
Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
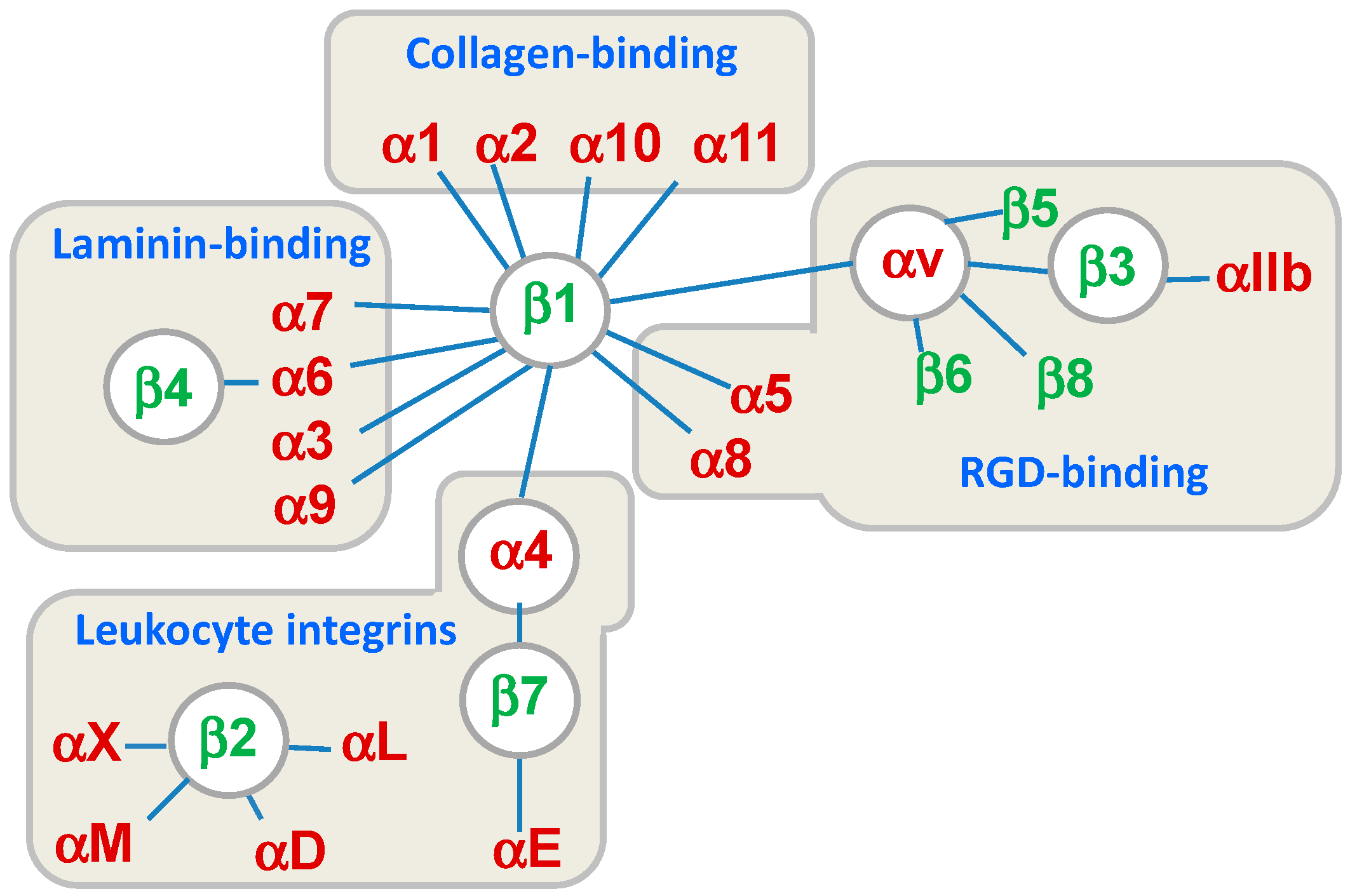
:1. Introduction
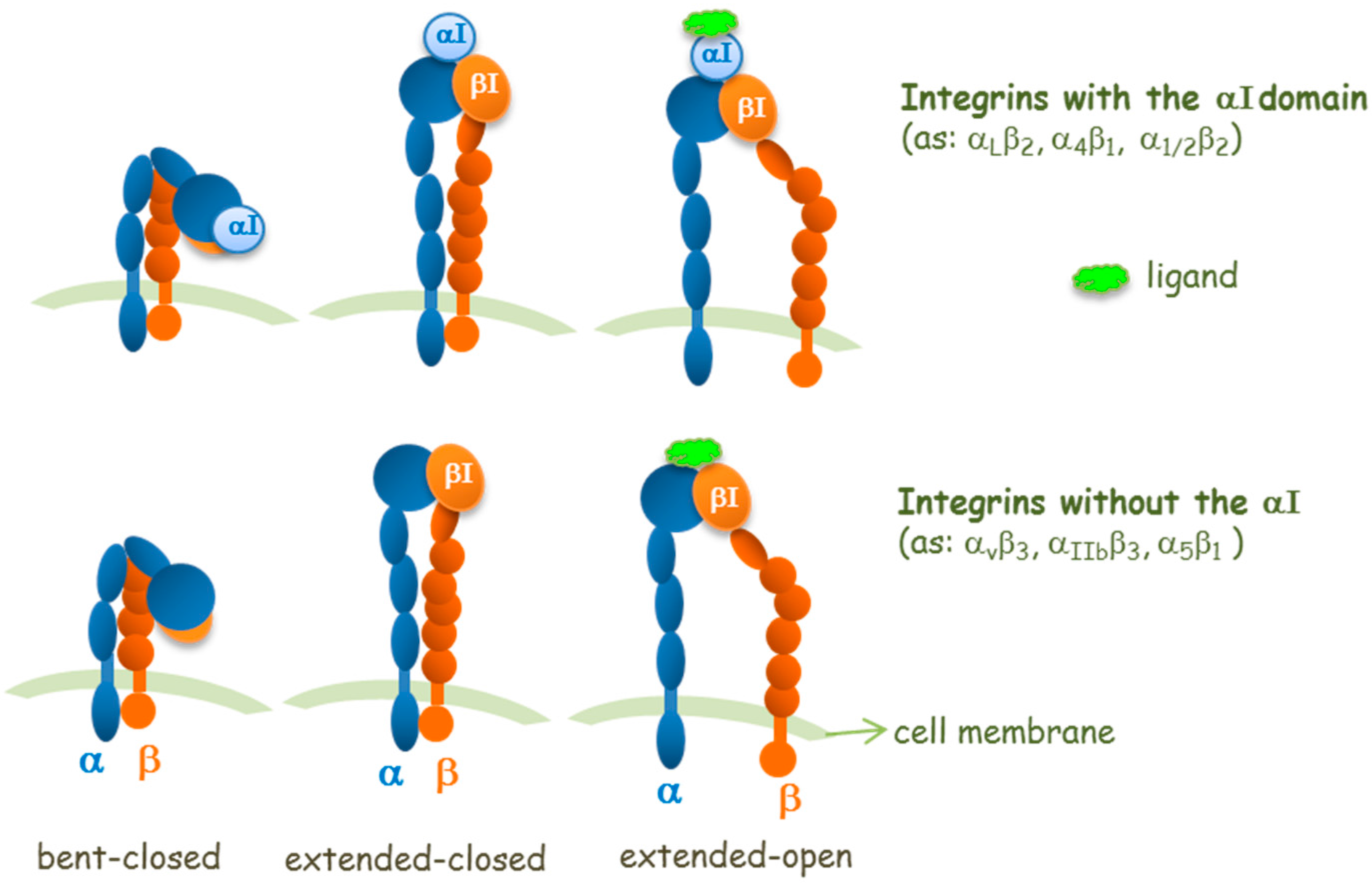
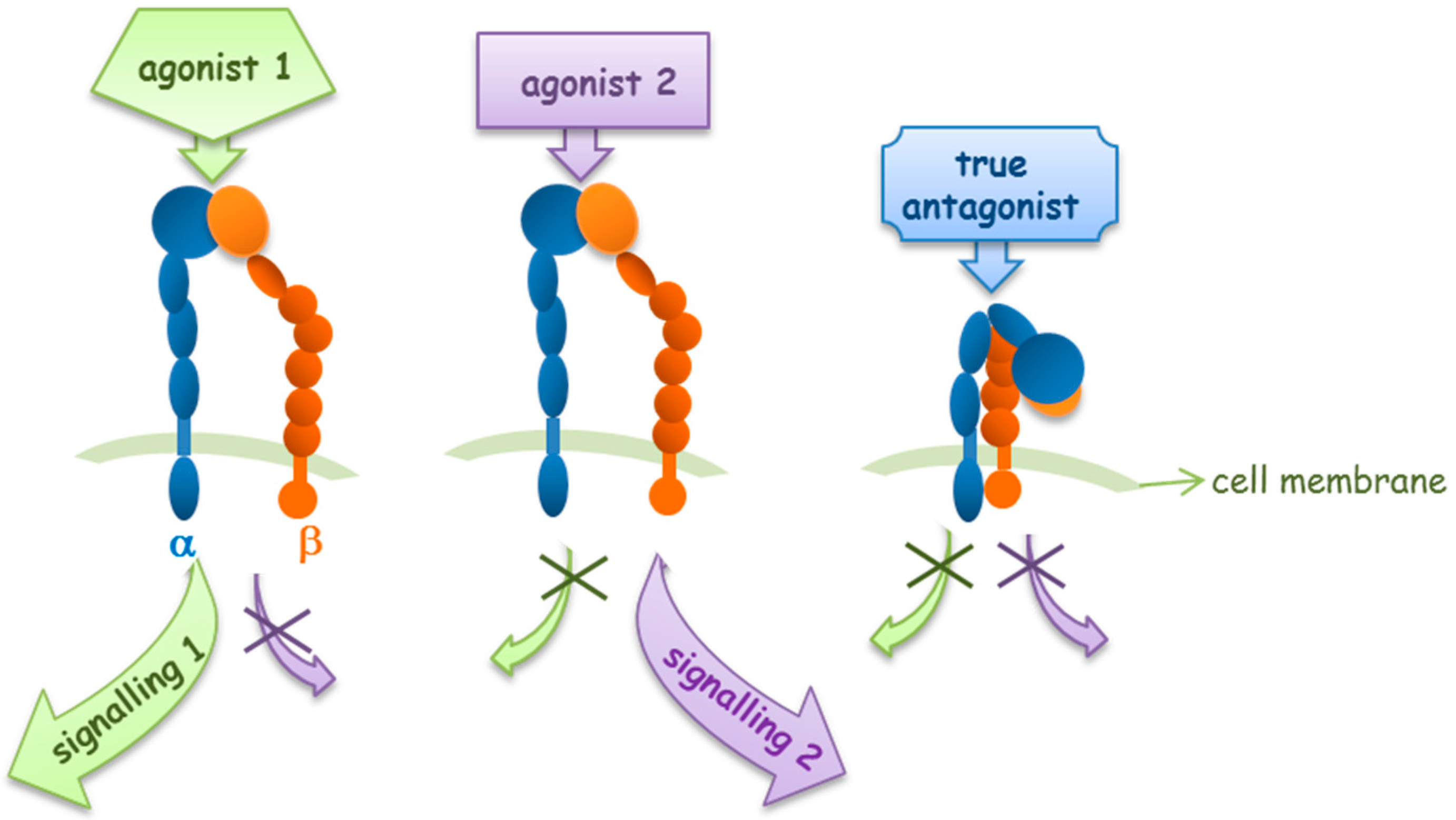
2. Agonists or Antagonists: That Is the Question
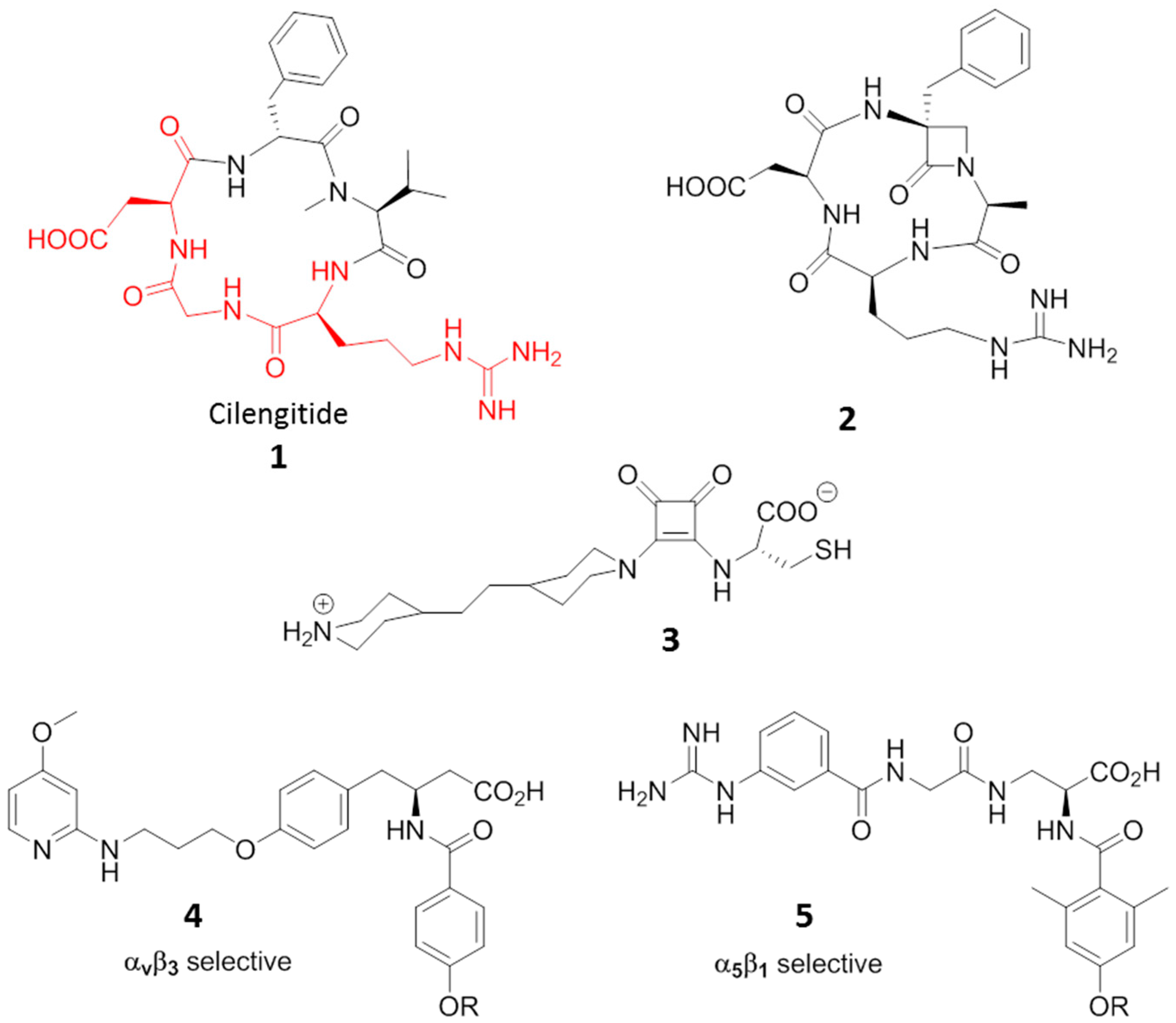
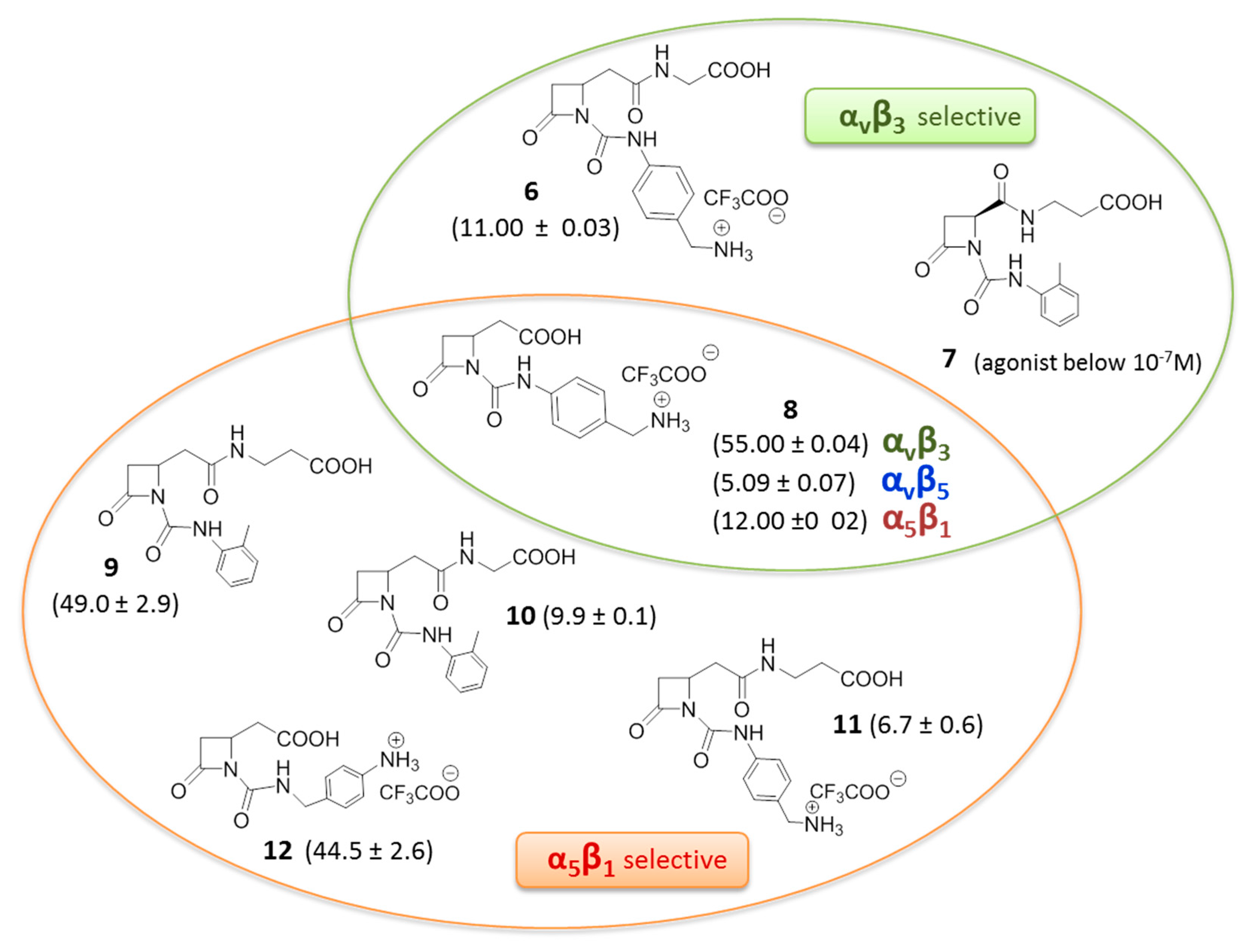
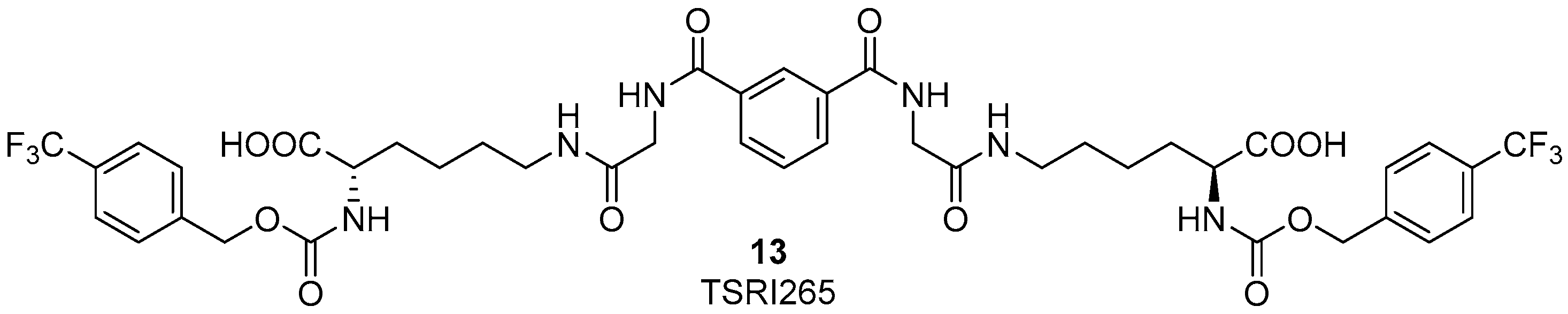
3. RGD-Binding Integrin Agonists
4. Leukocyte Integrins
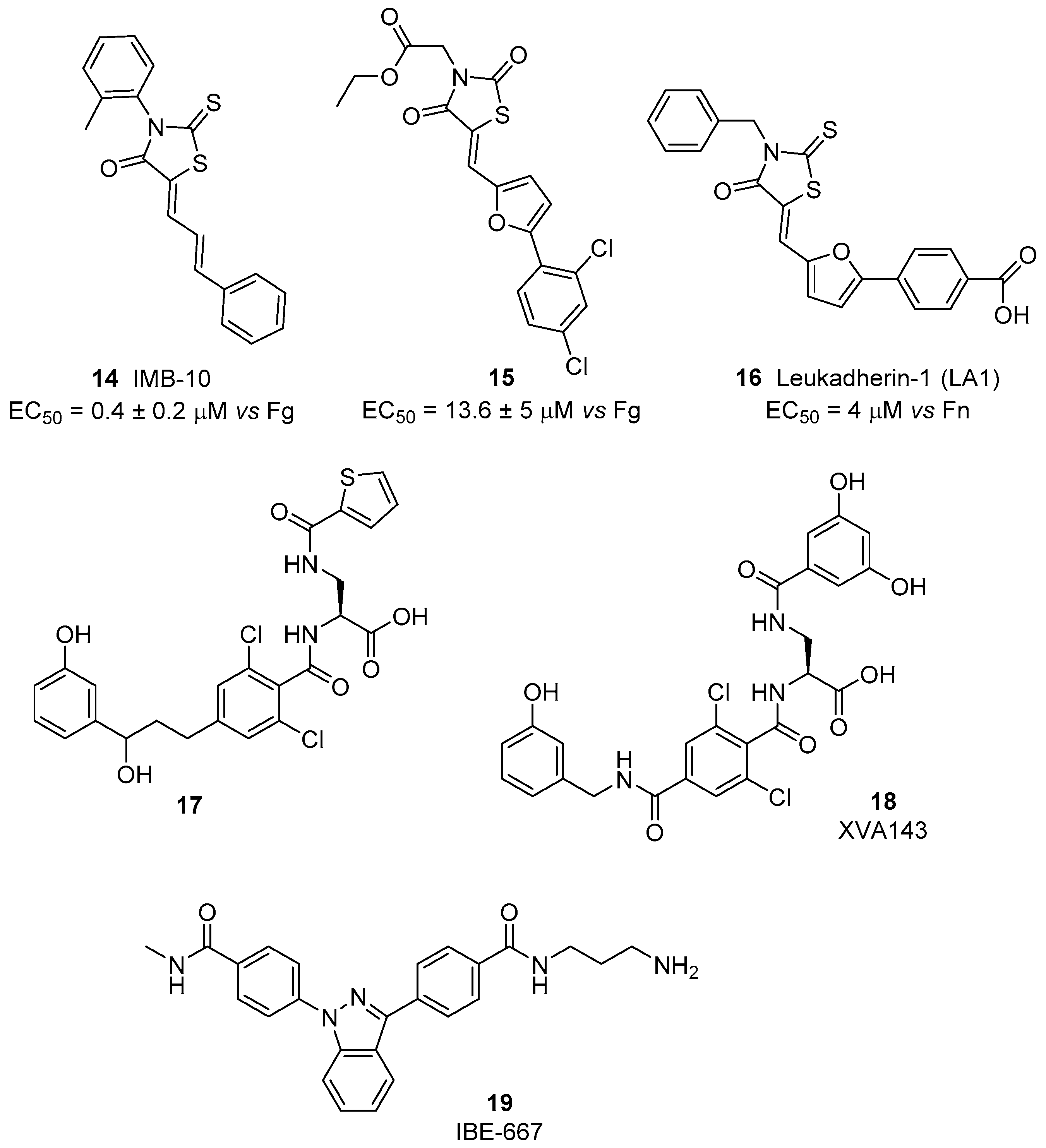
4.1. β2 Integrin Agonists
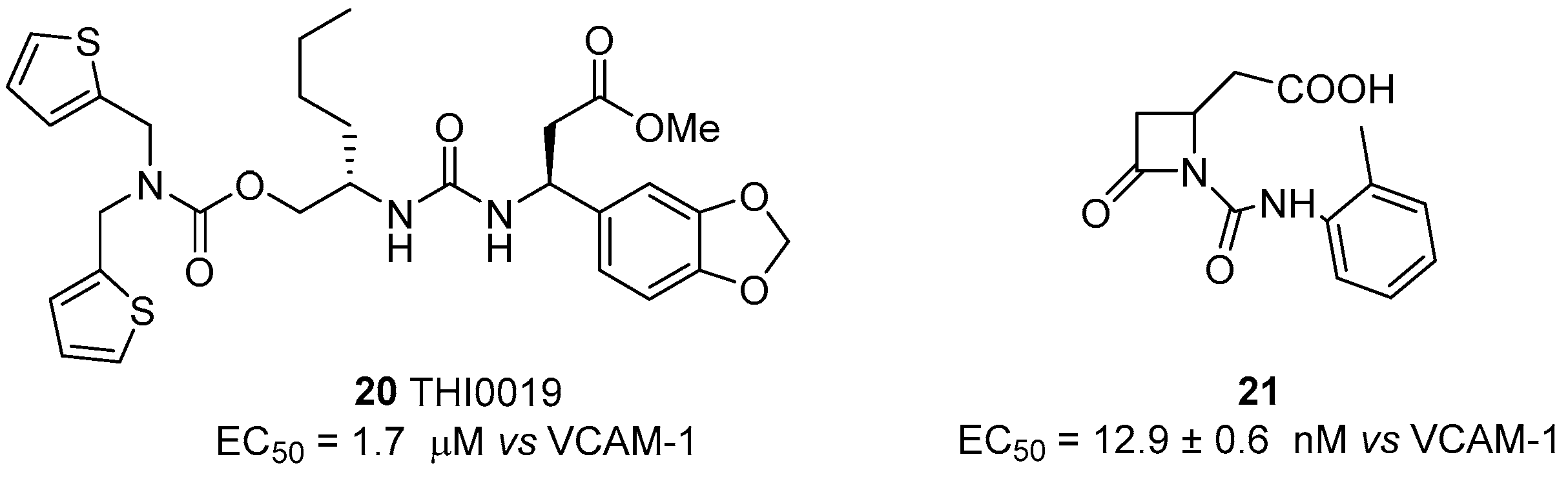
4.2. α4β1 Integrin Agonists
5. Laminin-Binding Integrins
6. Collagen-Binding Integrins
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Humphries, M.J. Integrin structure. Biochem. Soc. Trans. 2000, 28, 311–339. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signalling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.D.; Humphries, M.J. Integrin Structure, Activation, and Interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based Therapeutics: Biological Basis, Clinical Use and New Drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ye, F.; Ginsberg, M.H. Regulation of integrin activation. Annu. Rev. Cell Dev. Bi. 2011, 27, 321–345. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.-H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A.; Dustin, M.L. Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 2012, 24, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Beglova, N.; Blacklow, S.C.; Takagi, J.; Springer, T.A. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 2002, 9, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Adair, B.D.; Xiong, J.-P.; Maddock, C.; Goodman, S.L.; Arnaout, M.A.; Yeager, M. Three-dimensional EM structure of the ectodomain of integrin αvβ3 in a complex with fibronectin. J. Cell Biol. 2005, 168, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, J.; Negri, A.; Provasi, D.; Filizola, M.; Coller, B.S.; Springer, T.A. Closed headpiece of integrin αIIbβ3 and its complex with an αIIbβ3-specific antagonist that does not induce opening. Blood 2010, 116, 5050–5059. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, J.; Springer, T.A. Complete integrin headpiece opening in eight steps. J. Cell Biol. 2013, 201, 1053–1068. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Hellinga, H.W. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat. Struct. Biol. 2001, 8, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef]
