Ammonium Salts Catalyzed Acetalization Reactions in Green Ethereal Solvents

 , , , , , , and
, , , , , , and 
Abstract
:
1. Introduction
2. Results and Discussion
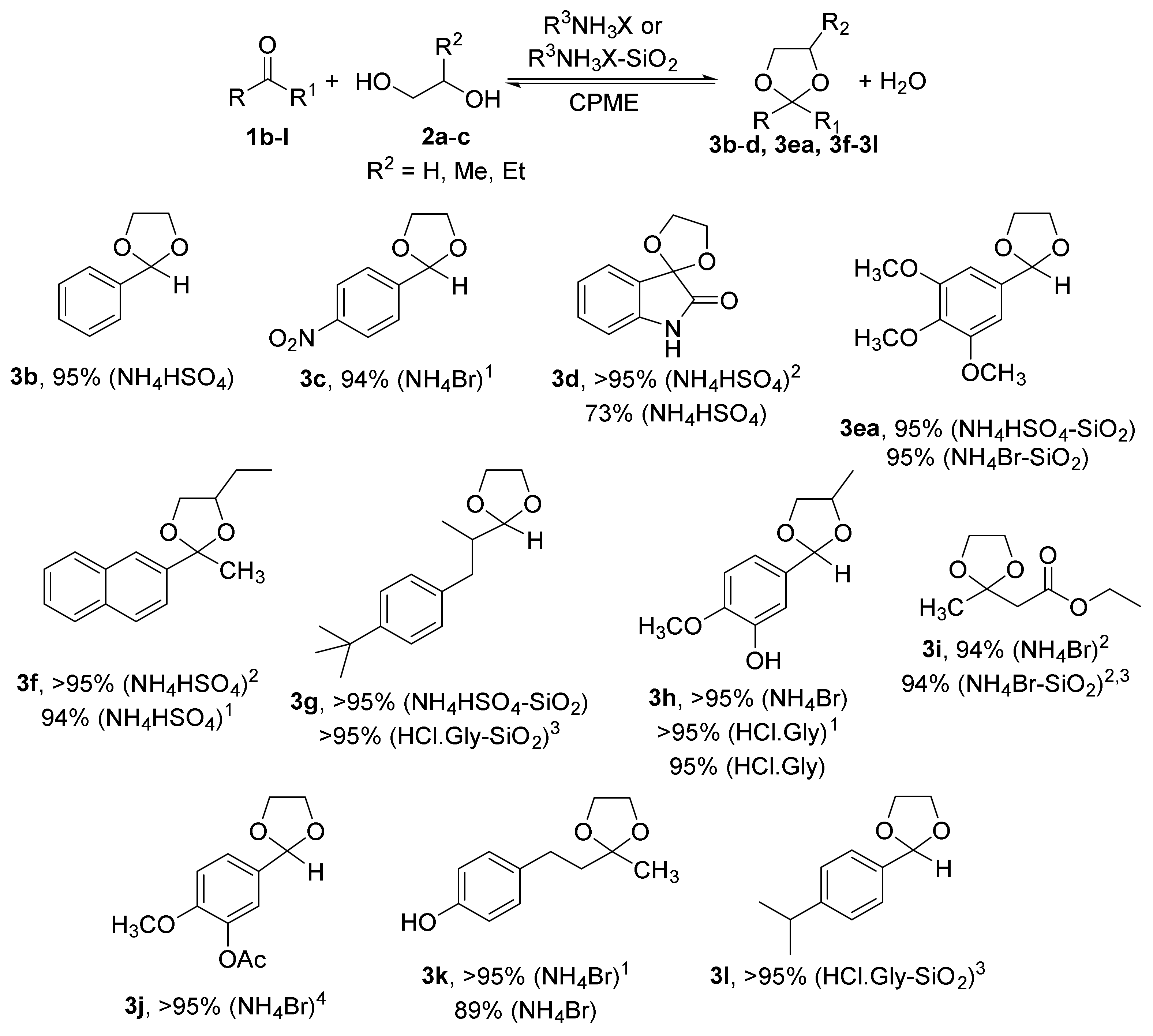
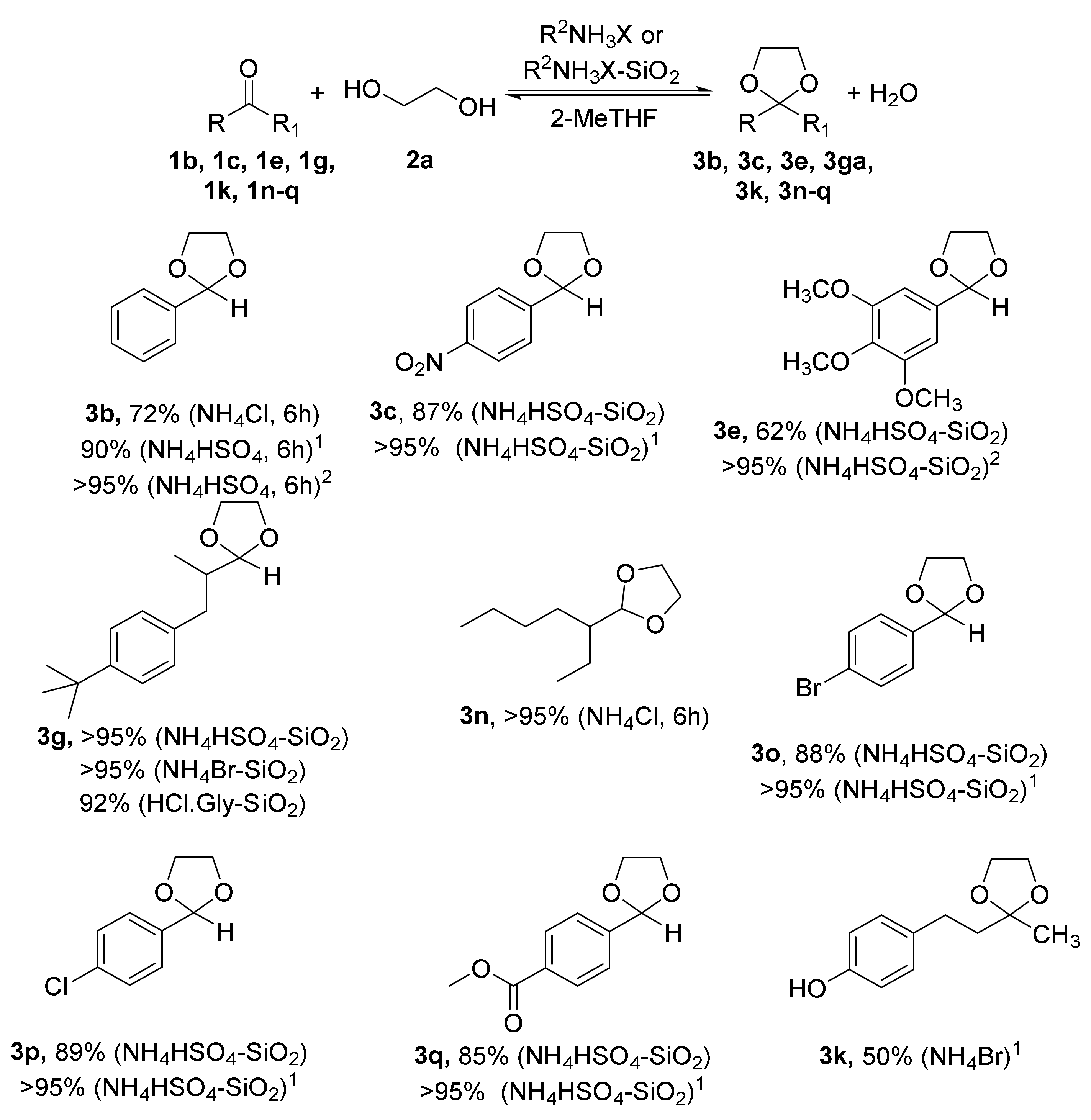
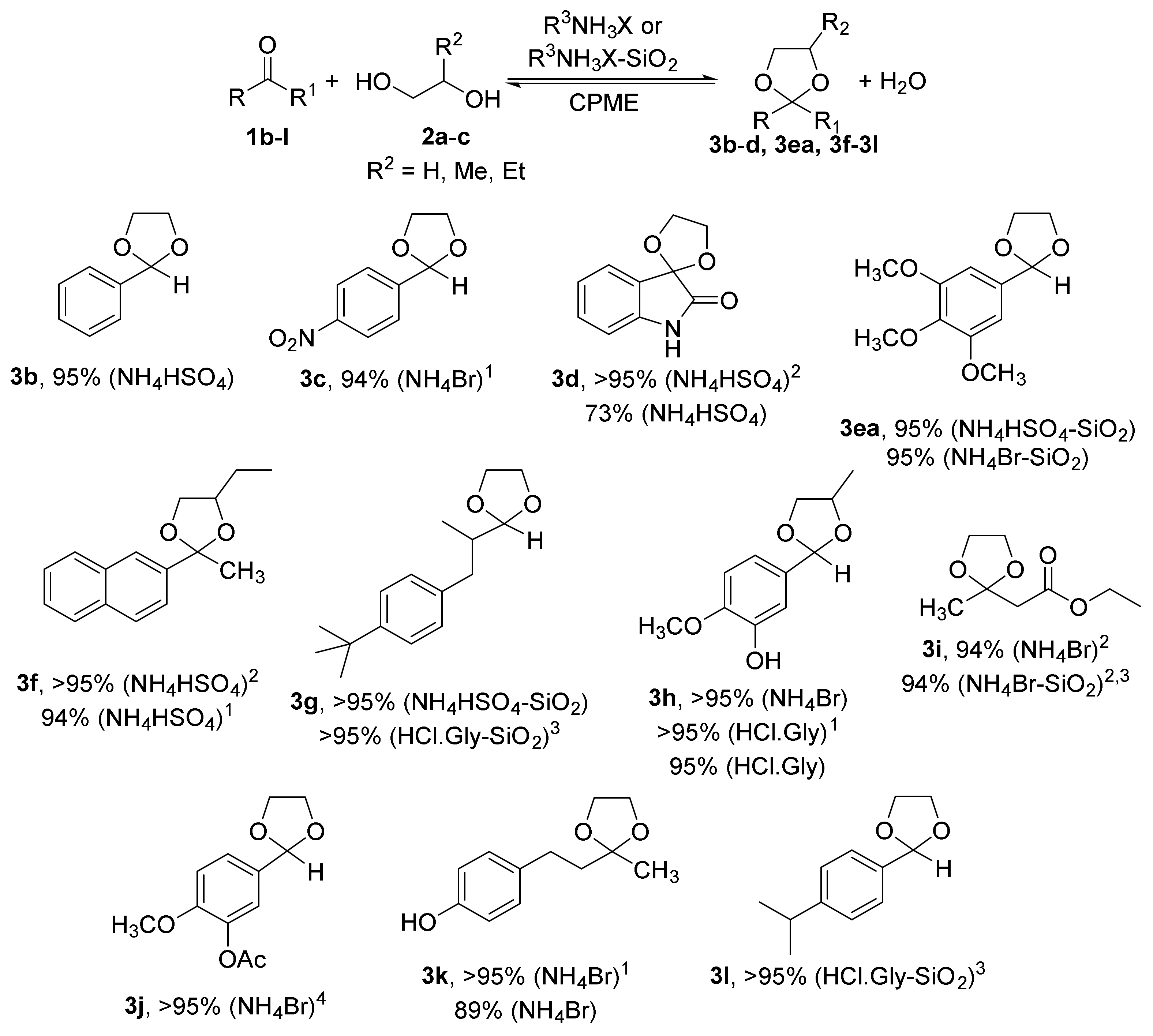
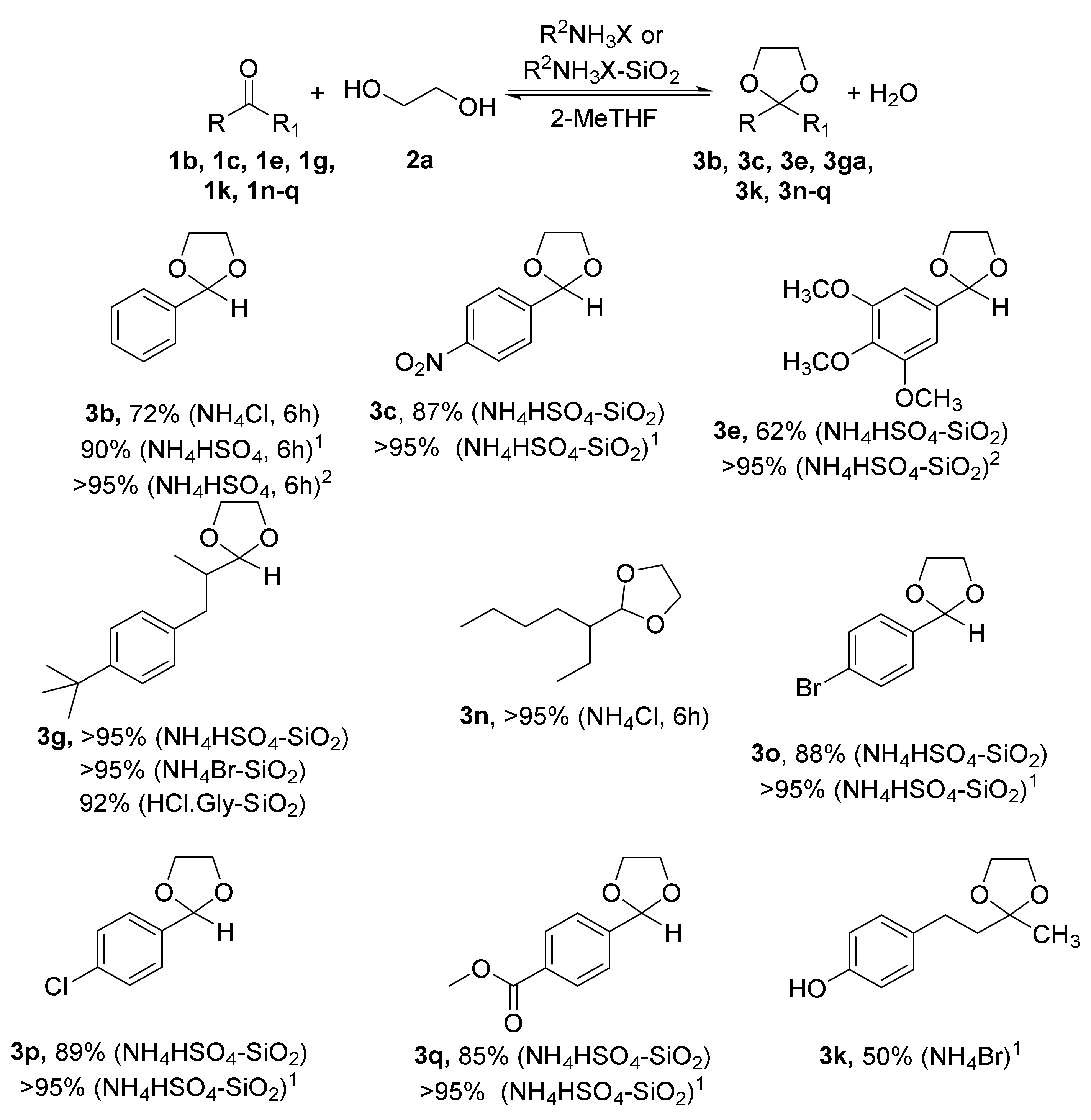
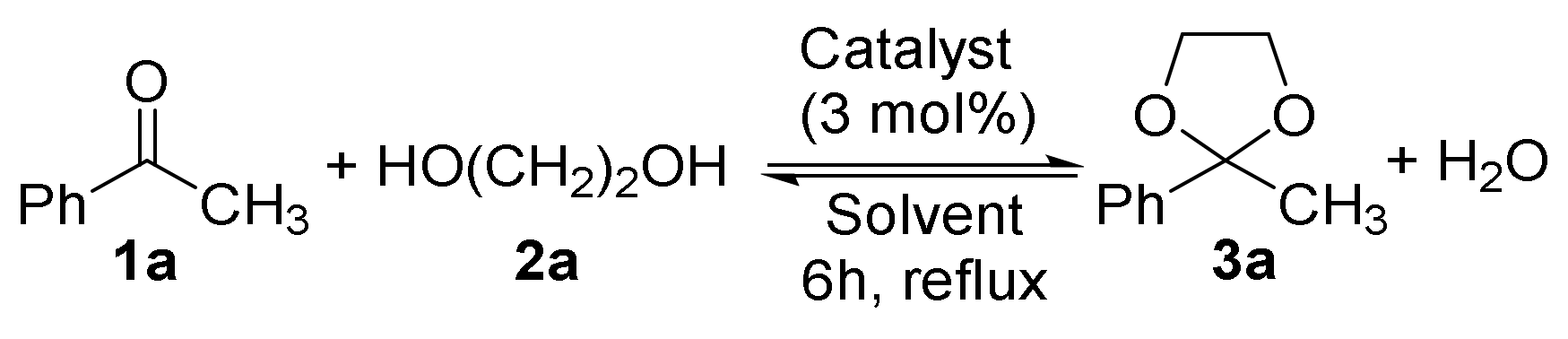
2.1. Synthesis of Acetals of Aldehydes and Ketones





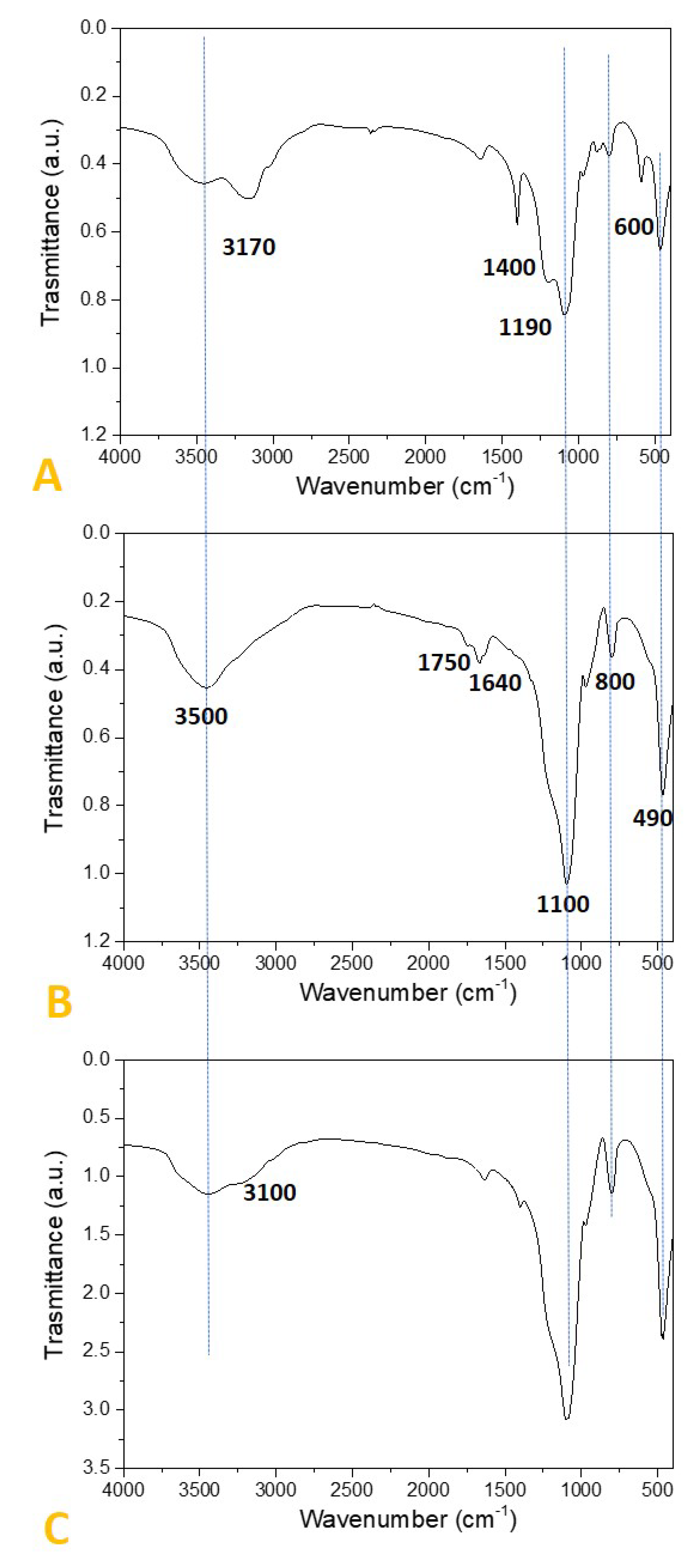
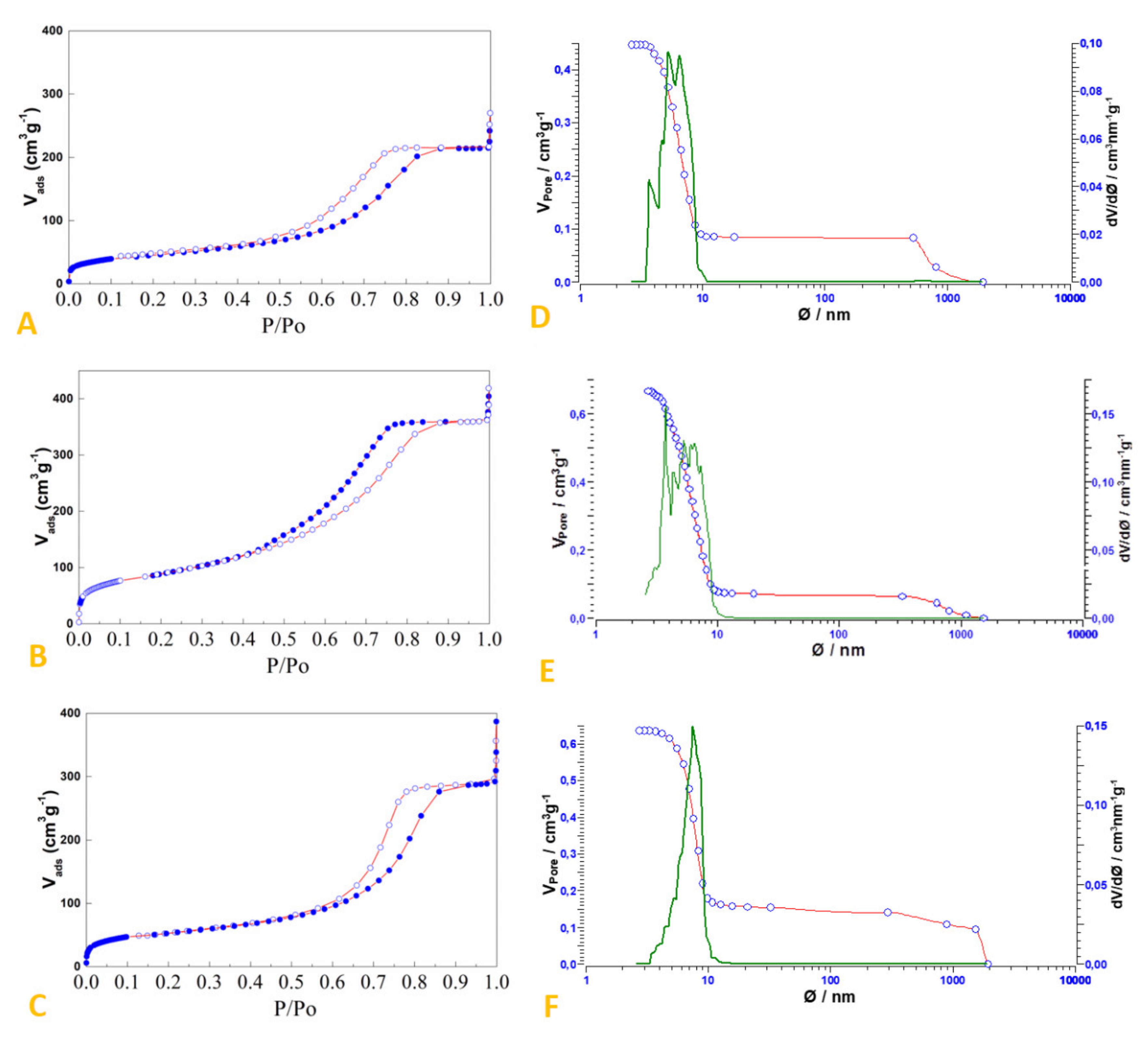
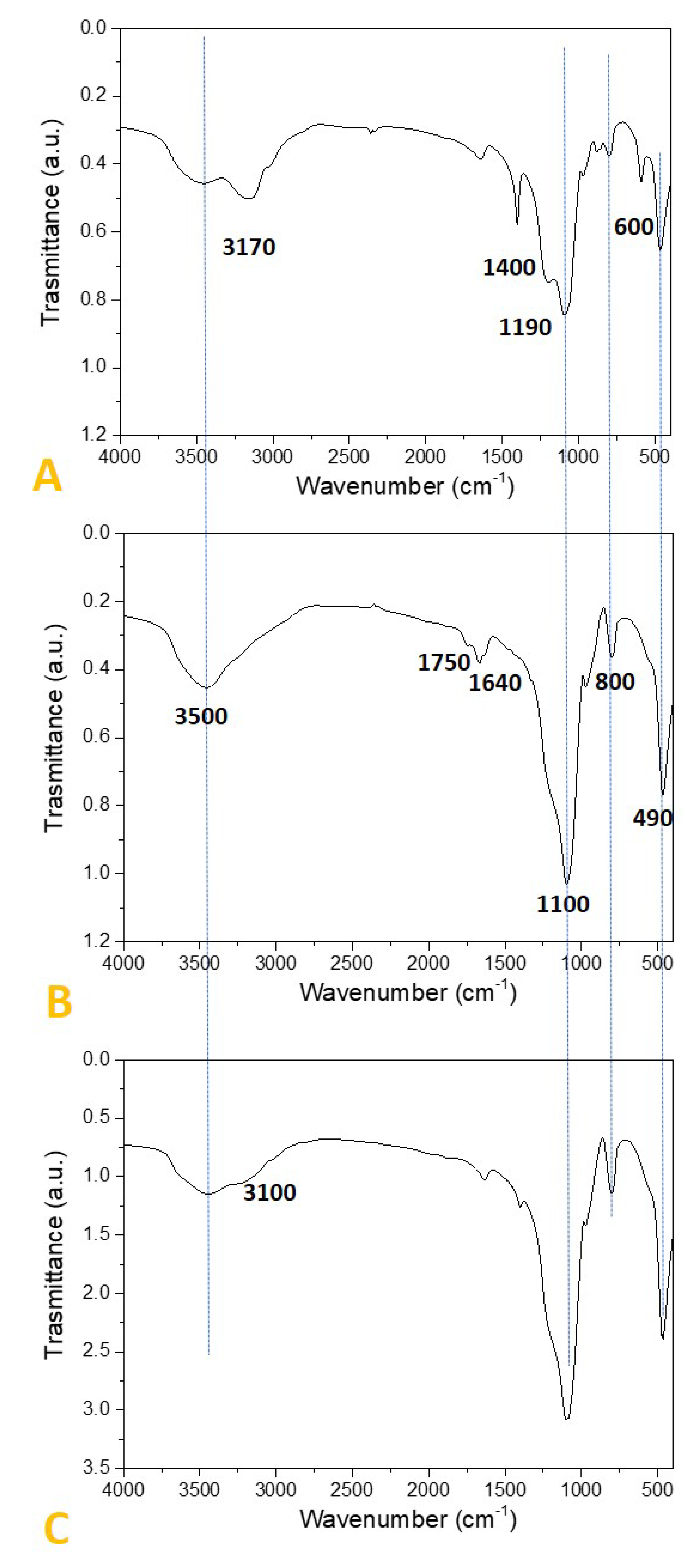
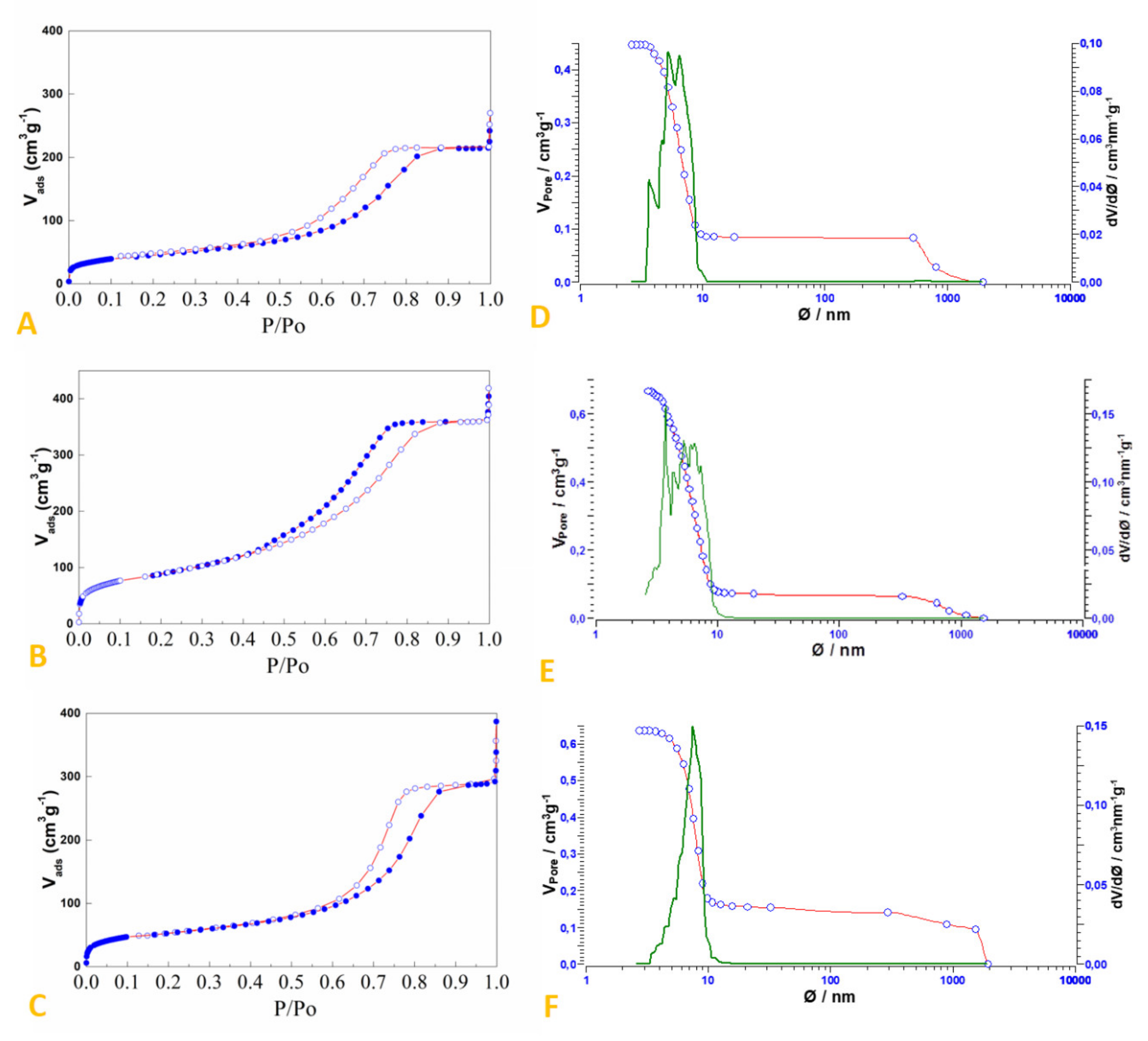
2.2. Preparation and Characterization of SiO2-Supported Ammonium Salts
2.3. Green Chemistry Metrics
3. Materials and Methods
3.1. General Procedure for the Acetalization Reaction
3.2. Preparation and Characterization of the SiO2-Supported Ammonium Salts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C.R.; Abou-Shehada, S.; Dunne, P.J. CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Henderson, R.K.; Jiménez-González, C.; Constable, D.J.C.; Alston, S.R.; Inglis, G.G.A.; Fisher, G.; Sherwood, J.; Binks, S.P.; Curzons, A.D. Expanding GSK’s solvent selection guide—Embedding sustainability into solvent selection starting at medicinal chemistry. Green Chem. 2011, 13, 854–862. [Google Scholar] [CrossRef]
- Raymond, M.J.; Slater, C.S.; Savelski, M.J. LCA approach to the analysis of solvent waste issues in the pharmaceutical industry. Green Chem. 2010, 12, 1826–1834. [Google Scholar] [CrossRef]
- Alfonsi, K.; Colberg, J.; Dunn, P.J.; Fevig, T.; Jennings, S.; Johnson, T.A.; Kleine, H.P.; Knight, C.; Nagy, M.A.; Perry, D.A.; et al. Green chemistry tools to influence a medicinal chemistry and research chemistry based organisation. Green Chem. 2008, 10, 31–36. [Google Scholar] [CrossRef]
