
Towards Targeting PI3K-Dependent Regulation of Gene Expression in Brain Cancer


{kind=link}
Abstract
:1. Introduction
2. The PI3K Pathway in Cancer
2.1. The Pi3k Pathway in Tumor Heterogeneity
2.2. The PI3K Pathway in High-Grade Brain Cancer
2.2.1. Current Treatments for Malignant Brain Cancer Are Inadequate
2.2.2. Promising Pre-Clinical and Clinical Trial Outcomes Using PI3K Pathway Inhibitors
2.3. The PI3K Pathway and Transcriptional Regulation via FOXO, NFκB and CREB
2.3.1. FOXO
2.3.2. NFκB
2.3.3. CREB
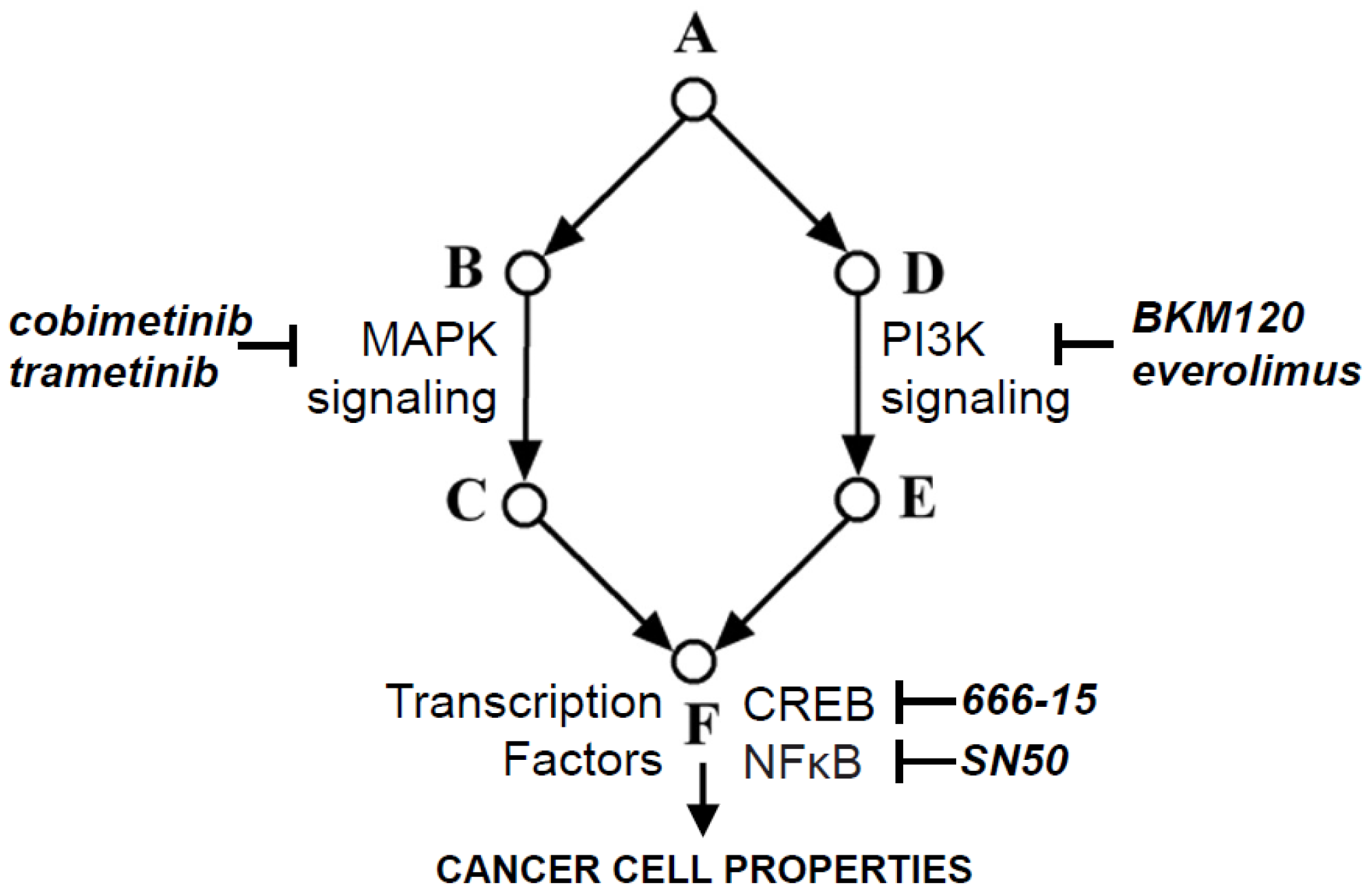
2.4. Relevance of PI3K Signaling-Transcriptional Networks to Novel Therapeutic Strategies in Brain Cancer
2.4.1. Targeting PI3K and NFκB Signaling
2.4.2. Targeting PI3K and CREB Signaling
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Schwindinger, W.F.; Robishaw, J.D. Heterotrimeric g-protein betagamma-dimers in growth and differentiation. Oncogene 2001, 20, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Schafer, B.; Gschwind, A.; Ullrich, A. Multiple g-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene 2004, 23, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, L.M. Brain tumors. N. Engl. J. Med. 2001, 344, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.J.; Campbell, I.G.; Leet, C.; Vincan, E.; Rockman, S.P.; Whitehead, R.H.; Thomas, R.J.; Phillips, W.A. The phosphatidylinositol 3'-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001, 61, 7426–7429. [Google Scholar] [PubMed]
- Phillips, W.A.; Russell, S.E.; Ciavarella, M.L.; Choong, D.Y.; Montgomery, K.G.; Smith, K.; Pearson, R.B.; Thomas, R.J.; Campbell, I.G. Mutation analysis of PIK3CA and PIK3CB in esophageal cancer and barrett's esophagus. Int. J. Cancer 2006, 118, 2644–2646. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.G.; Russell, S.E.; Choong, D.Y.; Montgomery, K.G.; Ciavarella, M.L.; Hooi, C.S.; Cristiano, B.E.; Pearson, R.B.; Phillips, W.A. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004, 64, 7678–7681. [Google Scholar] [CrossRef] [PubMed]