- Luo, B.H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Yednock, T.A.; Cannon, C.; Vandevert, C.; Goldbach, E.G.; Shaw, G.; Ellis, D.K.; Liaw, C.; Fritz, L.C.; Tanner, L.I. α4β1 Integrin-dependent cell adhesion is regulated by a low affinity receptor pool that is conformationally responsive to ligand. J. Biol. Chem. 1995, 270, 28740–28750. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Hemler, M.E. Are changes in integrin affinity and conformation overemphasized? Trends Biochem. Sci. 1998, 23, 30–34. [Google Scholar] [CrossRef]
- Hou, S.; Hang, Q.; Isaji, T.; Lu, J.; Fukuda, T.; Gu, J. Importance of membrane-proximal N-glycosylation on integrin b1 in its activation and complex formation. FASEB J. 2016, 30, 4120–4131. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Su, Y.; Xia, W.; Qin, Y.; Humphries, M.J.; Vestweber, D.; Cabañas, C.; Lu, C.; Springer, T.A. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. 2017, 36, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Roggiani, F.; Mezzanzanica, D.; Rea, K.; Tomassetti, A. Guidance of signaling activations by cadherins and integrins in epithelial ovarian cancer cells. Int. J. Mol. Sci. 2016, 17, 1387–1404. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.; Portioli, E.; Battistini, L.; Calorini, L.; Pupi, A.; Vacondio, F.; Arosio, D.; Bianchini, F.; Zanardi, F. Synthesis of novel c(AmpRGD)-Sunitinib dual conjugates as molecular tools targeting the αvβ3 integrin/VEGFR2 couple and impairing tumor-associated angiogenesis. J. Med. Chem. 2017, 60, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A.; McRoberts, K.; Coyner, M.; Andarawewa, K.L.; Frierson, H.F., Jr.; Sanders, J.M.; Swenson, S.; Markland, M.; Conaway, M.R.; Theodorescu, D. Integrin agonists as adjuvants in chemotherapy for melanoma. Clin. Cancer Res. 2008, 14, 6193–6197. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.J. Integrin cell adhesion receptors and the concept of agonism. Trends Pharmacol. Sci. 2000, 21, 29–32. [Google Scholar] [CrossRef]
- Simon, D.I. Opening the field of integrin biology to “biased agonism”. Circ. Res. 2011, 109, 1199–1201. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; Lefkowitz, R.J. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007, 28, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Faridi, M.H.; Altintas, M.M.; Gomez, C.; Duque, J.C.; Vazquez-Padron, R.I.; Gupta, V. Small molecule agonists of integrin CD11b/CD18 do not induce global conformational changes and are significantly better than activating antibodies in reducing vascular injury. Biochim. Biophys. Acta 2013, 1830, 3696–3710. [Google Scholar] [CrossRef] [PubMed]
