1. Introduction
Machine learning (ML) has been integrated into material-related fields over the past decade, suggesting the possibility of a powerful statistical technique that can be used to aid and/or replace costly simulations and hard-to-setup experiments, and leading the advances in a variety of areas. Accelerating simulations from quantum to continuum scales has been made possible, since ML is exploited to identify the most important features of a system and create simplified models that can be simulated much more quickly. In this way, property calculation and prediction are feasible, even when experiments or simulations are hard to perform. Data science and ML can be used to learn from historical data and make predictions about the properties of materials [
1,
2,
3], even those that have not yet been studied, and even suggest symbolic equations to describe them [
4,
5,
6].
The specific ML techniques that have been used in material-related fields include supervised learning, unsupervised learning, and reinforcement learning [
7]. Supervised learning has been the most widely investigated field for regression or classification tasks. It refers to labeled data, which is associated with known parameters that affect a property of interest and tries to predict either interpolated or extrapolated values. Unsupervised ML aims to discover interconnections inside unlabeled data, either with clustering techniques (e.g., the k-means algorithm) or by applying dimensionality reduction in high-dimensional data (e.g., proper orthogonal decomposition, POD, and principal component analysis, PCA). Reinforcement learning is exploited in cases in which interaction with the environment is significant, and is based on an observation/rewarding scheme employed to find the best scenario for a process [
8].
Depending on the number of computational layers included in an ML model, another categorization looks to shallow learning (SL) and deep learning (DL). Widely used SL algorithms include linear (or multi-linear) regression, LASSO, ridge, support vector machine (SVM), and decision-based algorithms (e.g., Decision Trees and Random Forest) [
9], among others. As these algorithms may perform better for specific parts of a dataset, ensemble and stack methods have also emerged in which groups of more than one algorithm are interconnected serially or in parallel, and in most cases, these present enhanced output results [
10]. However, in complex systems, such as turbulent fluid flows [
11], more complex computational layers (neural networks, NNs) are exploited, constructing a deeper architecture better able to deal with the ‘big data’ in DL models [
12].
In the field of molecular simulation, where the property extraction of materials is of paramount importance, ML has gained a central role [
13]. Atomic-scale simulations, with molecular dynamics (MD) being the most popular method, involve costly (in time and hardware resources) simulations which accurately calculate dynamical properties of the materials in all phases (solid, liquid, and gas). They have oftentimes been used in place of experiments when experiments have been difficult to perform. Moreover, they have opened a new pathway for the calculation of properties that cannot be extracted by theoretical or numerical simulations at the macroscale. The transport properties of fluids, specifically, shear viscosity and thermal conductivity, are two of the most computationally intensive properties to deal with in atomistic simulations, involving particle interactions, positions, and velocities in multiple time-frames [
14]. Molecular dynamics simulation, either in an equilibrium (EMD) or non-equilibrium (NEMD) manner, has, to this point, been incorporated to provide transport property data. However, data-driven approaches have much to offer in this direction. Current research efforts have already reported methods that combine physics-based and data-driven modeling [
15].