- Kinross, K.M.; Montgomery, K.G.; Kleinschmidt, M.; Waring, P.; Ivetac, I.; Tikoo, A.; Saad, M.; Hare, L.; Roh, V.; Mantamadiotis, T.; et al. An activating PIK3CA mutation coupled with pten loss is sufficient to initiate ovarian tumorigenesis in mice. J. Clin. Investig. 2012, 122, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Stawiski, E.; Janakiraman, V.; Chan, E.; Durinck, S.; Edgar, K.A.; Kljavin, N.M.; Rivers, C.S.; Gnad, F.; Roose-Girma, M.; et al. Conditional activation of PIK3CA(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene 2013, 32, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohee, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Ozawa, T.; Aldape, K.D.; Deen, D.F.; Berger, M.S.; Pieper, R.O. AKT pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 2001, 61, 6674–6678. [Google Scholar] [PubMed]
- Kita, D.; Yonekawa, Y.; Weller, M.; Ohgaki, H. PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 2007, 113, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Pelloski, C.E.; Lin, E.; Zhang, L.; Yung, W.K.; Colman, H.; Liu, J.L.; Woo, S.Y.; Heimberger, A.B.; Suki, D.; Prados, M.; et al. Prognostic associations of activated mitogen-activated protein kinase and AKT pathways in glioblastoma. Clin. Cancer Res. 2006, 12, 3935–3941. [Google Scholar] [CrossRef] [PubMed]
- Broderick, D.K.; Di, C.; Parrett, T.J.; Samuels, Y.R.; Cummins, J.M.; McLendon, R.E.; Fults, D.W.; Velculescu, V.E.; Bigner, D.D.; Yan, H. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004, 64, 5048–5050. [Google Scholar] [CrossRef] [PubMed]
- Duerr, E.M.; Rollbrocker, B.; Hayashi, Y.; Peters, N.; Meyer-Puttlitz, B.; Louis, D.N.; Schramm, J.; Wiestler, O.D.; Parsons, R.; Eng, C.; et al. Pten mutations in gliomas and glioneuronal tumors. Oncogene 1998, 16, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Trejo, C.L.; Green, S.; Marsh, V.; Collisson, E.A.; Iezza, G.; Phillips, W.A.; McMahon, M. Mutationally activated PIK3CA(H1047R) cooperates with BRAF(V600E) to promote lung cancer progression. Cancer Res. 2013, 73, 6448–6461. [Google Scholar] [CrossRef] [PubMed]
- Hare, L.M.; Phesse, T.J.; Waring, P.M.; Montgomery, K.G.; Kinross, K.M.; Mills, K.; Roh, V.; Heath, J.K.; Ramsay, R.G.; Ernst, M.; et al. Physiological expression of the PI3K-activating mutation PIK3CAH1047R combines with APC loss to promote development of invasive intestinal adenocarcinomas in mice. Biochem. J. 2014, 458, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Mantamadiotis, T.; University of Melbourne, Parkville, Victoria, Australia. Unpublished data. 2017.