- Dozynkiewicz, M.A.; Jamieson, N.B.; Macpherson, I.; Grindlay, J.; van den Berghe, P.V.; von Thun, A.; Morton, J.P.; Gourley, C.; Timpson, P.; Nixon, C.; et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell. 2012, 22, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.R.; Allen, J.L.; Chapman, A.; Morlan-Mairal, M.; Zindy, E.; Jacquemet, G.; Fernandez del Ama, L.; Ferizovic, N.; Green, D.M.; Howe, J.D.; et al. α5β1 integrin recycling promotes Arp2/3-independent cancer cell invasion via the formin FHOD3. J. Cell Biol. 2015, 210, 1013–1031. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate. Design, Synthesis and Clinical Evaluation Anticancer Agents. Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Millard, M.; Odde, S.; Neamati, N. Integrin Targeted Therapeutics. Theranostics 2011, 1, 154–188. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.M.; Pritchard, J.M.; Macdonald, S.J.F.; Jamieson, C.; Watson, A.J.B. Emergence of Small-Molecule Non-RGD-Mimetic Inhibitors for RGD Integrins. J. Med. Chem. 2017, 60, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Tolomelli, A.; Juaristi, E.; Gentilucci, L. Integrin Ligands with α/β-Hybrid Peptide Structure: Design, Bioactivity, and Conformational Aspects. Med. Res. Rev. 2016, 36, 389–424. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMTpromoter (CENTRIC EORTC 26071–22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Reynolds, A.R.; Hart, I.-R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by lowconcentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Wiedle, G.F.; Ross, F.P.; Imhof, B.A. Superactivation of integrin αvβ3 by low antagonist Concentrations. J. Cell Sci. 2001, 114, 1545–1553. [Google Scholar] [PubMed]
- Paladino, A.; Civera, M.; Belvisi, L.; Colombo, G. High affinity vs. native fibronectin in the modulation of αvβ3 integrin conformational dynamics: Insights from computational analyses and implications for molecular design. PLoS Comput. Biol. 2017, 13, e1005334. [Google Scholar] [CrossRef] [PubMed]
