Enzymatic Bioreactors: An Electrochemical Perspective


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
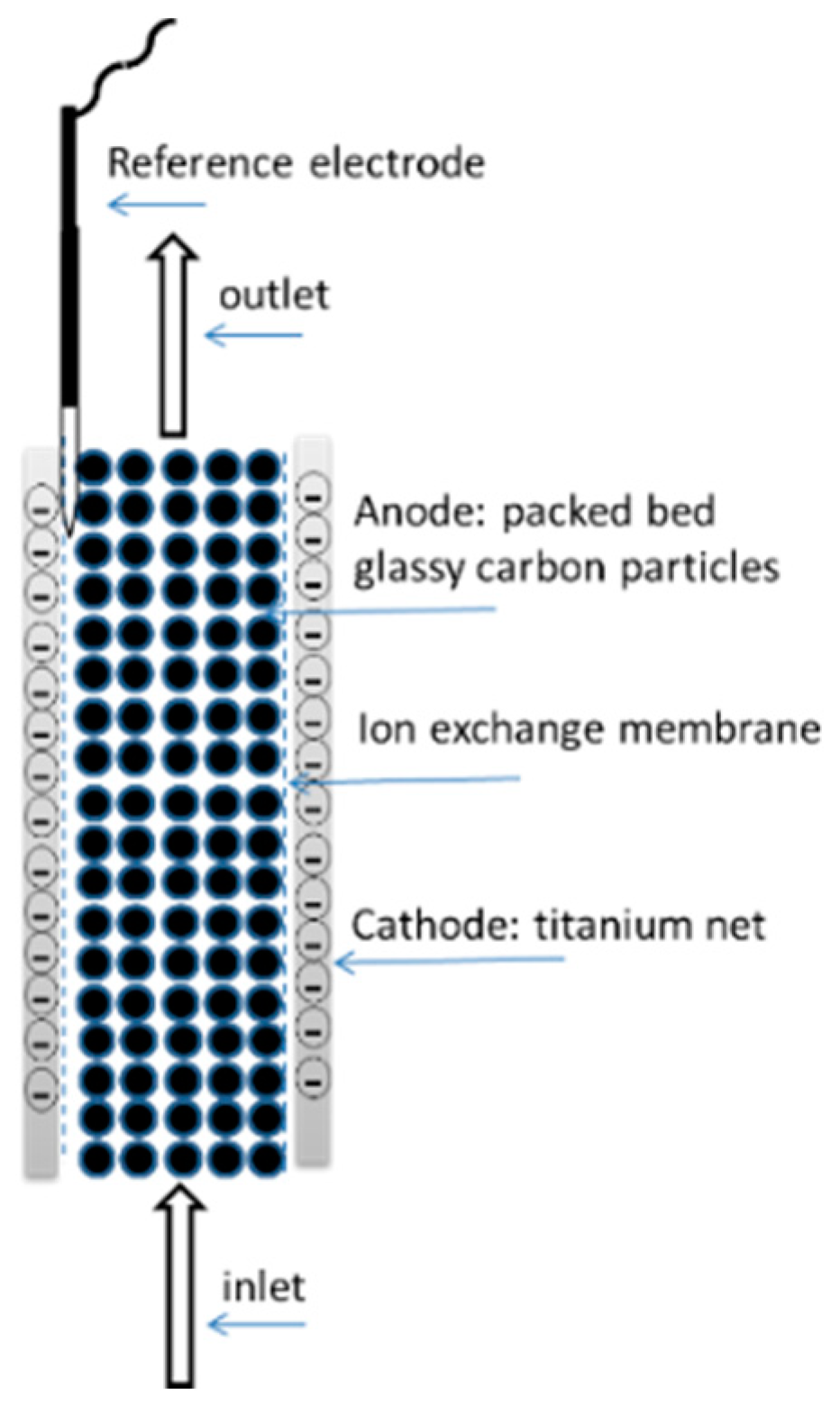{kind=link}
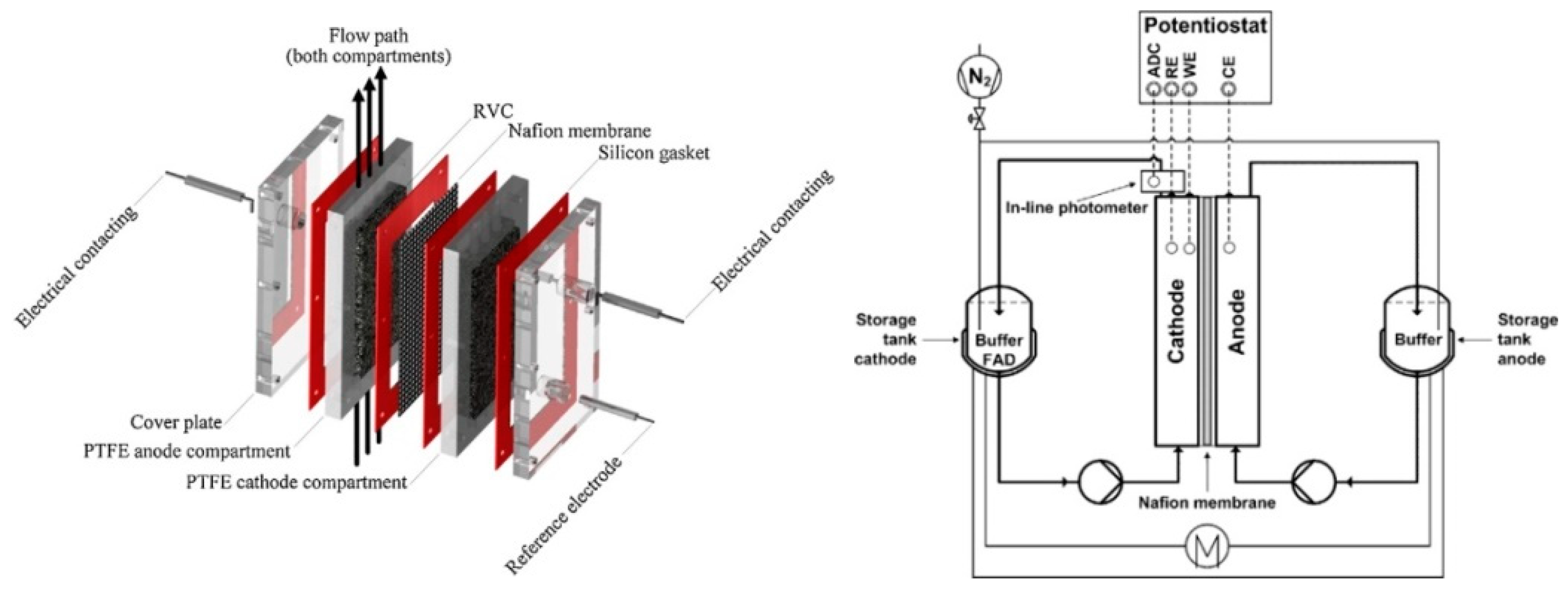{kind=link}