- Gunther, W.; Pawlak, E.; Damasceno, R.; Arnold, H.; Terzis, A.J. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br. J. Cancer 2003, 88, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Schiff, D. Treatment considerations for MGMT-unmethylated glioblastoma. Curr. Neurol. Neurosci. Rep. 2015, 15, 507. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/AKT/mtor signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed]
- Koul, D.; Fu, J.; Shen, R.; LaFortune, T.A.; Wang, S.; Tiao, N.; Kim, Y.W.; Liu, J.L.; Ramnarian, D.; Yuan, Y.; et al. Antitumor activity of NVP-BKM120—A selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin. Cancer Res. 2012, 18, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Pitz, M.W.; Eisenhauer, E.A.; MacNeil, M.V.; Thiessen, B.; Easaw, J.C.; Macdonald, D.R.; Eisenstat, D.D.; Kakumanu, A.S.; Salim, M.; Chalchal, H.; et al. Phase II study of PX-866 in recurrent glioblastoma. Neurooncology 2015, 17, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neurooncology 2015, 17, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Burgering, B.M.; Kops, G.J. Cell cycle and death control: Long live forkheads. Trends Biochem. Sci. 2002, 27, 352–360. [Google Scholar] [CrossRef]
- Biggs, W.H., 3rd; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase B/AKT-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. AKT promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Perrot, V.; Rechler, M.M. The coactivator p300 directly acetylates the forkhead transcription factor FOXO1 and stimulates foxo1-induced transcription. Mol. Endocrinol. (Baltimore, Md.) 2005, 19, 2283–2298. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Tanaka, K.; Fukamizu, A. Insulin-induced phosphorylation of FKHR (FOXO1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. USA 2003, 100, 11285–11290. [Google Scholar] [CrossRef] [PubMed]
- Plas, D.R.; Thompson, C.B. AKT activation promotes degradation of tuberin and FOXO3A via the proteasome. J. Biol. Chem. 2003, 278, 12361–12366. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like forkhead transcription factors mediate cell-cycle regulation by RAS and PKB through P27KIP1. Nature 2000, 404, 782–787. [Google Scholar] [PubMed]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.; Lam, E.W.; Burgering, B.M.; Medema, R.H. Cell cycle inhibition by FOXO forkhead transcription factors involves downregulation of cyclin d. Mol. Cell Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.; Malla, R.R.; Gondi, C.S.; Alapati, K.; Fassett, D.; Klopfenstein, J.D.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Co-depletion of cathepsin B and UPAR induces G0/G1 arrest in glioma via FOXO3A mediated p27 upregulation. PLoS ONE 2010, 5, e11668. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.J.; Koty, Z.; Nalbantoglu, J. Differential response of glioma cells to FOXO1-directed therapy. Cancer Res. 2009, 69, 5433–5440. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.V.; Shen, L.; Anderson, S.A.; Massague, J. Integration of SMAD and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Dynamic FOXO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Dansen, T.B.; Burgering, B.M. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008, 18, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Ren, L.; Wu, D.; Yang, X.; Zhou, Z.; Nie, Q.; Jiang, G.; Xue, S.; Weng, W.; Qiu, Y.; et al. Overexpression of FOXO3A is associated with glioblastoma progression and predicts poor patient prognosis. Int. J. Cancer 2017, 140, 2792–2804. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhang, Z.; Pei, H.; Wang, H.; Li, L.; Xia, Q. FOXO3A induces temozolomide resistance in glioblastoma cells via the regulation of beta-catenin nuclear accumulation. Oncol. Rep. 2017, 37, 2391–2397. [Google Scholar] [PubMed]