- Aizpurua, J.M.; Ganboa, J.L.; Palomo, C.; Loinaz, I.; Oyarbide, J.; Fernandez, X.; Balentovà, E.; Fratila, R.M.; Jiménez, A.; Miranda, J.I.; et al. Cyclic RGD beta-lactam peptidomimetics induce differential gene expression in human endothelial cells. ChemBioChem 2011, 12, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.K.; Sejwal, P.; Zhu, S.; Luk, Y. Enhanced cell adhesion andmature intracellular structure promoted by squaramide-based RGD mimics on bioinert surfaces. Bioorg. Med. Chem. 2013, 21, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Hersel, U.; Dahmen, C.; Kessler, H. RGD modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomaterials 2003, 24, 4385–4441. [Google Scholar] [CrossRef]
- Fraioli, R.; Rechenmacher, F.; Neubauer, S.; Manero, J.M.; Gil, J.; Kessler, H.; Mas-Moruno, C. Mimicking bone extracellular matrix: Integrin-binding peptidomimetics enhance osteoblast-like cells adhesion, proliferation, and differentiation on titanium. Colloids Surf. B 2015, 128, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Galletti, P.; Soldati, R.; Pori, M.; Durso, M.; Tolomelli, A.; Gentilucci, L.; Dattoli, S.D.; Baiula, M.; Spampinato, S.M.; Giacomini, D. Targeting integrins αvβ3 and α5β1 with new β-lactam derivatives. Eur. J. Med. Chem. 2014, 83, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Baiula, M.; Galletti, P.; Martelli, G.; Soldati, R.; Belvisi, L.; Civera, M.; Dattoli, S.D.; Spampinato, S.M.; Giacomini, D. New β-lactam derivatives modulate cell adhesion and signaling mediated by RGD-binding and leukocyte integrins. J. Med. Chem. 2016, 59, 9721–9742. [Google Scholar] [CrossRef] [PubMed]
- Tolomelli, A.; Gentilucci, L.; Mosconi, E.; Viola, A.; Dattoli, S.D.; Baiula, M.; Spampinato, S.; Belvisi, L.; Civera, M. Development of isoxazoline-containing peptidomimetics as dual αvβ3 and α5β1 integrin ligands. ChemMedChem. 2011, 6, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Strömblad, S.; Sanders, L.C.; von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; Cheresh, D.A. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 1996, 85, 683. [Google Scholar] [CrossRef]
- Boger, D.L.; Goldberg, J.; Silletti, S.; Kessler, T.; Cheresh, D.A. Identification of a novel class of small-molecule antiangiogenic agents through the screening of combinatorial libraries which function by inhibiting the binding and localization of proteinase MMP2 to integrin αvβ3. J. Am. Chem. Soc. 2001, 123, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Silletti, S.; Kessler, T.; Goldberg, J.; Boger, D.L.; Cheresh, D.A. Disruption of matrix metalloproteinase 2 binding to integrin αvβ3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc. Natl. Acad. Sci. USA 2001, 2, 119–124. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S. Why cancer and inflammation? Yale J. Biol. Med. 2006, 79, 123–130. [Google Scholar] [PubMed]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Middle, S.A.; Coupland, S.E.; Taktak, A.; Kidgell, V.; Slupsky, J.R.; Pettitt, A.R.; Till, K.J. Immunohistochemical analysis indicates that the anatomical location of B-cell non-Hodgkin’s lymphoma is determined by differentially expressed chemokine receptors, sphingosine-1-phosphate receptors and integrins. Exp. Hematol. Oncol. 2015, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Tao, J. The B-cell receptor orchestrates environment-mediated lymphoma survival and drug resistance in B-cell malignancies. Oncogene 2014, 33, 4107–4113. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, T.; Mocevicius, P.; Deduchovas, O.; Salnikova, O.; Castro-Santa, E.; Büchler, M.W.; Schmidt, J.; Ryschich, E. αLβ2 Integrin is indispensable for CD81 T-cell recruitment in experimental pancreatic and hepatocellular cancer. Int. J. Cancer 2012, 130, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Suojanen, J.; Salo, T.; Sorsa, T.; Koivunen, E. αMβ2 Integrin modulator exerts antitumor activity in vivo. Anticancer Res. 2007, 27, 3775–3782. [Google Scholar] [PubMed]
- Lee, J.O.; Rieu, P.; Arnout, M.A.; Liddington, R.C. Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18). Cell 1995, 80, 631–638. [Google Scholar] [CrossRef]
- Lee, J.O.; Bunkston, L.A.; Arnout, M.A.; Liddington, R.C. Two conformations of the integrin A-domain (I-domain): A pathway for activation? Structure 1995, 3, 1333–1340. [Google Scholar] [CrossRef]
- Shimaoka, M.; Salas, A.; Yang, W.; Weitz-Schmidt, G.; Springer, T.A. Small molecule integrin antagonists that bind to the β-2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity 2003, 19, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; Mul, E.P.; Bolm, M.; Kovach, N.L.; Gaeta, F.C.; Tollefson, V.; Elices, M.J.; Harlan, J.M. Freezing adhesion molecules in a state of high-avidity binding blocks eosinophil migration. J. Exp. Med. 1993, 178, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Björklund, M.; Aitio, O.; Stefanidakis, M.; Suojanen, J.; Salo, T.; Sorsa, T.; Koivunen, E. Stabilization of the activated αMβ2 integrin by a small molecule inhibits leukocyte migration and recruitment. Biochemistry 2006, 45, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Arnaout, M.A.; Gupta, V. A Simple, no-wash cell adhesion–based high-throughput assay for the discovery of small-molecule regulators of the integrin CD11b/CD18. J. Biomol. Screen. 