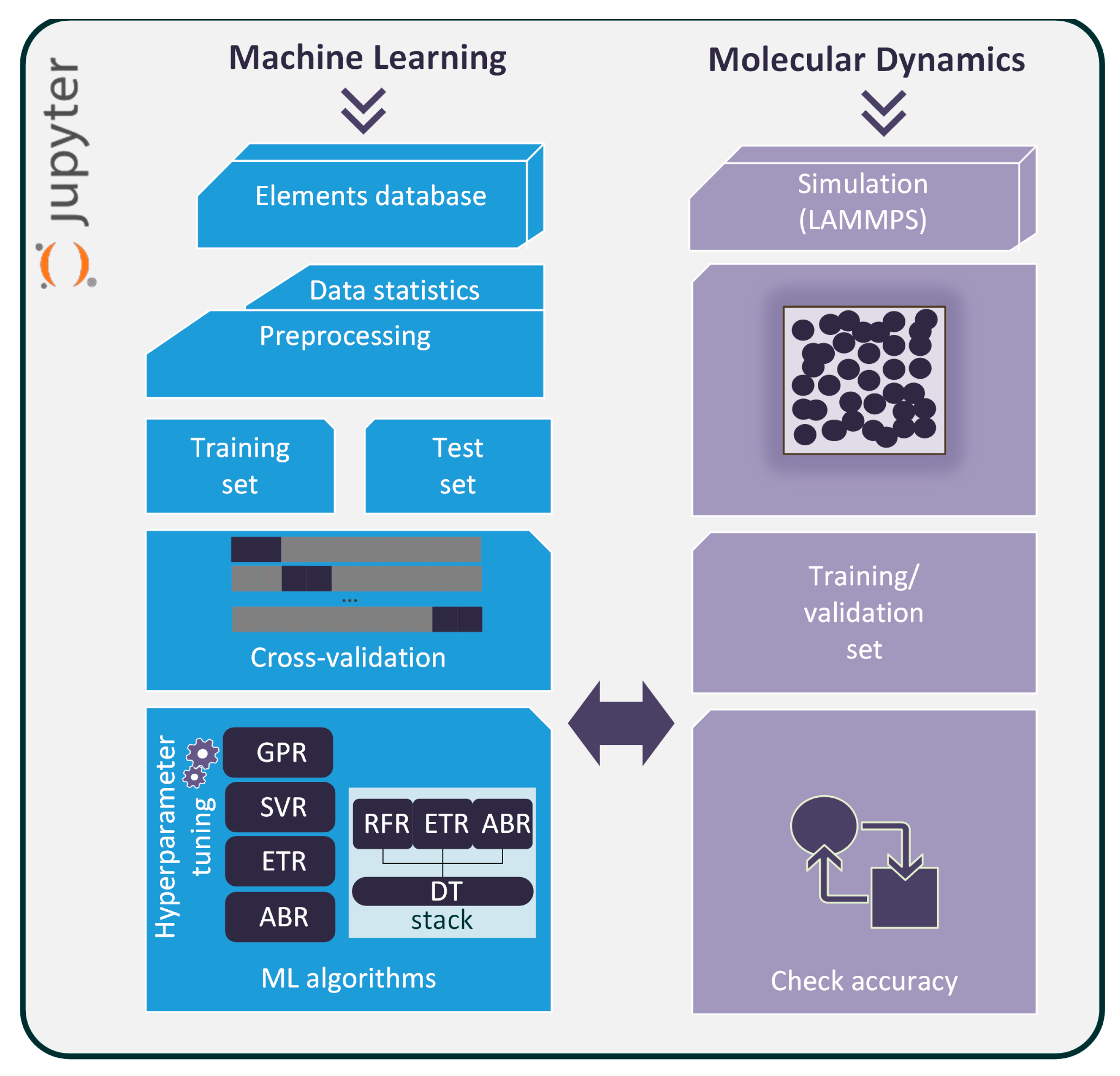
Therefore, it would be beneficial to embed novel ML methods into molecular simulations. In this paper, a combined MD simulation/ML prediction scheme has been constructed to accurately derive the transport properties, such as viscosity and thermal conductivity, of basic elements (argon, krypton, nitrogen, and oxygen) at the bulk state. The framework is embedded in a Jupyter Lab environment [
16]. It starts by analyzing historical data from the literature [
17], performs property prediction with available data, and functions in parallel to an MD computational flow that calculates the transport properties in phase space points where no data is available. The ML model is further fed with MD-extracted points, and it is then retrained, achieving increased accuracy in most cases. The fast ML platform is capable of extracting new phase-state points while remaining linked to the MD simulations, ensuring that results are accurate and bound to physical laws. Of equal importance is the enrichment of the data in the current literature with new values of viscosity and thermal conductivity which can be further processed by the research community.