- Capello, C.; Fischer, U.; Hungerbühler, K. What is a green solvent? A comprehensive framework for the environmental assessment of solvents. Green Chem. 2007, 9, 927–934. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green solvents for sustainable organic synthesis: State of the art. Green Chem. 2005, 7, 267–272. [Google Scholar] [CrossRef]
- Surburg, H.; Panten, J. Common Fragrance and Flavour Materials, 5th ed.; Wiley VCH: Weinheim, Germany, 2006; pp. 12–17, 38–44, 111–119. [Google Scholar]
- Hermann, A. Profragrances and properfumes. In The Chemistry and Biology of Volatiles; Hermann, A., Ed.; Wiley: Chichester, UK, 2010; pp. 333–362. [Google Scholar]
- Sivik, M.R. Profragrance Cyclic Acetals. WO Patent 99/00377, 26 June 1998. [Google Scholar]
- de Carvalho, D.C.; Oliveira, A.C.; Ferreira, O.P.; Filho, J.M.; Tehuacanero-Cuapa, S.; Oliveira, A.C. Titanate nanotubes as acid catalysts for acetalization of glycerol with acetone: Influence of the synthesis time and the role of structure on the catalytic performance. Chem. Eng. J. 2017, 313, 1454–1467. [Google Scholar] [CrossRef]
- Zhang, F.; Jin, Y.; Shi, J.; Zhong, Y.; Zhu, W.; El-Shall, M.S. Polyoxometalates confined in the mesoporous cages of metal–organic framework MIL-100(Fe): Efficient heterogeneous catalysts for esterification and acetalization reactions. Chem. Eng. J. 2015, 269, 236–244. [Google Scholar] [CrossRef]
- Gonzalez-Arellano, C.; Deb, S.; Luque, R. Selective glycerol transformations to high value-added products catalysed by aluminosilicate-supported iron oxide nanoparticles. Catal. Sci. Technol. 2014, 4, 4242–4249. [Google Scholar] [CrossRef]