2007, 12, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Faridi, M.H.; Maiguel, D.; Barth, C.J.; Stoub, D.; Day, R.; Schürer, S.; Gupta, V. Identification of novel agonists of the integrin CD11b/CD18. Bioorg. Med. Chem. Lett. 2009, 19, 6902–6906. [Google Scholar] [CrossRef] [PubMed]
- Maiguel, D.; Faridi, M.H.; Wei, C.; Kuwano, Y.; Balla, K.M.; Hernandez, D.; Barth, C.J.; Lugo, G.; Donnelly, M.; Nayer, A.; et al. Small Molecule–Mediated Activation of the Integrin CD11b/CD18 Reduces Inflammatory Disease. Sci. Signal 2011, 4, ra57. [Google Scholar] [CrossRef] [PubMed]
- Celik, E.; Faridi, M.H.; Kumar, V.; Deep, S.; Moy, V.T.; Gupta, V. Agonist leukadherin-1 increases cd11b/cd18-dependent adhesion via membrane tethers. Biophys. J. 2013, 105, 2517–2527. [Google Scholar] [CrossRef] [PubMed]
- Jagarapu, J.; Kelchtermans, J.; Rong, M.; Chen, S.; Hehre, D.; Hummler, S.; Farisi, M.H.; Gupta, V.; Wu, S. Efficacy of leukadherin-1 in the prevention of hyperoxia-induced lung injury in neonatal rats. Am. J. Respir. Cell Mol. Biol. 2015, 53, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Q.; Guo, L.; Cimbaluk, D.J.; Elshabrawy, H.; Faridi, M.H.; Jolly, M.; George, J.F.; Agarwal, A.; Gupta, V. A small molecule β2 integrin agonist improves chronic kidney allograft survival by reducing leukocyte recruitment and accompanying vasculopathy. Front. Med. 2014, 1, 45. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Carman, C.V.; Kim, M.; Salas, A.; Shimaoka, M.; Springer, T. A Small molecule agonist of an integrin, αLβ2. J. Biol. Chem. 2006, 281, 37904–37912. [Google Scholar] [CrossRef] [PubMed]
- Gadek, T.R.; Burdick, D.J.; McDowell, R.S.; Stanley, M.S.; Marsters, J.C.; Paris, K.J.; Oare, D.A.; Reynolds, M.E.; Ladner, C.; Zioncheck, K.A.; et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science 2002, 295, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.V.; Welzenbach, K.; Steinberger, P.; Krähenbühl, S.; Weitz-Schmidt, G. Downstream effect profiles discern different mechanisms of integrin αLβ2 inhibition. Biochem. Pharm. 