3. Results and Discussion
With a series of parallel MD simulations, viscosity and thermal conductivity values for (P-T) state points missing from the elements database have been calculated. We have to note here that the base MD program has been verified on the specific database values. To validate the results of the simulations, we first calculated the values of η and λ provided in the database (i.e., the existing data points), for which we have taken the same or nearly the same values, within statistical accuracy, and next, we proceeded to calculate the unknown data points.
Next, characteristic algorithmic implementations concerning the MD code were described; all calculated transport properties values are given in the respective Tables. All of this data is embedded in the initial elements database and the predictive ability of our ML algorithms is evaluated.
3.1. MD Programming
Both thermal conductivity and viscosity components are reported as running averages over the whole simulation time [
60]. Due to the sophisticated computational techniques needed to automatically extract MD-calculated values for viscosity and thermal conductivity, a hypercomputer system (HPC) has been deployed. Computational techniques incorporated to manage data files for each element, for every (
P-
T) pair and for every simulation instance (as discussed above, we have performed 10 parallel simulations to extract one average value for statistical reasons), are embedded in a Jupyter Lab Python environment.
First, we prepare the input files and place them in separate folders for each available case that runs on LAMMPS, and for each element (4 elements), temperature (20 different temperature values), and pressure (10 different pressure values) range. This is performed automatically for the
simulation instances; the procedure is shown in Algorithm 1.
| Algorithm 1. Prepare and run MD simulations |
| 1: | Open generic LAMMPS bulk fluid simulation file |
| 2: | Define element masses list, m = [·] |
| 3: | Create file path for every element |
| 4: | Define temperature range list, T = [·] |
| 5: | Create file path for every temperature value |
| 6: | Define pressure range list, P = [·] |
| 7: | Create file path for every pressure value |
| 8: | for i, j, k in mi = [·], Tj = [·], Pk = [·] |
| 9: | Create LAMMPS input files |
Algorithm 2 depicts the MD program flow in LAMMPS. After proper initialization, the code runs in 10 parallel instances to achieve better statistical accuracy and calculates the properties of interest. This is performed for every case constructed by Algorithm 1. In the final stage of a simulation, calculated values for viscosity and thermal conductivity are stored in a Pandas DataFrame and, finally, in .csv files. All values are embedded in the initial elements database.
| Algorithm 2. Backbone of a bulk fluid MD simulation |
| 1: | Define simulation box, units, atom_style |
| 2: | Setup simulation variables (P, T, ρ, rc, lattice constant) |
| 3: | Setup LAMMPS computes: temperature and pressure |
| 4: | Initialization run for t = 106 timesteps |
| 5: | Give random initial velocity values to particles |
| 6: | Define pair styles and pair coefficients |
| 7: | Begin NPT simulation |
| 8: | for i = 1:10 |
| 9: | ith production run: |
| 10: | Calculate kinetic and potential energy per atom |
| 11: | Calculate stress tensors and heat flux components |
| 12: | Perform time auto-correlations |
| 13: | Calculate pair correlation function |
| 14: | Calculate viscosity and thermal conductivity |
| 15: | Statistical averaging of the results |
| 16 | Store calculated values to pandas dataframe |
| 17: | Store calculated values to .csv in the respective folder |
3.2. MD Simulations
Turning our attention to the transport properties extracted from the MD simulations now, we present calculated viscosity and thermal conductivity values for Ar in
Table 3 and
Table 4, for Kr in
Table 5 and
Table 6, for N in
Table 7 and
Table 8, and for O in
Table 9 and
Table 10, respectively. These values are embedded in the initial property database (taken from the literature) and employed for transport property prediction in the ML model constructed. We have to note that tabulated values shown here are taken far from the critical points, and their values are close to neighboring, validated results found in the literature-only database.