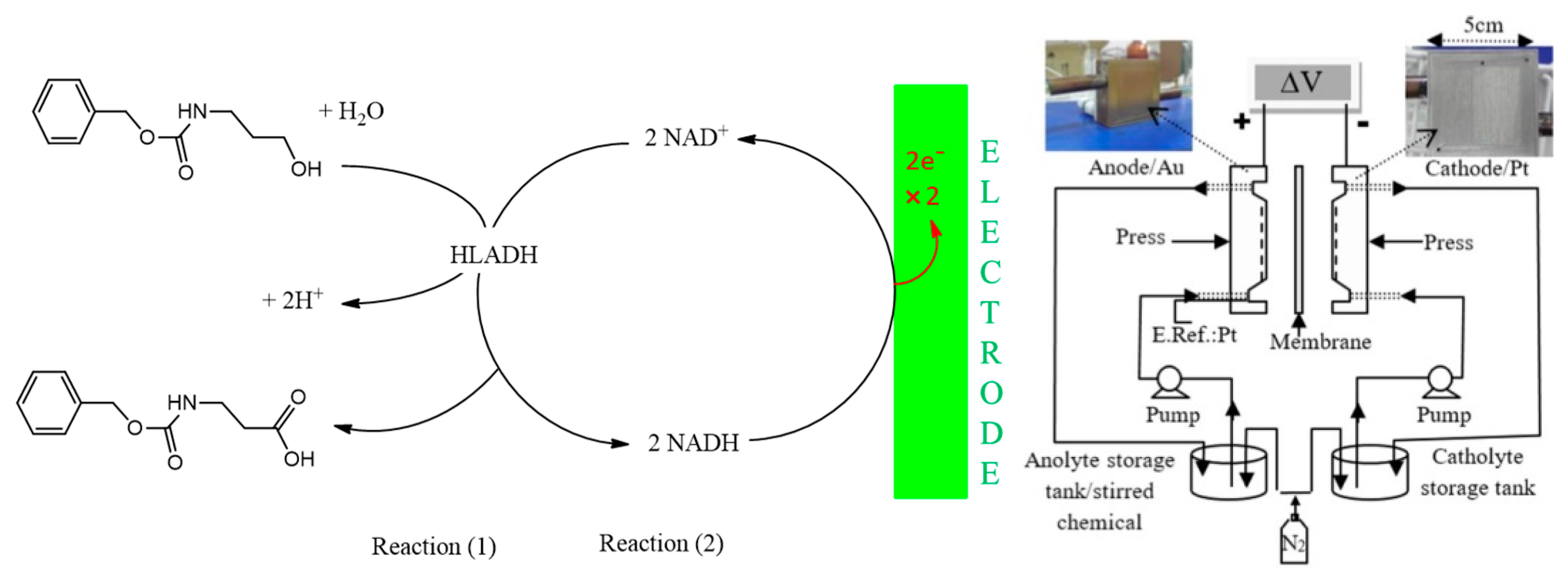{kind=link}
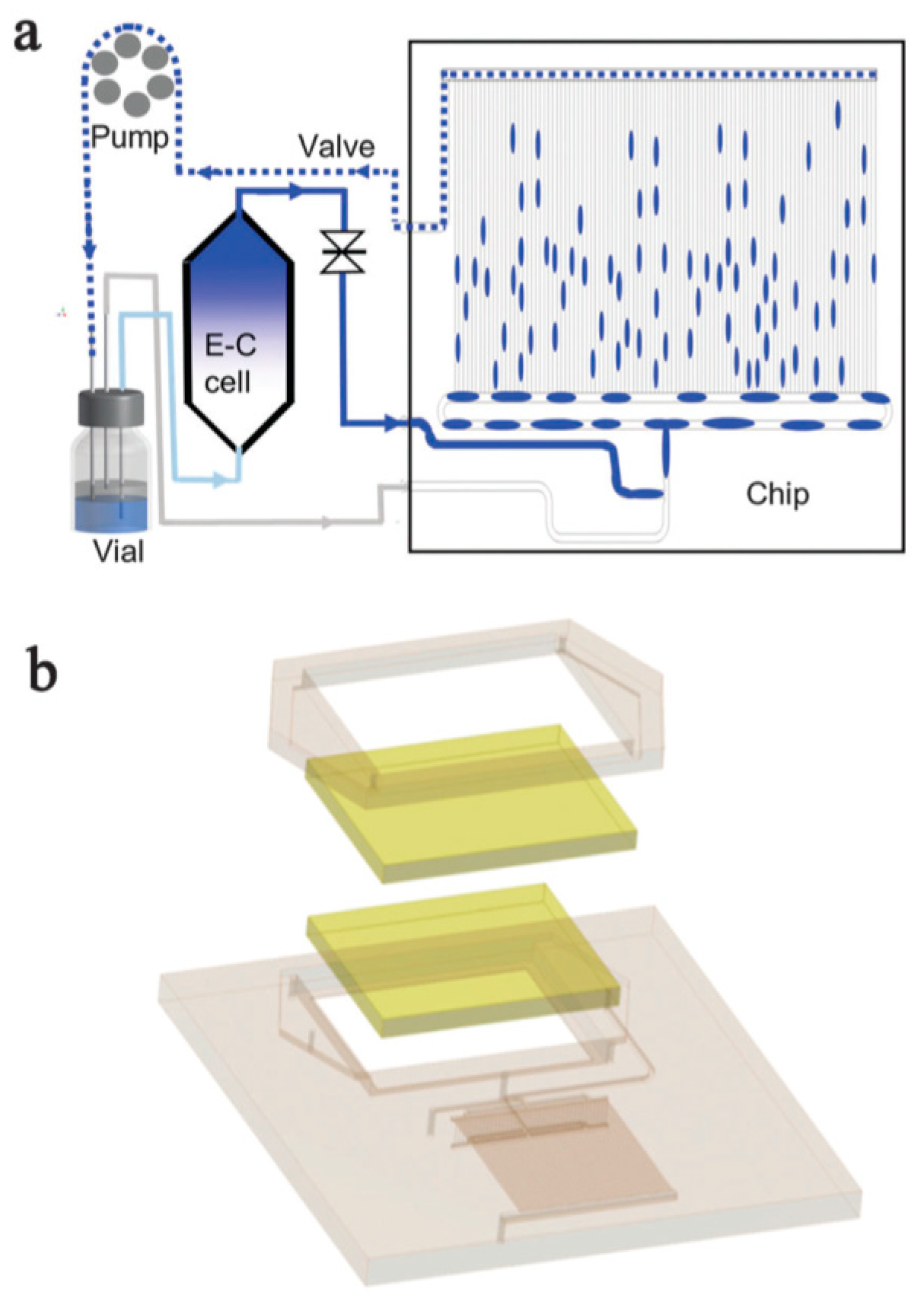{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