2016, 119, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Hintersteiner, M.; Kallen, J.; Schmied, M.; Graf, C.; Jung, T.; Mudd, G.; Shave, S.; Gstach, H.; Auer, M. Identification and X-ray Co-crystal Structure of a Small-Molecule Activator of LFA-1-ICAM-1 Binding. Angew. Chem. Int. Ed. 2014, 53, 4322–4326. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhu, J.; Mi, L.Z.; Walz, T.; Sun, H.; Chen, J.; Springer, T.A. Structural specializations of α4β7, an integrin that mediates rolling adhesion. J. Cell. Biol. 2012, 196, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Vanderslice, P.; Biediger, R.J.; Woodside, D.G.; Brown, W.S.; Khounlo, S.; Warier, N.D.; Gundlach, C.W., IV; Caivano, A.R.; Bornmann, W.G.; Maxwell, D.S.; et al. Small molecule agonist of very late antigen-4 (VLA-4) integrin induces progenitor cell adhesion. J. Biol. Chem. 2013, 288, 19414–19428. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Narsinh, K.H.; Lan, F.; Wang, L.; Nguyen, P.K.; Hu, S.; Lee, A.; Han, L.; Gong, Y.; Huang, M.; et al. Early stem cell engraftment predicts late cardiac functional recovery: Preclinical insights from molecular imaging. Circ. Cardiovasc. Imaging 2012, 5, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Vrtovec, B.; Poglajen, G.; Lezaic, L.; Sever, M.; Domanovic, D.; Cernelc, P.; Socan, A.; Schrepfer, S.; Torre-Amione, G.; Haddad, F.; Wu, J.C. Effects of intracoronary CD34+ stem cell transplantation in nonischemic dilated cardiomyopathy patients: 5-year follow up. Circ. Res. 2013, 112, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Ido, H.; Sanzen, N.; Hayashi, M.; Sato-Nishiuchi, R.; Futaki, S.; Sekiguchi, K. The C-terminal region of laminin beta chains modulates the integrin binding affinities of laminins. J. Biol. Chem. 2009, 284, 7820–7831. [Google Scholar] [CrossRef] [PubMed]
- Ramovs, V.; Te Molder, L.; Sonnenberg, A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. 2017, 57–58, 213–243. [Google Scholar] [CrossRef] [PubMed]
- Subbaram, S.; Dipersio, C.M. Integrin α3β1 as a breast cancer target. Expert Opin. Ther. Targets 2011, 15, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Khan, S.Q.; Faridi, M.H.; Wei, C.; Tardi, N.J.; Altintas, M.M.; Elshabrawy, H.A.; Mangos, S.; Quick, K.L.; Sever, S.; et al. A podocyte-based automated screening assay identifies protective small molecules. J. Am. Soc. Nephrol. 2015, 26, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, X.; Hahm, H.S.; Wei, W.; Hao, E.; Hayek, A.; Ding, S. Revealing a core signalling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molucules. Proc. Natl. Acad. Sci. USA 2010, 107, 8129–8134. [Google Scholar] [CrossRef] [PubMed]