- Mallesham, B.; Sudarsanam, P.; Raju, G.; Reddy, B.M. Design of highly efficient Mo and W-promoted SnO2 solid acids for heterogeneous catalysis: Acetalization of bio-glycerol. Green. Chem. 2013, 15, 478–489. [Google Scholar] [CrossRef]
- Miao, J.; Wan, H.; Shao, Y.; Guan, G.; Xu, B. Acetalization of carbonyl compounds catalyzed by acidic ionic liquid immobilized on silica gel. J. Mol. Cat. A Chem. 2011, 348, 77–82. [Google Scholar] [CrossRef]
- Deutsch, J.; Martin, A.; Lieske, H. Investigations on heterogeneously catalysed condensations of glycerol to cyclic acetals. J. Catal. 2007, 245, 428–435. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Velty, A. Synthesis of hyacinth, vanilla, and blossom orange fragrances: The benefit of using zeolites and delaminated zeolites as catalysts. Appl. Catal. A Gen. 2004, 263, 155–161, and references therein. [Google Scholar] [CrossRef]
- Sartori, G.; Ballini, R.; Bigi, R.; Bosica, G.; Maggi, R.; Righi, P. Protection (and deprotection) of functional groups in organic synthesis by heterogeneous catalysis. Chem. Rev. 2004, 104, 199–250. [Google Scholar] [CrossRef]
- Palaniappan, S.; Narander, P.; Saravanan, C.; Rao, V.J. Polyaniline-supported sulfuric acid salt as a powerful catalyst for the protection and deprotection of carbonyl compounds. Synlett 2003, 1793–1796. [Google Scholar] [CrossRef]
- Green, T.W.; Wuts, P.G.M. Protection of the carbonyl group. In Protective Groups in Organic Synthesis, 3rd ed.; Wiley: New York, NY, USA, 1999; pp. 293–329. [Google Scholar]
- Azzena, U.; Carraro, M.; Modugno, G.; Pisano, L.; Urtis, L. Heterogeneous acidic catalysts for the tetrahydropyranylation of alcohols and phenols in green ethereal solvents. Beilstein J. Org. Chem. 2018, 14, 1655–1659. [Google Scholar] [CrossRef] [Green Version]