- Aggarwal, B.B. Nuclear factor-kappaB: The enemy within. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. Nf-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. Nf-kappaB activation by tumour necrosis factor requires the AKT serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [PubMed]
- Kane, L.P.; Mollenauer, M.N.; Xu, Z.; Turck, C.W.; Weiss, A. AKT-dependent phosphorylation specifically regulates COT induction of NF-kappa B-dependent transcription. Mol. Cell Biol. 2002, 22, 5962–5974. [Google Scholar] [CrossRef] [PubMed]
- Madrid, L.V.; Mayo, M.W.; Reuther, J.Y.; Baldwin, A.S., Jr. AKT stimulates the transactivation potential of the rela/p65 subunit of NF-kappa b through utilization of the ikappa B kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 2001, 276, 18934–18940. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, N.; Leung, S.; Stark, G.R. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappab p65/rela subunit. Mol. Cell Biol. 1999, 19, 4798–4805. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Domon-Dell, C.; Kang, J.; Chung, D.H.; Freund, J.N.; Evers, B.M. Down-regulation of the tumor suppressor pten by the tumor necrosis factor-alpha/nuclear factor-kappaB (NF-kappaB)-inducing kinase/NF-kappab pathway is linked to a default ikappaB-alpha autoregulatory loop. J. Biol. Chem. 2004, 279, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Astort, F.; Repetto, E.M.; Rocha-Viegas, L.; Mercau, M.E.; Puch, S.S.; Finkielstein, C.V.; Pecci, A.; Cymeryng, C.B. Role of CREB on heme oxygenase-1 induction in adrenal cells: Involvement of the PI3K pathway. J. Mol. Endocrinol. 2016, 57, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Liptay, S.; Weber, C.K.; Ludwig, L.; Wagner, M.; Adler, G.; Schmid, R.M. Mitogenic and antiapoptotic role of constitutive NF-kappaB/REL activity in pancreatic cancer. Int. J. Cancer 2003, 105, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, G.S.; Zhan, Y.; Johnson, G.R.; O'Rourke, D.M. Distinct domains in the SHP-2 phosphatase differentially regulate epidermal growth factor receptor/nf-kappab activation through GAB1 in glioblastoma cells. Mol. Cell Biol. 2004, 24, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Shimamura, T.; Barbera, S.; Bejcek, B.E. Nf-kappab controls growth of glioblastomas/ astrocytomas. Mol. Cell Biochem. 2008, 307, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Mechanisms and consequences of activation of protein kinase B/AKT. Curr. Opin. Cell. Biol. 1998, 10, 262–267. [Google Scholar] [CrossRef]
- Bredel, M.; Scholtens, D.M.; Yadav, A.K.; Alvarez, A.A.; Renfrow, J.J.; Chandler, J.P.; Yu, I.L.; Carro, M.S.; Dai, F.; Tagge, M.J.; et al. NFkBIA deletion in glioblastomas. N. Engl. J. Med. 2011, 364, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.; Ruiz-Ontanon, P.; Vazquez-Barquero, A.; Moris, F.; Fernandez-Luna, J.L. The NFkappaB pathway: A therapeutic target in glioblastoma. Oncotarget 2011, 2, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Filiz, G.; Brown, D.V.; Hollande, F.; Gonzales, M.; D'Abaco, G.; Papalexis, N.; Phillips, W.A.; Malaterre, J.; Ramsay, R.G.; et al. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis 2014, 3, e108. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jiang, J.; Badawi, M.; Schmittgen, T.D. Mir-221 regulates CD44 in hepatocellular carcinoma through the PI3K-AKT-MTOR pathway. Biochem. Biophys. Res. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, S.; Heath, J.K.; deJong-Curtain, T.A.; Hogan, B.M.; Lieschke, G.J.; Malaterre, J.; Ramsay, R.G.; Mantamadiotis, T. CREB activity modulates neural cell proliferation, midbrain-hindbrain organization and patterning in zebrafish. Dev. Biol. 2007, 307, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, S.; Malaterre, J.; Hollande, F.; Darcy, P.K.; Ramsay, R.G.; Mantamadiotis, T. CAMP response element binding protein is required for mouse neural progenitor cell survival and expansion. Stem Cells 2009, 27, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.R. The molecular biology of memory: CAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 2012, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Wiskott, L.; Gage, F.H. Functional significance of adult neurogenesis. Curr. Opin. Neurobiol. 2004, 14, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Mantamadiotis, T.; Lemberger, T.; Bleckmann, S.C.; Kern, H.; Kretz, O.; Martin Villalba, A.; Tronche, F.; Kellendonk, C.; Gau, D.; Kapfhammer, J.; et al. Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 2002, 31, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Shankar, D.B.; Sakamoto, K.M. The role of Cyclic-AMP binding protein (CREB) in leukemia cell proliferation and acute leukemias. Leuk. Lymphoma 2004, 45, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch, R.; Tavor, E.; Jacob-Hirsch, J.; Zeira, E.; Amariglio, N.; Pappo, O.; Rechavi, G.; Galun, E.; Honigman, A. A pivotal role of cyclic AMP-responsive element binding protein in tumor progression. Cancer Res. 2004, 64, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Kim, S.W.; Ryu, S.H.; Chung, W.C.; Koo, J.S. Growth suppression of lung cancer cells by targeting cyclic AMP response element-binding protein. Cancer Res. 2008, 68, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, A.; Fernando, H.; Watkins, G.; Mansel, R.E.; Jiang, W.G. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol. Rep. 2007, 18, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.S.; Liu, D.D.; Bekele, B.N.; Kim, M.K.; Pisters, K.; Lippman, S.M.; Wistuba, I.I.; Koo, J.S. Cyclic amp response element-binding protein overexpression: A feature associated with negative prognosis in never smokers with non-small cell lung cancer. Cancer Res. 2008, 68, 6065–6073. [Google Scholar] [CrossRef] [PubMed]
- Gubbay, O.; Rae, M.T.; McNeilly, A.S.; Donadeu, F.X.; Zeleznik, A.J.; Hillier, S.G. CAMP response element-binding (CREB) signalling and ovarian surface epithelial cell survival. J. Endocrinol. 2006, 191, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Peltier, J.; O'Neill, A.; Schaffer, D.V. PI3K/AKT and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 2007, 67, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Zhang, Z.; Wang, J.; Guo, J.; Shen, W.H.; Yin, Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011, 71, 2821–2825. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Le Joncour, V.; Tonon, M.C.; Anouar, Y.; Proust, F.; Morin, F.; Gandolfo, P.; Joly, F.; Hilber, P.; Castel, H. Evaluation of the impact of the cancer therapy everolimus on the central nervous system in mice. PLoS ONE 2014, 9, e113533. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.J.; Ramsay, R.G. Directly targeting transcriptional dysregulation in cancer. Nat. Rev. Cancer 2015, 15, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Borgohain, M.P.; Lahkar, M.; Ahmed, S.; Chowdhury, L.; Kumar, S.; Pant, R.; Choubey, A. Small molecule inhibiting nuclear factor-kB ameliorates oxidative stress and suppresses renal inflammation in early stage of alloxan-induced diabetic nephropathy in rat. Basic Clin. Pharmacol. Toxicol. 2017, 120, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Kanak, M.A.; Shahbazov, R.; Yoshimatsu, G.; Levy, M.F.; Lawrence, M.C.; Naziruddin, B. A small molecule inhibitor of NFkappaB blocks er stress and the NLRP3 inflammasome and prevents progression of pancreatitis. J. Gastroenterol. 