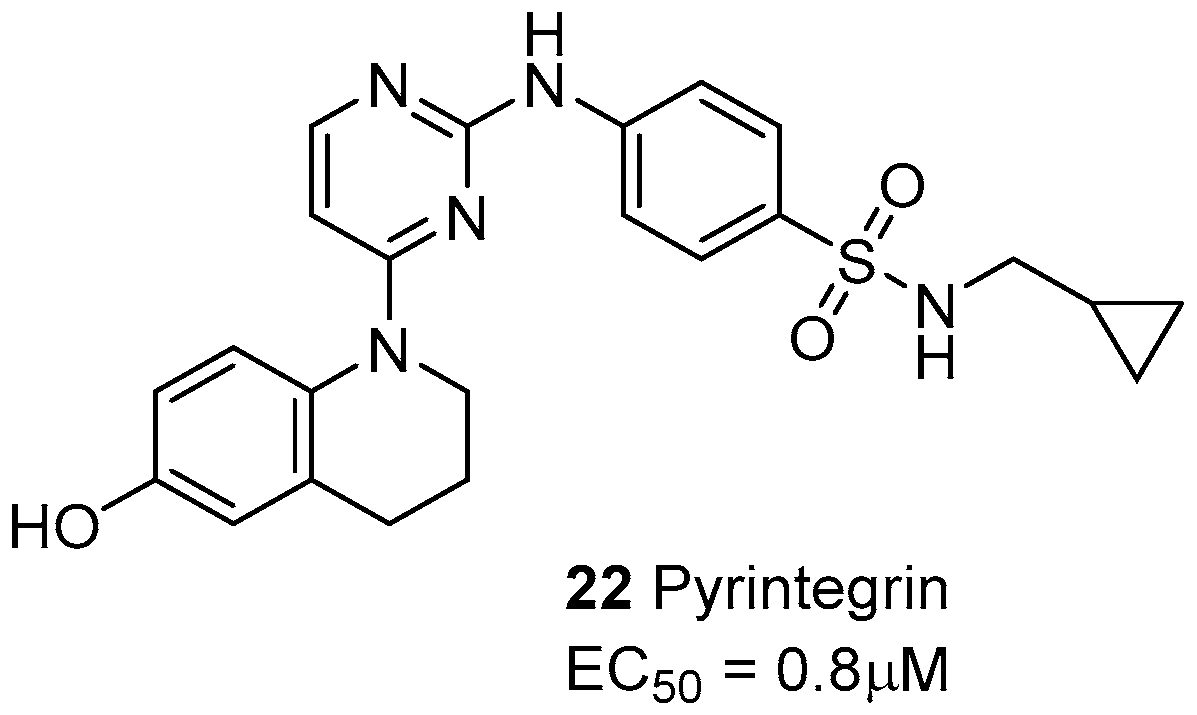
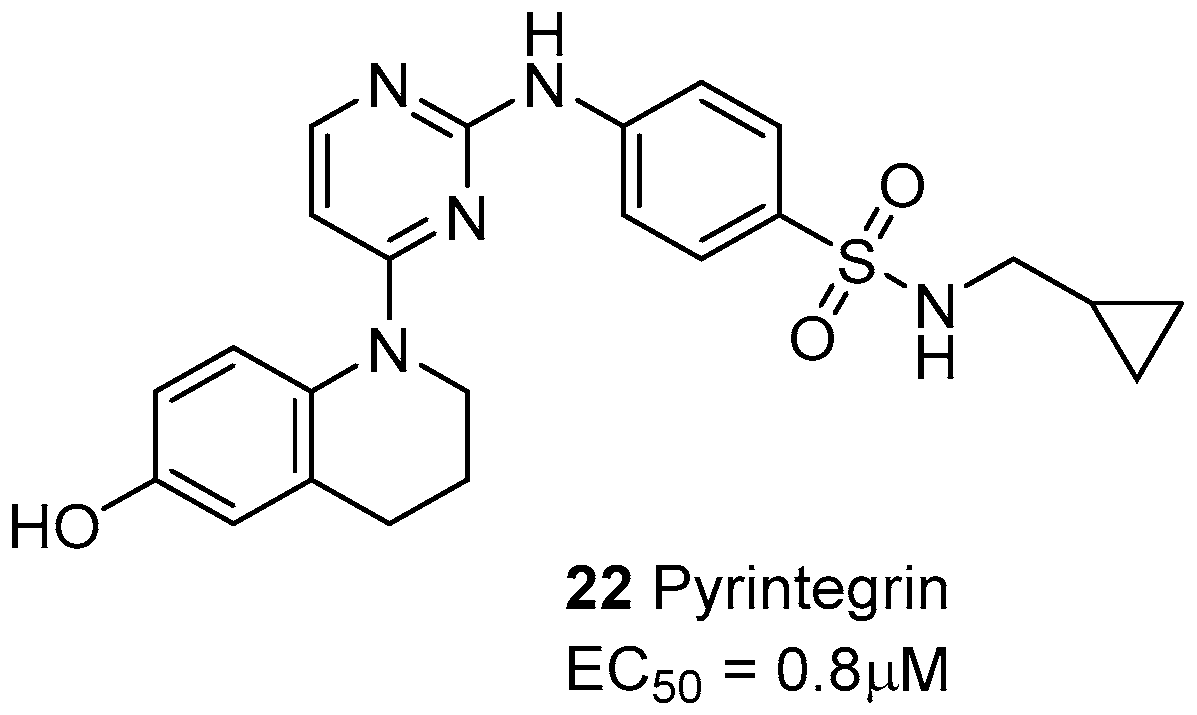
- Shah, B.S.; Chen, M.; Suzuki, T.; Embree, M.; Kong, K.; Lee, C.H.; He, L.; Xiang, L.; Ahn, J.A.; Ding, S.; Mao, J.J. Pyrintegrin induces soft tissue formation by transplanted or endogenous cells. Sci. Rep. 2017, 7, 36402. [Google Scholar] [CrossRef] [PubMed]
- Zeltz, C.; Gullberg, D. The integrin–collagen connection—a glue for tissue repair? J. Cell Sci. 2016, 129, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-H.; Lin, K.-T.; Chang, C.-H.; Peng, H.-C.; Hua, T.-F. The integrin α2β1 agonist, aggretin, promotes proliferation and migration of VSMC through NF-kB translocation and PDGF production. Brit. J. Pharmacol. 2009, 156, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-H.; Chang, C.-H.; Hsu, C.C.; Lin, K.T.; Peng, H.-C.; Hua, T.-F. Aggretin Venom Polypeptide as a Novel Anti-angiogenesis Agent by Targeting Integrin α2β1. Sci. Rep. 2017, 7, 43612. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.D.; Carvajal, N.; Jin, A.; Matsumoto, K.; Yamada, K.M. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ritsma, L.; Dey-Guha, I.; Talele, N.; Salony, X.S.; Chowdhury, J.; Ross, K.N.; Ramaswamy, S. Integrin β1 activation induces an antimelanoma host response. PLoS ONE 2017, 12, e0175300. [Google Scholar] [CrossRef] [PubMed]

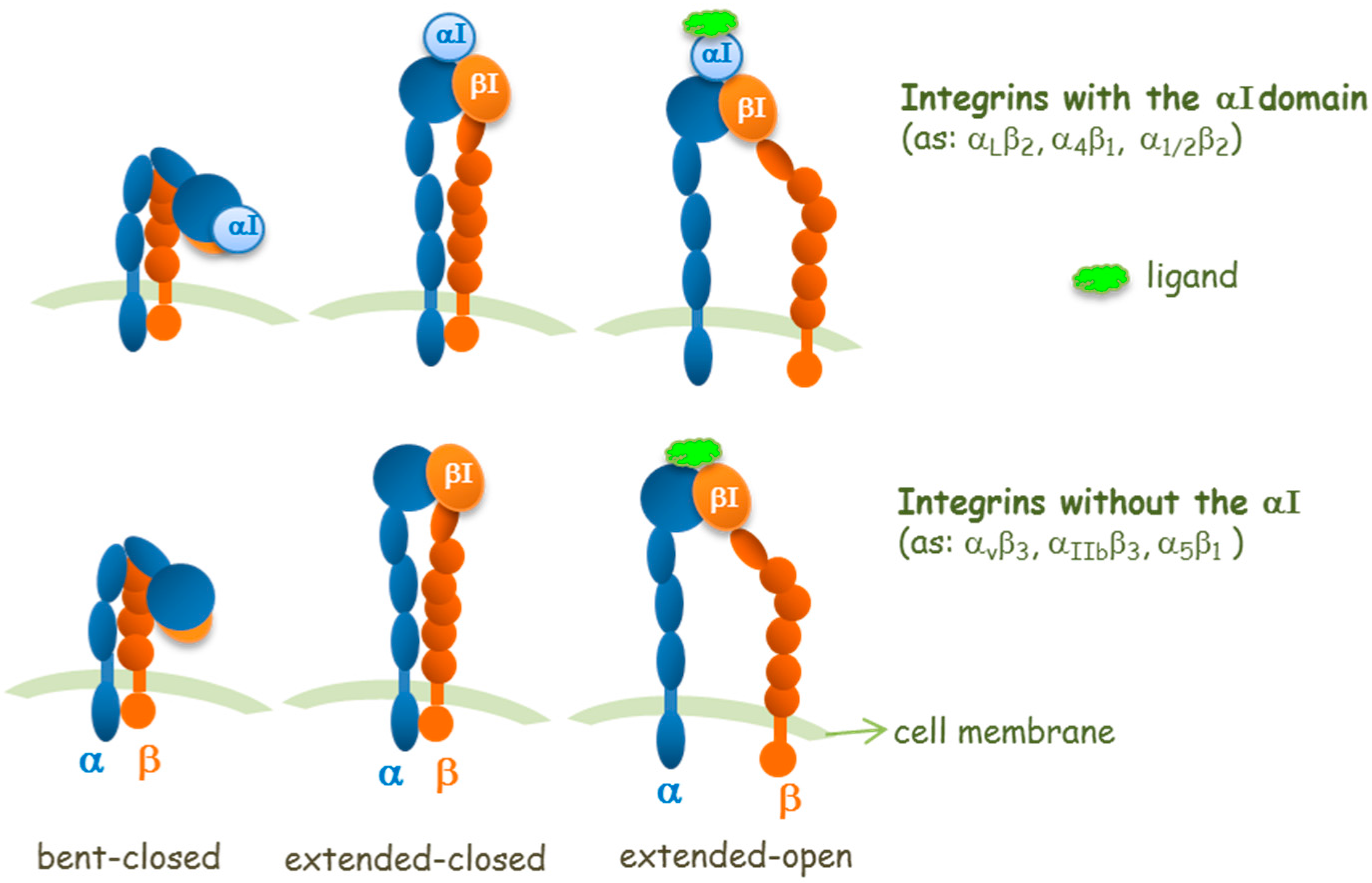
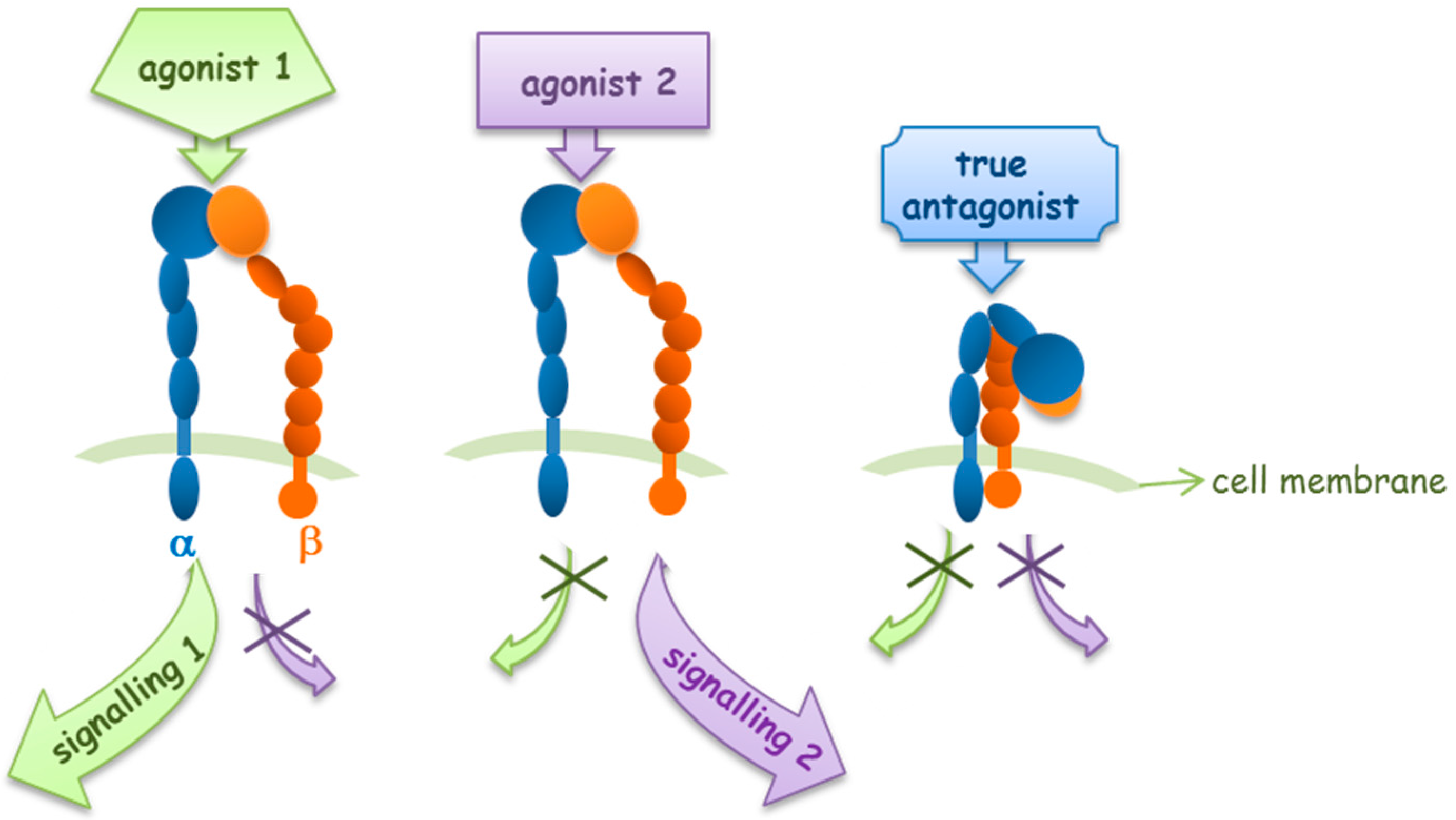
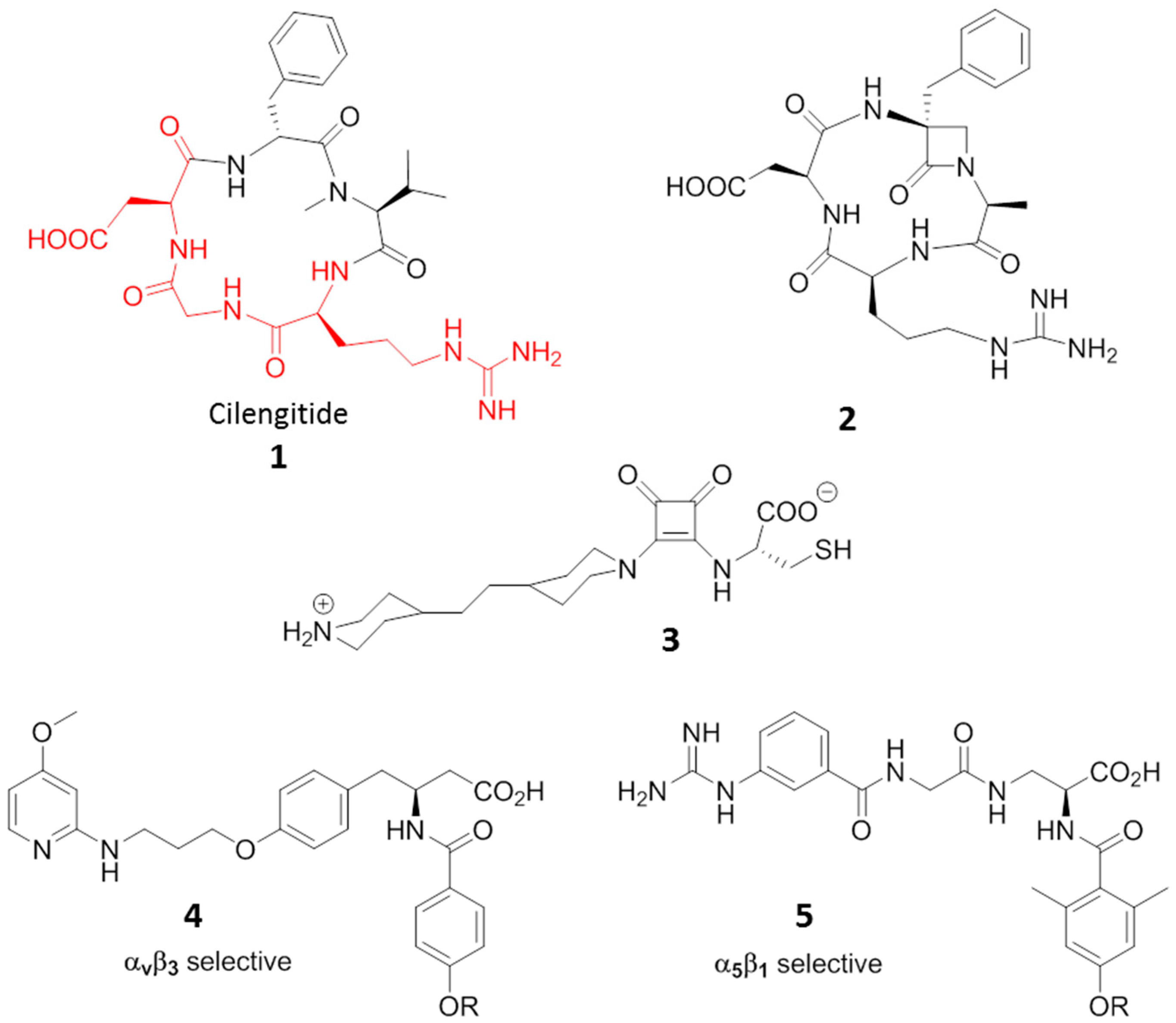
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolomelli, A.; Galletti, P.; Baiula, M.; Giacomini, D. Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators. Cancers 2017, 9, 78. https://doi.org/10.3390/cancers9070078
Tolomelli A, Galletti P, Baiula M, Giacomini D. Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators. Cancers. 2017; 9(7):78. https://doi.org/10.3390/cancers9070078
Chicago/Turabian StyleTolomelli, Alessandra, Paola Galletti, Monica Baiula, and Daria Giacomini. 2017. "Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators" Cancers 9, no. 7: 78. https://doi.org/10.3390/cancers9070078
APA StyleTolomelli, A., Galletti, P., Baiula, M., & Giacomini, D. (2017). Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators. Cancers, 9(7), 78. https://doi.org/10.3390/cancers9070078