Bioelectrochemistry and Bioelectrocatalysts
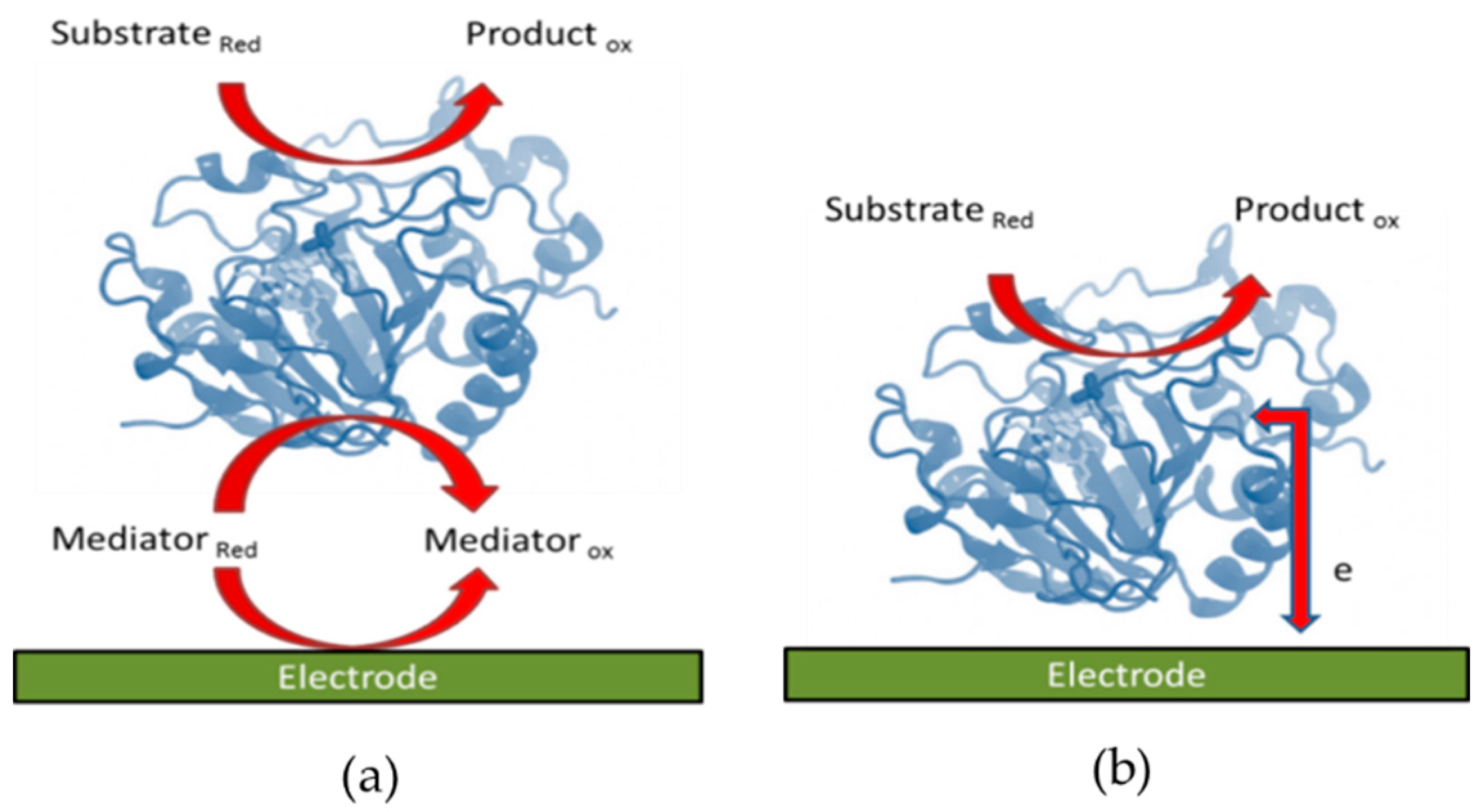
