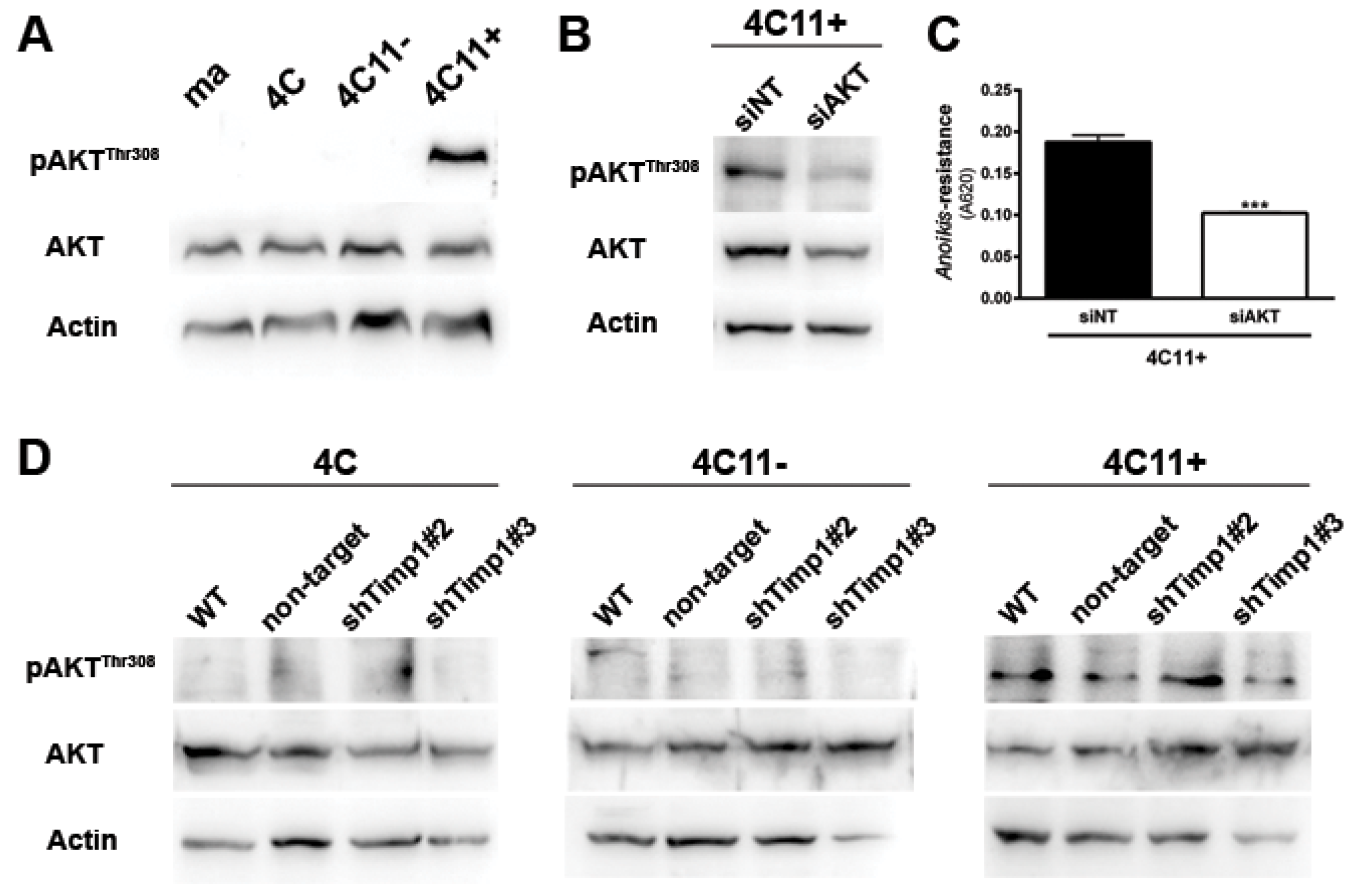
2.3. AKT Contributes to Anoikis Resistance in Metastatic Melanoma Cell Line Independently of Timp1
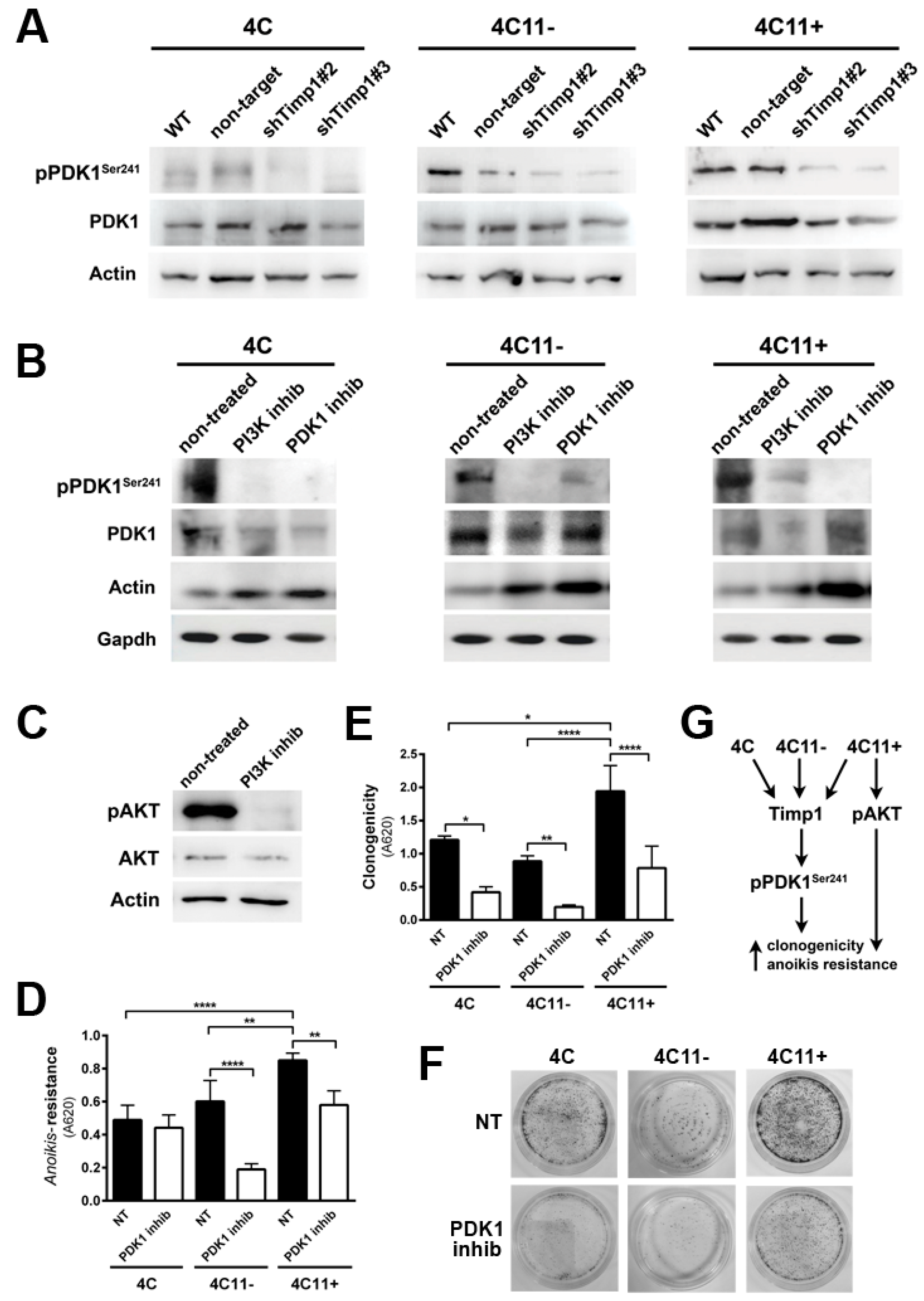
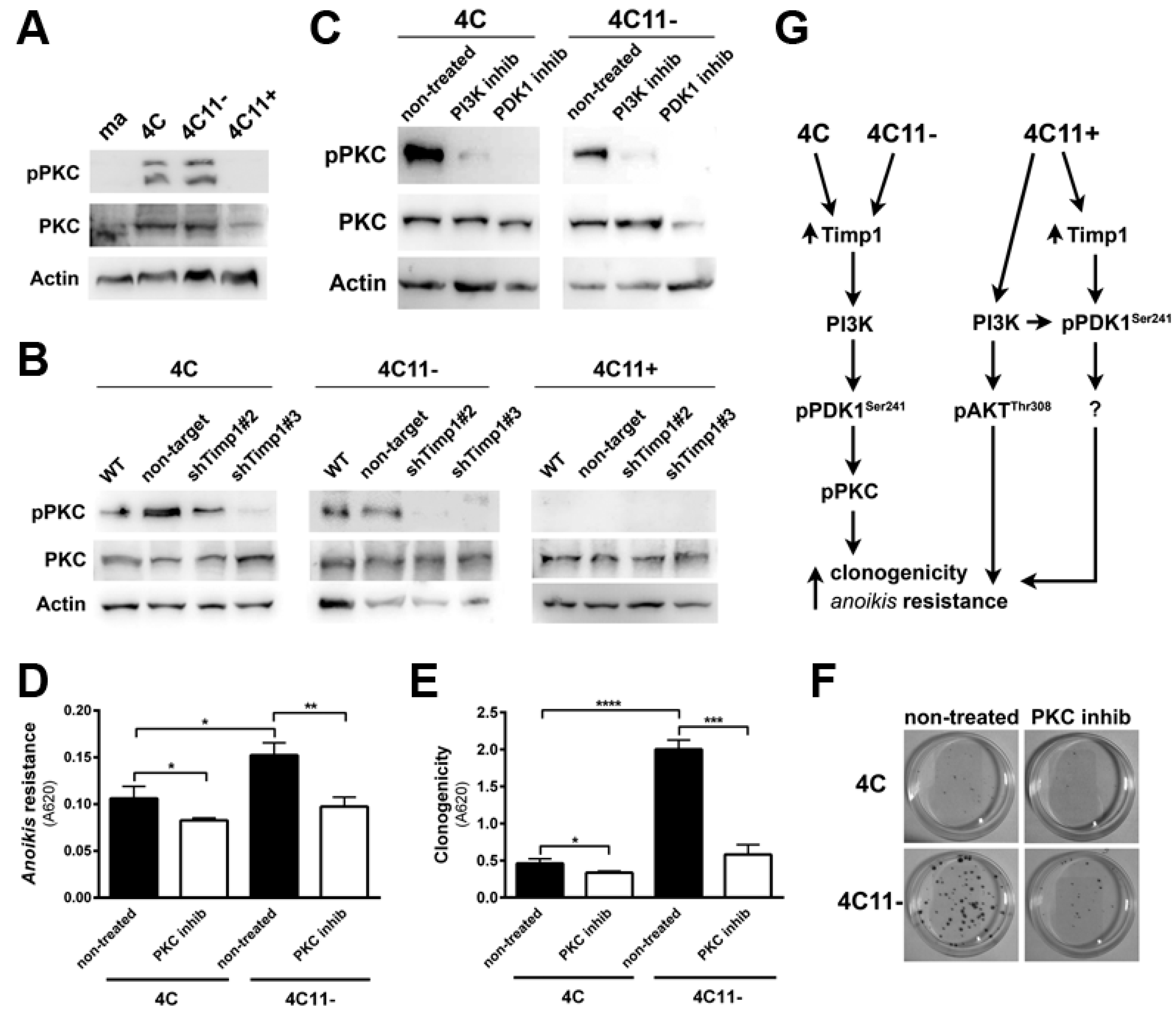
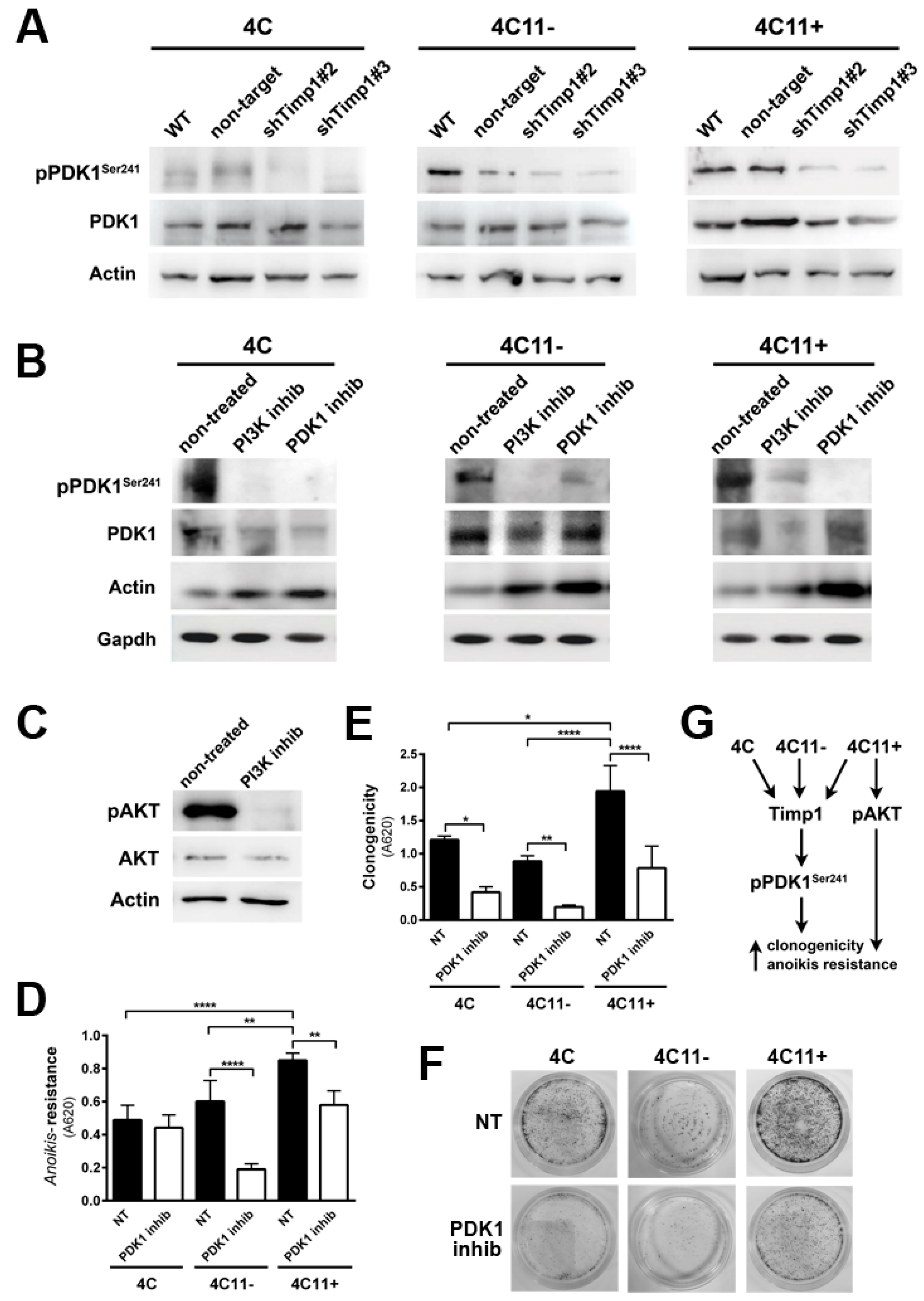
2.4. PDK1 Is Activated by Timp1 along Melanoma Progression and Contributes to Cell Survival
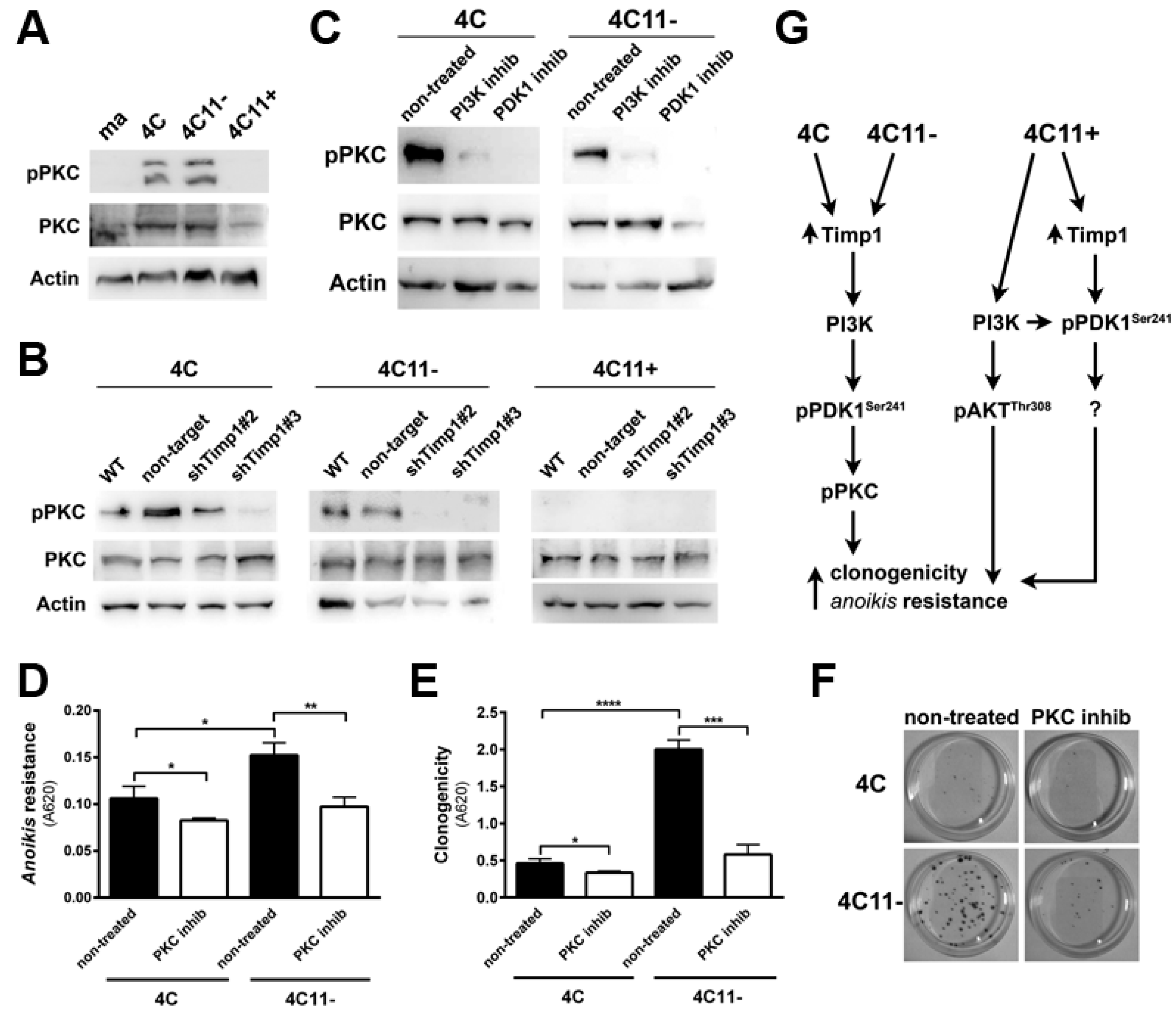
2.5. Timp1 Modulates the PKC Activation via PDK1 in the Early Stages of Melanoma
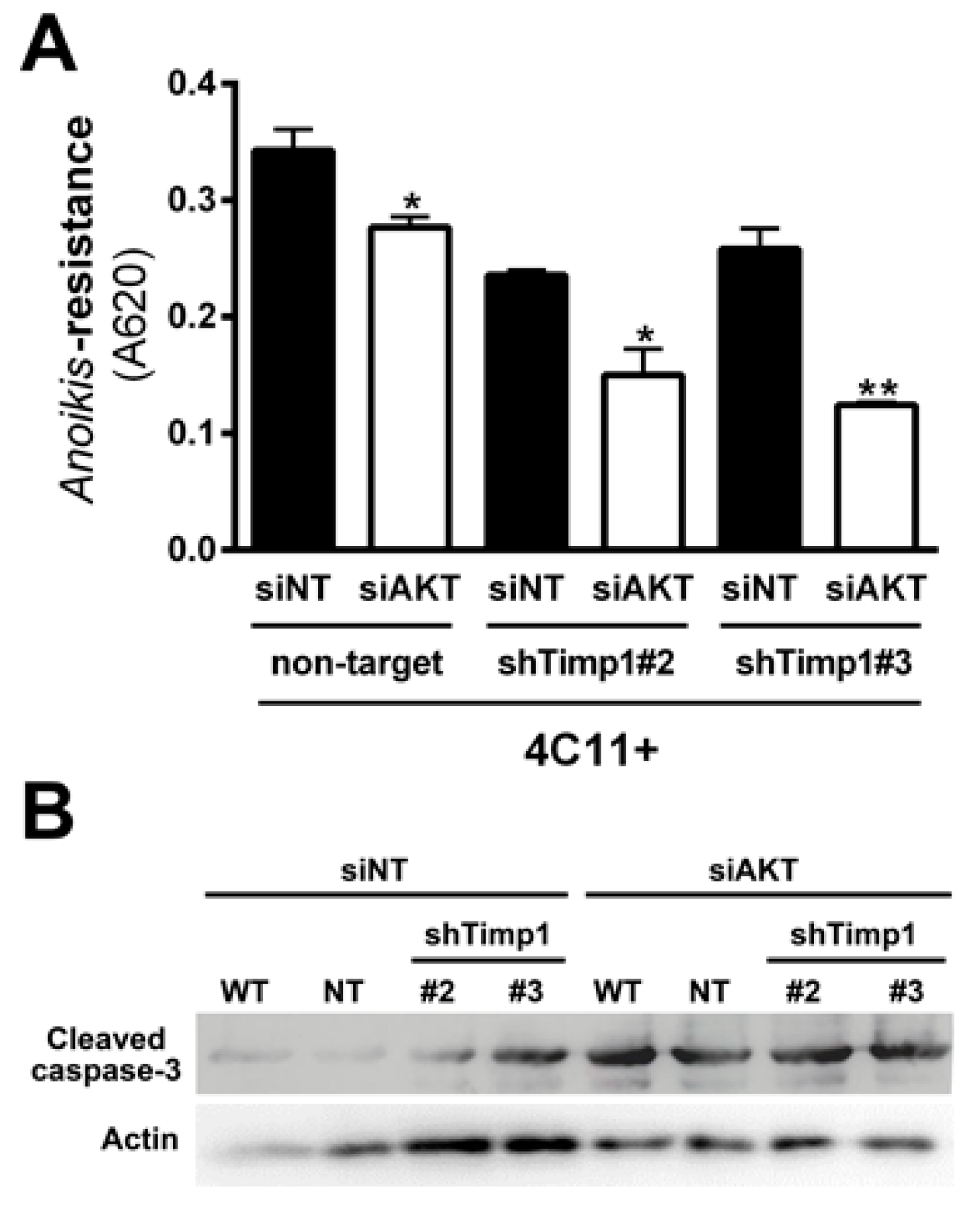
2.6. Simultaneous Inhibition of Timp1 and AKT Impairs More Efficiently the Survival of Metastatic Melanoma Cells
3. Discussion
4. Materials and Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berwick, M.; Buller, D.B.; Cust, A.; Gallagher, R.; Lee, T.K.; Meyskens, F.; Pandey, S.; Thomas, N.E.; Veierod, M.B.; Ward, S. Melanoma