- Azzena, U. 1,2-Diaryl-1,2-disodioethanes—Versatile, highly reactive, and tunable synthetic equivalents of sodium metal. Aust. J. Chem. 2017, 70, 647–651. [Google Scholar] [CrossRef]
- Azzena, U.; Carraro, M.; Mamuye, A.D.; Murgia, I.; Pisano, L.; Zedde, G. Cyclopentyl methyl ether–NH4X—A novel solvent—Catalyst system for low impact acetalization reactions. Green Chem. 2015, 17, 3281–3284. [Google Scholar] [CrossRef]
- Azzena, U.; Kondrot, F.; Pisano, L.; Pittalis, M. A green solvent approach to the chemistry of 1,2-Diaryl-1,2-disodioethanes. Appl. Organometal. Chem. 2012, 26, 180–184. [Google Scholar] [CrossRef]
- Watanabe, K.; Yamagiwa, N.; Torisawa, Y. Cyclopentyl methyl ether as a new and alternative process solvent. Org. Proc. Res. Dev. 2007, 11, 251–258. [Google Scholar] [CrossRef]
- Sakamoto, S. Contribution of cyclopentyl methyl ether (CPME) to green chemistry. Chim. Oggi 2013, 31, 24–27. [Google Scholar]
- Azzena, U.; Carraro, M.; Pisano, L.; Monticelli, S.; Bartolotta, R.; Pace, V. Cyclopentyl methyl ether—An elective eco-friendly ethereal solvent in classical and modern organic chemistry. ChemSusChem 2019, 12, 40–70. [Google Scholar] [CrossRef] [Green Version]
- de Gonzalo, G.; Alcántara, A.R.; Dominguez de María, P. Cyclopentyl methyl ether (CPME): A versatile eco-friendly solvent for applications in biotechnology and biorefineries. ChemSusChem 2019, 12, 2083–2097. [Google Scholar] [CrossRef]
- Alcántara, A.R.; Domínguez de María, P. Recent advances on the use of 2-methyltetrahydrofuran (2-MeTHF) in biotransformations. Curr. Green Chem. 2018, 5, 86–103. [Google Scholar] [CrossRef]