The initial database for Kr is significantly smaller compared to that for Ar (see
Table 1). The Kr MD simulations have given many outlier values; in order to ensure that our ML model performs in an equally fine manner, we have kept only the simulation values close to neighboring ones. Also, a similar strategy has been followed for the N and O elements.
3.3. Machine-Learning Predictions
The ability of the implied ML algorithms to predict the transport properties of the elements is captured by the accuracy measures shown in
Table 11. The mean absolute error (
MAE) is given by
where
for the
ith data point; index real, the database value; pred, the ML predicted value.
The root mean squared error (
RMSE) is
the mean absolute percentage error (
MAPE) is
and, finally, the coefficient of determination,
R2, is given by
In
Table 11, only the initial database (without MD-extracted values) has been considered for the calculations. In general, the algorithms incorporated here are capable of providing accurate predictions for all element-transport properties. As expected, the stacked algorithm (SA) has given the most accurate results for all elements. However, the ETR has given almost similar and in some cases, even more accurate results than SA. The algorithms that exploit kernels, such as SVR and GPR, are also good choices, although there may be some increased computational cost, as compared to the tree-based algorithms. Nevertheless, this overhead is not important in small and medium-sized datasets, such as the one that has been incorporated into this paper.
Furthermore, the initial database contains Kr values that have been calculated based on the principle of the corresponding states [
61]. This is an indirect method used to obtain the properties of the elements, but we do not expect it to be of the same accuracy as an experimental or a proper simulation technique. Taking also into consideration that the Kr data points only number 516 (see
Table 1), we believe that these are the two reasons why the accuracy measures for Kr are smaller, as compared to all other elements.
In
Table 12, the MD viscosity and thermal conductivity data have also been employed in the training and validation process, and every ML algorithm has been retrained. The accuracy of each algorithm is similar to those shown in
Table 11, but some error metrics here have slightly increased. This deviation is small; it falls within the range of statistical accuracy. The new MD data points may be considered accurate, but, on the other hand, they come from a different method, computational—not experimental, and this may cause a kind of deviation from the real values. In any case, the deviations are small for Ar and N, and they increase a small amount more for O and Kr.
4. Conclusions
The transport properties of Ar, Kr, N, and O are examined in this paper. The main objectives are to calculate and predict viscosity and thermal conductivity. Experimental and theoretical data from the literature, after being pre-processed, has been initially exploited to train various machine-learning algorithms.
Ensemble, classical, kernel-based, and stacked algorithmic techniques have been proven effective in predicting the transport properties of fluid elements, achieving high levels of accuracy and low levels of error. Notwithstanding their performance, we have shown that they can be used in parallel with classical molecular dynamics simulations, in a twofold framework capable of exchanging information between the atomistic simulation and the machine-learning statistical backbone. The molecular dynamics framework incorporated is capable of producing training data automatically over a broad range of simulation conditions. Taking also into consideration that the observed accuracy of the developed machine-learning methods is enhanced, we expect that this twofold computational model will lead to substantial improvements in simulation applications, where possible.
This could open new directions in dealing with and calculating material properties. In cases where molecular dynamics (or another material-focused method’s) simulations are too time-consuming or expensive, machine learning can be incorporated as a faster and cost-effective alternative. Therefore, we believe that the bulk element simulation presented here can be upscaled in the future to deal with more complex fluids and confined geometries, even within wide ranges of temperatures and pressure conditions.
While the purpose of machine learning is not to create new physics, machine learning can be exploited to accelerate traditional computational methods. The future challenge is to become more interpretable, transparent, and explainable. This will allow scientists to better understand how the models work, and it will make these processes easier to apply in new and innovative ways. Incorporating machine learning into the field of fluid mechanics still has much to offer, and there is great potential for this technology to revolutionize the way that fluids are studied, modeled, and manufactured.



 ,
, 




{kind=link}
{kind=link}
{kind=link}