2017, 52, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Kastrati, I.; Siklos, M.I.; Calderon-Gierszal, E.L.; El-Shennawy, L.; Georgieva, G.; Thayer, E.N.; Thatcher, G.R.; Frasor, J. Dimethyl fumarate inhibits the nuclear factor kappaB pathway in breast cancer cells by covalent modification of p65 protein. J. Biol. Chem. 2016, 291, 3639–3647. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Lafuente, N.; Alcaraz-Garcia, M.J.; Sebastian-Ruiz, S.; Gomez-Espuch, J.; Funes, C.; Moraleda, J.M.; Garcia-Garay, M.C.; Montes-Barqueros, N.; Minguela, A.; Alvarez-Lopez, M.R.; et al. The gene expression response of chronic lymphocytic leukemia cells to Il-4 is specific, depends on ZAP-70 status and is differentially affected by an nfkappab inhibitor. PLoS OME 2014, 9, e109533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ren, X.; Cheng, Y.; Liu, X.; Allen, J.E.; Zhang, Y.; Yuan, Y.; Huang, S.Y.; Yang, W.; Berg, A.; et al. The NFkappaB inhibitor, SN50, induces differentiation of glioma stem cells and suppresses their oncogenic phenotype. Cancer Biol. Ther. 2014, 15, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Zanotto-Filho, A.; Braganhol, E.; Battastini, A.M.; Moreira, J.C. Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/AKT and NFkappaB pathways, mitochondrial dysfunction, and activation of p38-JNK1/2 signaling. Investig. New Drugs 2012, 30, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Zanotto-Filho, A.; Braganhol, E.; Schroder, R.; de Souza, L.H.; Dalmolin, R.J.; Pasquali, M.A.; Gelain, D.P.; Battastini, A.M.; Moreira, J.C. NFkappaB inhibitors induce cell death in glioblastomas. Biochem. Pharmacol. 2011, 81, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Suppression of the invasive potential of glioblastoma cells by mtor inhibitors involves modulation of NFkappaB and PKC-alpha signaling. Sci. Rep. 2016, 6, 22455. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Li, B.X.; Kassenbrock, A.; Xue, C.; Wang, X.; Qian, D.Z.; Sears, R.C.; Xiao, X. Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J. Med. Chem. 2015, 58, 5075–5087. [Google Scholar] [CrossRef] [PubMed]
- Li, B.X.; Gardner, R.; Xue, C.; Qian, D.Z.; Xie, F.; Thomas, G.; Kazmierczak, S.C.; Habecker, B.A.; Xiao, X. Systemic inhibition of CREB is well-tolerated in vivo. Sci. Rep. 2016, 6, 34513. [Google Scholar] [CrossRef] [PubMed]
- Mitton, B.; Chae, H.D.; Hsu, K.; Dutta, R.; Aldana-Masangkay, G.; Ferrari, R.; Davis, K.; Tiu, B.C.; Kaul, A.; Lacayo, N.; et al. Small molecule inhibition of cAMP response element binding protein in human acute myeloid leukemia cells. Leukemia 2016, 30, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, H.S.; Park, S.A.; Ryu, S.H.; Meng, W.; Jurgensmeier, J.M.; Kurie, J.M.; Hong, W.K.; Boyer, J.L.; Herbst, R.S.; et al. A novel small-molecule inhibitor targeting CREB-CBP complex possesses anti-cancer effects along with cell cycle regulation, autophagy suppression and endoplasmic reticulum stress. PLoS ONE 2015, 10, e0122628. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanomas. Pharmacol. Res. 2017, 117, 20–31. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantamadiotis, T. Towards Targeting PI3K-Dependent Regulation of Gene Expression in Brain Cancer. Cancers 2017, 9, 60. https://doi.org/10.3390/cancers9060060
Mantamadiotis T. Towards Targeting PI3K-Dependent Regulation of Gene Expression in Brain Cancer. Cancers. 2017; 9(6):60. https://doi.org/10.3390/cancers9060060
Chicago/Turabian StyleMantamadiotis, Theo. 2017. "Towards Targeting PI3K-Dependent Regulation of Gene Expression in Brain Cancer" Cancers 9, no. 6: 60. https://doi.org/10.3390/cancers9060060
APA StyleMantamadiotis, T. (2017). Towards Targeting PI3K-Dependent Regulation of Gene Expression in Brain Cancer. Cancers, 9(6), 60. https://doi.org/10.3390/cancers9060060