- Monticelli, S.; Castoldi, L.; Murgia, I.; Senatore, R.; Mazzeo, E.; Wackerlig, J.; Urban, E.; Langer, T.; Pace, V. Recent advancements on the use of 2-methyltetrahydrofuran in organometallic chemistry. Monatsch. Chem. 2017, 148, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Pace, V.; Hoyos, P.; Castoldi, L.; Dominguez de María, P.; Alcántara, A.R. 2-methyltetrahydrofuran (2-MeTHF): A biomass-derived solvent with broad application in organic chemistry. ChemSusChem 2012, 5, 1369–1379. [Google Scholar] [CrossRef]
- Aycock, D.F. Solvent applications of 2-methyltetrahydrofuran in organometallic and biphasic reactions. Org. Proc. Res. Dev. 2007, 11, 156–159. [Google Scholar] [CrossRef]
- Handbook of Chemistry and Physics, 57th ed.; Weast, R.C. (Ed.) CRC Press: Cleveland, OH, USA, 1976–1977; pp. D 1–D 36. [Google Scholar]
- Watanabe, K. The toxicological assessment of cyclopentyl methyl ether (CPME) as a green solvent. Molecules 2013, 18, 3183–3194. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, V.; Coleman, J.; Ferry, J.B.; Johnson, N.; Mathe, M.; Scott, J.P.; Xu, J. Toxicological assessment of 2-methyltetrahydrofuran and cyclopentyl methyl ether in support of their use in pharmaceutical chemical process development. Org. Proc. Res. Dev. 2011, 15, 939–941. [Google Scholar] [CrossRef]
- No conversion of the starting material was observed in the presence of 3 mol% of glycine.
- pKa (in H2O): HSO4− = 1.99 and NH4+ = 9.25: Lange’s Handbook of Chemistry, 15th ed.; Dean, J.A. (Ed.) McGraw-Hill: New York, NY, USA, 1999; Table 8.7. [Google Scholar]
- Poor conversions have already been observed for reactions run in the presence of (unactivated) zeolite ZSM-5 or (activated) molecular sieves (3 Ǻ); see ref. [22].