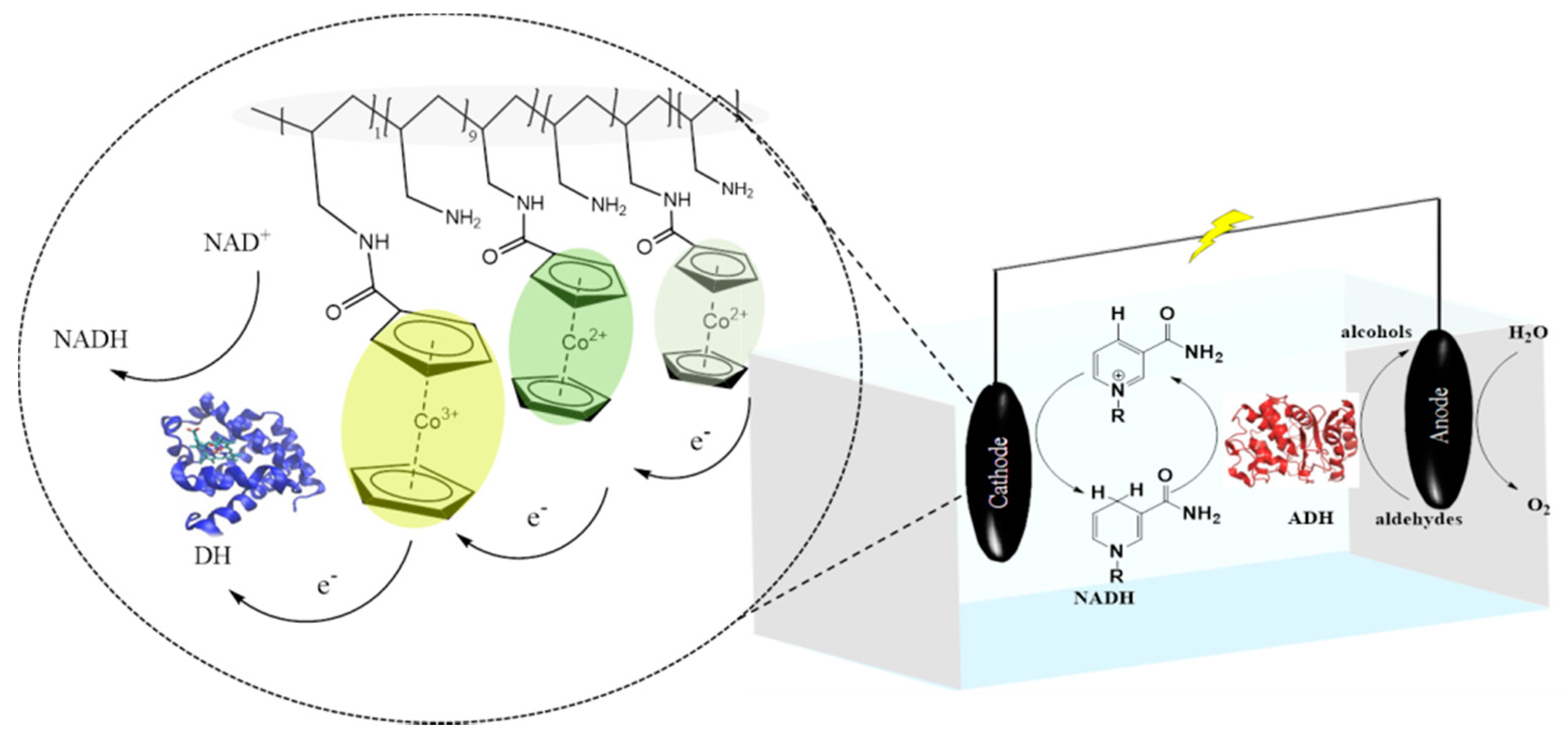
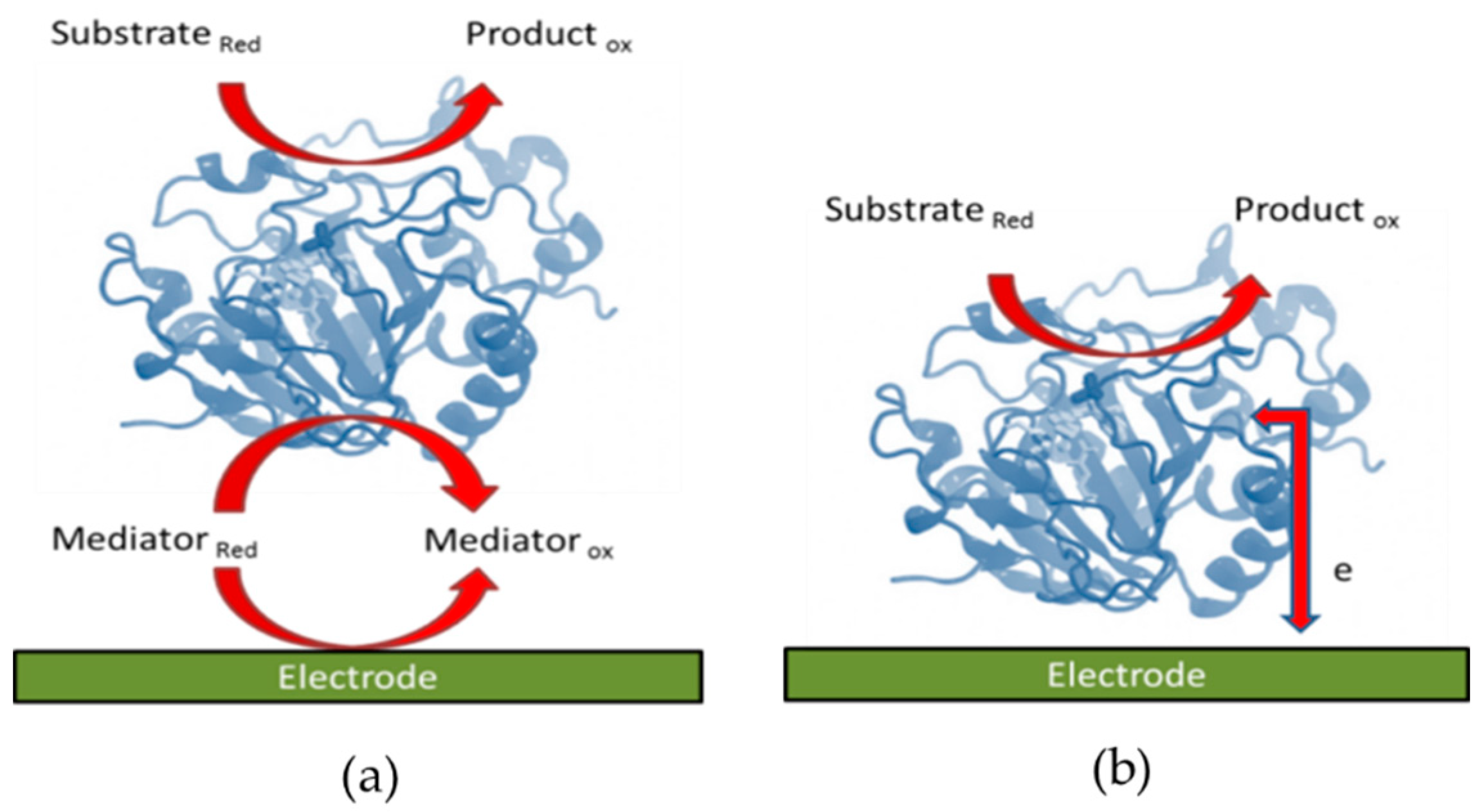
2.1. Mediated Electron Transfer
2.2. Direct Electron Transfer