Epidemiology and Prevention. Cancer Treat Res. 2016, 167, 17–49. [Google Scholar] [PubMed]
- Palmieri, G.; Ombra, M.; Colombino, M.; Casula, M.; Sini, M.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front. Oncol. 2015, 5, 183. [Google Scholar] [CrossRef] [PubMed]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated AKT3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Fyffe, C.; Falasca, M. 3-Phosphoinositide-dependent protein kinase-1 as an emerging target in the management of breast cancer. Cancer Manag. Res. 2013, 5, 271–280. [Google Scholar] [PubMed]
- Du, J.; Yang, M.; Chen, S.; Li, D.; Chang, Z.; Dong, Z. PDK1 promotes tumor growth and metastasis in a spontaneous breast cancer model. Oncogene 2015, 35, 1018–1020. [Google Scholar] [CrossRef] [PubMed]
- Oba-Shinjo, S.M.; Correa, M.; Ricca, T.I.; Molognoni, F.; Pinhal, M.A.; Neves, I.A.; Marie, S.K.; Sampaio, L.O.; Nader, H.B.; Chammas, R.; et al. Melanocyte transformation associated with substrate adhesion impediment. Neoplasia 2006, 8, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Ricca, T.I.; Liang, G.; Suenaga, A.P.; Han, S.W.; Jones, P.A.; Jasiulionis, M.G. Tissue inhibitor of metalloproteinase 1 expression associated with gene demethylation confers anoikis resistance in early phases of melanocyte malignant transformation. Transl. Oncol. 2009, 2, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. Tissue inhibitors of metalloproteinases in cell signaling: Metalloproteinase-independent biological activities. Sci. Signal. 