- As a note of caution, it is worth noting that reactions run under atmospheric conditions (CaCl2 valve) in 2-MeTHF (see Experimental Part and Supplementary Materials) led to the formation of minor amounts of peroxides (<5 mg/L) as semiquantitatively evaluated with a commercially available kit (Quantofix®, measuring range 05–25 mgL−1 H2O2). Indeed, the degradation mechanism of 2-MeTHF under free radical conditions, involving peroxide intermediates, was recently reported: Kobayashi, S.; Tamura, T.; Yoshimoto, S.; Kawakami, T.; Masuyama, A. 4-Methyltetrahydropyran (4-MeTHP): Application as an Organic Reaction Solvent. Chem. Asian J. 2019, 14, 3921–3937, Accordingly, such reactions can be more safely run under an inert atmosphere. [Google Scholar]
- For a solvent free SiO2 mediated synthesis of acetals under autogenous pressure, see: Rohand, T.; Savary, J.; Markó, I.E. Synthesis of dioxolanes and oxazolidines by silica gel catalysis. Monatsh. Chem. 2018, 149, 1429–1436. [Google Scholar]
- Vyglazov, O.G.; Chuiko, V.A.; Izotova, L.V.; Vintarskaya, Z.V.; Yudenko, R.Y. Synthesis and perfume characteristics of acetals containing an aromatic ring. Russ. J. Appl. Chem. 2001, 74, 1888–1891. [Google Scholar] [CrossRef]
- Kenya, I.; Takashi, A. Vanillin Acetals and Sensory Stimulation Composition Containing the Same. WO Patent 2007004740 (A1), 1 January 2007. [Google Scholar]
- Clement, M.J.; Corma, A.; Velty, A.; Susarte, M. Zeolites for the production of fine chemicals: Synthesis of the fructone fragrancy. J. Catal. 2000, 196, 345–351. [Google Scholar] [CrossRef]
- Perflavory. Available online: http://www.perflavory.com/docs/doc1035181.html (accessed on 31 August 2020).
- Perflavory. Available online: http://www.perflavory.com/docs/doc1020771.html (accessed on 31 August 2020).
- The Good Scents Company Information System. Available online: http://www.thegoodscentscompany.com/data/rw1004031.html (accessed on 31 August 2020).
- pKa (in H2O): P-TSA = −1.34: Serjeant, E.P.; Dempsey, B. Ionisation Constants of Organic Acids in Aqueous Solution; IUPAC Chemical Data Series No 23; Pergamon Press: New York, NY, USA, 1979. [Google Scholar]
- Harrowven, D.C.; Woodcock, T.; Howes, P.D. Total synthesis of cavicularin and riccardin C: Addressing the synthesis of an arene that adopts a boat configuration. Angew. Chem. Int. Ed. 2005, 44, 3899–3901. [Google Scholar] [CrossRef]
- Kadam, A.; Nguyen, M.; Kopach, M.; Richardson, P.; Gallou, F.; Wan, Z.-K.; Zhang, W. Comparative performance evaluation and systematic screening of solvents in a range of Grignard reactions. Green Chem. 2013, 15, 1880–1888. [Google Scholar] [CrossRef] [Green Version]
- For an in-deep investigation on the reactivity of Grignard reagents in CPME, see: Kobayashi, S.; Shibukawa, K.; Miyaguchi, Y.; Masuyama, A. Grignard reactions in cyclopentyl methyl ether. Asian J. Org. Chem. 2016, 5, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Breton, G.W. Selective monoacetylation of unsymmetrical diols catalyzed by silica gel-supported sodium hydrogen sulfate. J. Org. Chem. 1997, 62, 8952–8954. [Google Scholar] [CrossRef]
- Cui, Y.; Bu, X.; Zou, H.; Xu, X.; Zhou, D.; Liu, H.; Zhang, X.; Liu, Y.; Sun, H.; Jiang, J.; et al. A dual solvent evaporation route for preserving carbon nanoparticle fluorescence in silica gel and producing white light-emitting diodes. Mater. Chem. Front. 2017, 1, 387–393. [Google Scholar] [CrossRef]
- Shi, Y.-J.; Shu, H.; Zhang, Y.-H.; Fan, H.-M.; Zhang, Y.-P.; Yang, L.-J. Formation and decomposition of NH4HSO4 during selective catalytic reduction of NO with NH3 over V2O5-WO3/TiO2 catalysts. Fuel Process. Technol. 2016, 150, 141–147. [Google Scholar] [CrossRef]
- Cziczo, D.J.; Abbatt, J.P.D. Infrared observations of the response of NaCl, MgCl2, NH4HSO4, and NH4NO3 aerosols to changes in relative humidity from 298 to 238 K. J. Phys. Chem. A 2000, 104, 2038–2047. [Google Scholar] [CrossRef]
- Ye, D.; Qu, R.; Liu, S.; Zheng, C.; Gao, X. New insights into the decomposition behavior of NH4HSO4 on the SiO2-decorated SCR catalyst and its enhanced so2-resistant ability. ACS Omega 2019, 4, 4927–4935. [Google Scholar] [CrossRef] [Green Version]
- Fischer, G.; Cao, X.; Cox, N.; Francis, M. The FT-IR spectra of glycine and glycylglycine zwitterions isolated in alkali halide matrices. Chem. Phys. 2005, 313, 39–49. [Google Scholar] [CrossRef]
- Shukur, M.F.; Kadir, M.F.Z. Electrical and transport properties of NH4Br-doped cornstarch-based solid biopolymer electrolyte. Ionics 2015, 21, 111–124. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquérol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Liu, W.; Yin, P.; Liu, X.; Dong, X.; Zhang, J.; Xu, Q. Thermodynamics, kinetics, and isotherms studies for gold(III) adsorption using silica functionalized by diethylenetriaminemethylenephosphonic acid. Chem. Eng. Res. Des. 2013, 91, 2748–2758. [Google Scholar] [CrossRef]
- Sigma-Aldrich. Available online: https://www.sigmaaldrich.com/catalog/product/sial/227196?lang=it®ion=IT, (accessed on 31 August 2020).
- Yang, T.; Liu, Q.; Cheng, Y.; Cai, W.; Ma, Y.; Yang, L.; Wu, Q.; Orband-Miller, L.A.; Zhou, L.; Xiang, Z.; et al. Discovery of tertiary amine and indole derivatives as potent RORγt inverse agonists. ACS Med. Chem. Lett. 2014, 5, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Qiu, R.; Gong, J.; Tian, J.; Huang, Y. Tertiary Amine Derivative or Salt Thereof, Preparation Method and Application Thereof. CN Patent CN109206346 (A), 20 December 2019. The experimental procedure in English is reported in Reaxys®. [Google Scholar]
- Cliffe, I.A.; Lien, E.L.; Mansell, H.L.; Steiner, K.E.; Todd, R.S.; White, A.C.; Black, R.M. Oral hypoglycemic agents. Pyrimido [1,2-a]indoles and related compounds. J. Med. Chem. 1992, 35, 1169–1175. [Google Scholar] [CrossRef]
- Wenkert, E.; Hudlický, T. Reactions of isatin dimethyl ketal and its ethyl imino ether with methyllithium. Synth. Commun. 1977, 7, 541–547. [Google Scholar] [CrossRef]
- Xu, G.; Lei, H.; Zhou, G.; Zhang, C.; Xie, L.; Zhang, W.; Cao, R. Boosting hydrogen evolution by using covalent frameworks of fluorinated cobalt porphyrins supported on carbon nanotubes. Chem. Commun. 2019, 55, 12647–12650. [Google Scholar] [CrossRef]
- Nishi, H.; Namari, T.; Kobatake, S. Photochromic polymers bearing various diarylethene chromophores as the pendant: Synthesis, optical properties, and multicolor photochromism. J. Mater. Chem. 2011, 21, 17249–17258. [Google Scholar] [CrossRef]
- Curzons, A.D.; Constable, D.J.C.; Mortimer, D.N.; Cunningham, V.L. So you think your process is green, how do you know?—Using principles of sustainability to determine what is green—A corporate perspective. Green Chem. 2001, 3, 1–6. [Google Scholar] [CrossRef]