2008. [Google Scholar] [CrossRef] [PubMed]
- Toricelli, M.; Melo, F.H.; Peres, G.B.; Silva, D.C.; Jasiulionis, M.G. Timp1 interacts with beta-1 integrin and CD63 along melanoma genesis and confers anoikis resistance by activating PI3-K signaling pathway independently of Akt phosphorylation. Mol. Cancer 2013, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, H.; Glazer, R.I. Transformation of mammary epithelial cells by 3-phosphoinositide-dependent protein kinase-1 (PDK1) is associated with the induction of protein kinase Calpha. Cancer Res. 2002, 62, 3538–3543. [Google Scholar] [PubMed]
- Scortegagna, M.; Lau, E.; Zhang, T.; Feng, Y.; Sereduk, C.; Yin, H.; De, S.K.; Meeth, K.; Platt, J.T.; Langdon, C.G.; et al. PDK1 and SGK3 Contribute to the Growth of BRAF-Mutant Melanomas and Are Potential Therapeutic Targets. Cancer Res. 2015, 75, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Falasca, M. Targeting PDK1 in cancer. Curr. Med. Chem. 2011, 18, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.C.; Liu, X.W.; Najy, A.J.; Jung, Y.S.; Won, J.; Chai, K.X.; Fridman, R.; Kim, H.R. TIMP-1 via TWIST1 induces EMT phenotypes in human breast epithelial cells. Mol. Cancer Res. 2014, 12, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Dou, C.; Jia, Y.; Tu, K.; Zheng, X. TIMP-1 activated carcinoma-associated fibroblasts inhibit tumor apoptosis by activating SDF1/CXCR4 signaling in hepatocellular carcinoma. Oncotarget 2015, 6, 12061–12079. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, C.; Vinther, L.; Belling, K.C.; Wurtz, S.O.; Yadav, R.; Lademann, U.; Rigina, O.; Do, K.N.; Ditzel, H.J.; Lykkesfeldt, A.E.; et al. TIMP1 overexpression mediates resistance of MCF-7 human breast cancer cells to fulvestrant and down-regulates progesterone receptor expression. Tumour Biol. 2013, 34, 3839–3851. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Fan, X.; Hao, M.; Wang, J.; Zhou, X.; Sun, X. Higher levels of TIMP-1 expression are associated with a poor prognosis in triple-negative breast cancer. Mol. Cancer 2016, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Xu, S.; Zhang, H.; Wang, Y.; Xiao, C.; Jiang, T.; Wu, L.; Zhang, T.; Sun, X.; Zhong, L.; et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J. Exp. Clin. Cancer Res. 2016, 35, 148. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Hemmings, B.A. PIKKing on PKB: Regulation of PKB activity by phosphorylation. Curr. Opin. Cell Biol. 2009, 21, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Demel, H.R.; Feuerecker, B.; Piontek, G.; Seidl, C.; Blechert, B.; Pickhard, A.; Essler, M. Effects of topoisomerase inhibitors that induce DNA damage response on glucose metabolism and PI3K/Akt/mTOR signaling in multiple myeloma cells. Am. J. Cancer Res. 2015, 5, 1649–1664. [Google Scholar] [PubMed]
- Colaneri, G.N.; Cruz, A.T.; Silva, D.C.P.; Stilhano, R.S.; Han, S.W.; Jasiulionis, M.G.; Pharmacology Department, Universidade Federal de São Paulo, São Paulo, Brazil. Unpublished data. 2017.