- Jiménez-González, C.; Ponder, C.S.; Broxterman, Q.B.; Manley, J.B. Using the right green yardstick: Why process mass intensity is used in the pharmaceutical industry to drive more sustainable processes. Org. Process Res. Dev. 2011, 15, 912–917. [Google Scholar] [CrossRef]
- Li, J.; Simmons, E.M.; Eastgate, M.D. A data-driven strategy for predicting greenness scores, rationally comparing synthetic routes and benchmarking PMI outcomes for the synthesis of molecules in the pharmaceutical industry. Green Chem. 2017, 19, 127–139. [Google Scholar] [CrossRef]
- Andraos, J. Critical evaluation of published algorithms for determining material efficiency green metrics of chemical reactions and synthesis plans. ACS Sustain. Chem. Eng. 2016, 4, 1917–1933. [Google Scholar] [CrossRef]
- Radi, S.; Basbas, N.; Tighadouini, S.; Bacquet, M. New polysiloxane surfaces modified with ortho-, meta- or para-nitrophenyl receptors for copper adsorption. J. Surf. Eng. Mater. Adv. Technol. 2014, 4, 21–28. [Google Scholar]
- Burhan, M.; Shahzad, M.W.; Ng, K.C. A universal theoretical framework in material characterization for tailored porous surface design. Sci. Rep. 2019, 9, 8773. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organic Solvent | Bp (°C) | Composition (%) 1 |
|---|---|---|
| Toluene | 85.0 2 | 79.8:20.2 2 |
| Benzene | 69.4 2 | 91.1:8.9 2 |
| CPME | 83 3 | 83.7:16.3 3 |
| 2-MeTHF | 71 4 | 89.4:10.6 4 |
| Entry | 2a (Equivalent) | Solvent | Catalyst | Product Distribution 1a/3a (%) 2 |
|---|---|---|---|---|
| 1 | 1.1 | CPME | NH4Cl | 35/65 3 |
| 2 | 1.1 | CPME | NH4Br | 21/79 3 |
| 3 | 1.1 | CPME | HCl.Gly | 36/64 3 |
| 4 | 1.1 | CPME | (NH4)2SO4 | 68/32 |
| 5 | 1.1 | CPME | (NH4)HSO4 | 10/90 4 |
| 6 | 1.5 | CPME | (NH4)HSO4 | 5/95 |
| 7 | 2.0 | CPME | (NH4)HSO4 | <5/>95 3,5 |
| 8 | 2.0 | Toluene | (NH4)HSO4 | 20/80 |
| 9 | 1.1 | CPME | p-TSA | 13/87 |
| 10 | 2.0 | CPME | p-TSA | <5/>95 |
| 11 | 1.1 | CPME | Amberlyst 15® | 10/90 6 |
| 12 | 1.1 | CPME | Mont K10 7 | 46/54 |
| 13 | 1.1 | 2-MeTHF | (NH4)HSO4 | 60/40 |
| 14 | 2.0 | 2-MeTHF | (NH4)HSO4 | 37/63 5 |
| 15 | 1.1 | 2-MeTHF | Amberlyst 15® | 89/11 6 |
| 16 | 1.1 | 2-MeTHF | Mont K10 7 | 72/28 |
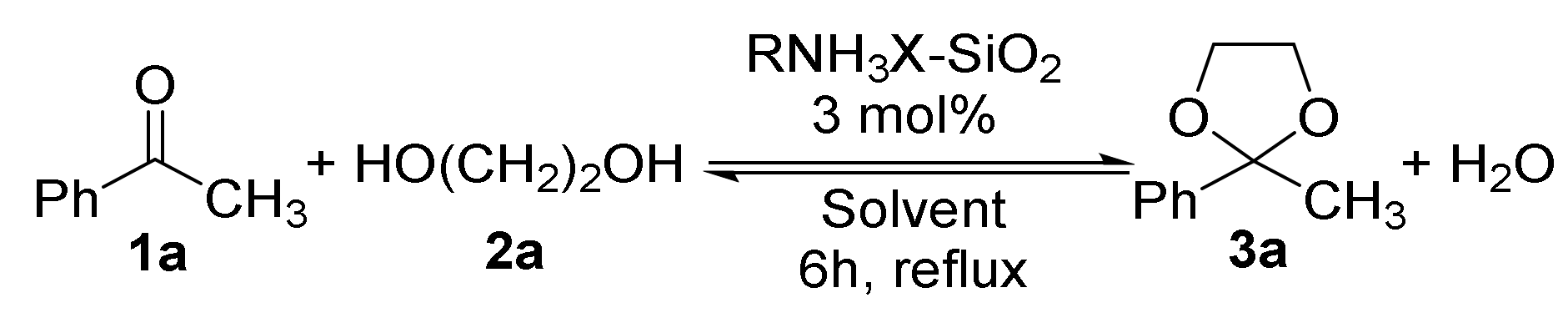
| Entry | 2a (Equivalent) | Solvent | Catalyst | Product Distribution 1a/3a (%) 2 |
|---|---|---|---|---|
| 1 | 1.1 | CPME | NH4Br-SiO2 | 25/75 |
| 2 | 1.1 | CPME | HCl.Gly-SiO2 | 35/65 3 |
| 3 | 1.1 | CPME | NH4HSO4-SiO2 | 14/86 4 |
| 4 | 2.0 | CPME | NH4HSO4-SiO2 | <5/>95 4 |
| 5 | 1.1 | CPME | SiO2 | 93/7 |
| 6 | 1.1 | 2-MeTHF | NH4HSO4-SiO2 | 51/49 |
| 7 | 2.0 | 2-MeTHF | NH4HSO4-SiO2 | 38/62 |
| System | Surface Area (m2/g) | Pore Volume (cm3/g) | Median Pore Width (nm) |
|---|---|---|---|
| Silica Gel (60 Å, 230–240 mesh) | 550 1 | ≈0.8 1 | 6.0 1 |
| HCl.Gly-SiO2 | 319 | 0.555 | 5.2 |
| NH4Br-SiO2 | 185 | 0.444 | 5.5 |
| NH4HSO4-SiO2 | 161 | 0.331 | 5.6 |
| Compound | Procedure [Ref] | Yield (%) | RME (%) | PMI |
|---|---|---|---|---|
| 3c | This work | 95 1 | 65 | 4.6 |
| 3c | Yang et al. [60] | 100 1 | 70 | 20.5 |
| 3c | Wang et al. [61] | 100 1 | 61 | 12.4 |
| 3d | This work | 95 1 | 67 | 3.9 |
| 3d | Cliffe et al. [62] | 99 2 | 48 | 63.4 |
| 3d | Wenkert et al. [63] | 90 2 | 82 | 44.7 |
| 3o | This work | 95 2 | 78 | 3.3 |
| 3o | Xu et al. [64] | 100 2 | 28 | 37.4 |
| 3o | Nishi et al. [65] | 95 2 | 25 | 20.4 |
| 3j | This work Toluene/p-TSA | 69 2 | 62 | 6.0 |
| 3j | This work CPME/NH4HSO4 | 63 2 | 57 | 3.2 |
| 3j | This work CPME/NH4Br | 78 2 | 71 | 2.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzena, U.; Carraro, M.; Corrias, M.; Crisafulli, R.; De Luca, L.; Gaspa, S.; Nuvoli, L.; Pintus, S.; Pisano, L.; Polese, R.; et al. Ammonium Salts Catalyzed Acetalization Reactions in Green Ethereal Solvents. Catalysts 2020, 10, 1108. https://doi.org/10.3390/catal10101108
Azzena U, Carraro M, Corrias M, Crisafulli R, De Luca L, Gaspa S, Nuvoli L, Pintus S, Pisano L, Polese R, et al. Ammonium Salts Catalyzed Acetalization Reactions in Green Ethereal Solvents. Catalysts. 2020; 10(10):1108. https://doi.org/10.3390/catal10101108
Chicago/Turabian StyleAzzena, Ugo, Massimo Carraro, Martina Corrias, Rosella Crisafulli, Lidia De Luca, Silvia Gaspa, Luca Nuvoli, Salvatore Pintus, Luisa Pisano, Riccardo Polese, and et al. 2020. "Ammonium Salts Catalyzed Acetalization Reactions in Green Ethereal Solvents" Catalysts 10, no. 10: 1108. https://doi.org/10.3390/catal10101108
APA StyleAzzena, U., Carraro, M., Corrias, M., Crisafulli, R., De Luca, L., Gaspa, S., Nuvoli, L., Pintus, S., Pisano, L., Polese, R., Sanna, M., Satta, G., Senes, N., Urtis, L., & Garroni, S. (2020). Ammonium Salts Catalyzed Acetalization Reactions in Green Ethereal Solvents. Catalysts, 10(10), 1108. https://doi.org/10.3390/catal10101108