- Foley, T.M.; Payne, S.N.; Pasch, C.A.; Yueh, A.E.; Van De Hey, D.R.; Korkos, D.P.; Clipson, L.; Maher, M.E.; Matkowskyj, K.A.; Newton, M.A.; et al. Dual PI3K/mTOR Inhibition in Colorectal Cancers with APC and PIK3CA Mutations. Mol. Cancer Res. 2017, 15, 317–327. [Google Scholar] [CrossRef]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Komander, D.; van Aalten, D.M.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bergami, P.; Kim, H.; Dewing, A.; Goydos, J.; Aaronson, S.; Ronai, Z. c-Jun regulates phosphoinositide-dependent kinase 1 transcription: Implication for Akt and protein kinase C activities and melanoma tumorigenesis. J. Biol. Chem. 2010, 285, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Ruller, C.; Feng, Y.; Lazova, R.; Kluger, H.; Li, J.L.; De, S.K.; Rickert, R.; Pellecchia, M.; Bosenberg, M.; et al. Genetic inactivation or pharmacological inhibition of Pdk1 delays development and inhibits metastasis of Braf(V600E)::Pten(-/-) melanoma. Oncogene 2014, 33, 4330–4339. [Google Scholar] [CrossRef] [PubMed]
- Iorns, E.; Lord, C.J.; Grigoriadis, A.; McDonald, S.; Fenwick, K.; Mackay, A.; Mein, C.A.; Natrajan, R.; Savage, K.; Tamber, N.; et al. Integrated functional, gene expression and genomic analysis for the identification of cancer targets. PLoS ONE 2009, 4, e5120. [Google Scholar] [CrossRef] [PubMed]
- Peifer, C.; Alessi, D.R. New anti-cancer role for PDK1 inhibitors: Preventing resistance to tamoxifen. Biochem. J. 2009, 417, e5–e7. [Google Scholar] [CrossRef] [PubMed]
- Denning, M.F. Specifying protein kinase C functions in melanoma. Pigment Cell Melanoma Res. 2012, 25, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Bekhite, M.M.; Finkensieper, A.; Binas, S.; Muller, J.; Wetzker, R.; Figulla, H.R.; Sauer, H.; Wartenberg, M. VEGF-mediated PI3K class IA and PKC signaling in cardiomyogenesis and vasculogenesis of mouse embryonic stem cells. J. Cell Sci. 2011, 124, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.S.; Sutton, C.R.; Rao, S. Protein kinase C in the immune system: From signalling to chromatin regulation. Immunology 2015, 146, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Carita, G.; Frisch-Dit-Leitz, E.; Dahmani, A.; Raymondie, C.; Cassoux, N.; Piperno-Neumann, S.; Nemati, F.; Laurent, C.; De Koning, L.; Halilovic, E.; et al. Dual inhibition of protein kinase C and p53-MDM2 or PKC and mTORC1 are novel efficient therapeutic approaches for uveal melanoma. Oncotarget 2016, 7, 33542–33556. [Google Scholar] [CrossRef]
- Halder, K.; Banerjee, S.; Bose, A.; Majumder, S.; Majumdar, S. Overexpressed PKCdelta downregulates the expression of PKCalpha in B16F10 melanoma: Induction of apoptosis by PKCdelta via ceramide generation. PLoS ONE 2014, 9, e91656. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, R.F.; Paul, M.A.; Valmaseda, A.; Francois, A.; Jabr, R.; Anjum, S.; Marber, M.S.; Budhram-Mahadeo, V.; Heads, R.J. Protein kinase Cepsilon-calcineurin cosignaling downstream of toll-like receptor 4 downregulates fibrosis and induces wound healing gene expression in cardiac myofibroblasts. Mol. Cell. Biol. 2014, 34, 574–594. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.C.; Cooper, P.J.; Hart, I.R. A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int. J. Cancer 1987, 39, 414–418. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toricelli, M.; Melo, F.H.M.; Hunger, A.; Zanatta, D.; Strauss, B.E.; Jasiulionis, M.G. Timp1 Promotes Cell Survival by Activating the PDK1 Signaling Pathway in Melanoma. Cancers 2017, 9, 37. https://doi.org/10.3390/cancers9040037
Toricelli M, Melo FHM, Hunger A, Zanatta D, Strauss BE, Jasiulionis MG. Timp1 Promotes Cell Survival by Activating the PDK1 Signaling Pathway in Melanoma. Cancers. 2017; 9(4):37. https://doi.org/10.3390/cancers9040037
Chicago/Turabian StyleToricelli, Mariana, Fabiana H. M. Melo, Aline Hunger, Daniela Zanatta, Bryan E. Strauss, and Miriam G. Jasiulionis. 2017. "Timp1 Promotes Cell Survival by Activating the PDK1 Signaling Pathway in Melanoma" Cancers 9, no. 4: 37. https://doi.org/10.3390/cancers9040037
APA StyleToricelli, M., Melo, F. H. M., Hunger, A., Zanatta, D., Strauss, B. E., & Jasiulionis, M. G. (2017). Timp1 Promotes Cell Survival by Activating the PDK1 Signaling Pathway in Melanoma. Cancers, 9(4), 37. https://doi.org/10.3390/cancers9040037