3. Immobilisation Strategies
4. Biocatalytic Reactors
4.1. Batch Reactors
4.2. Flow Reactors
4.2.1. Packed-Bed Reactors
4.2.2. Monolithic Reactors
4.2.3. Wall-Coated Reactors
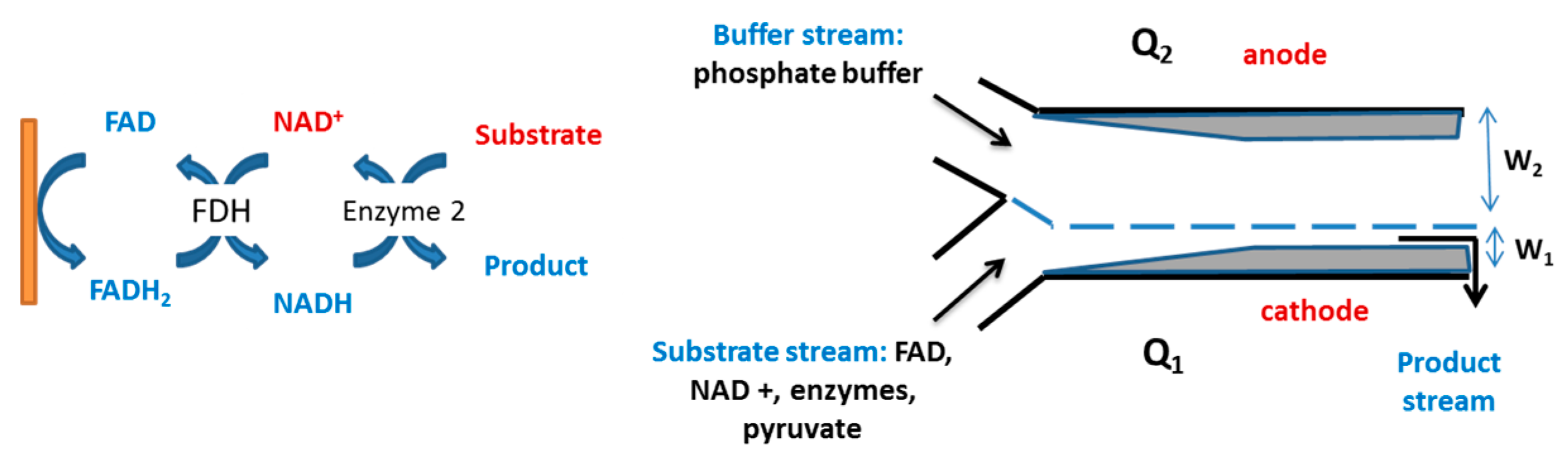
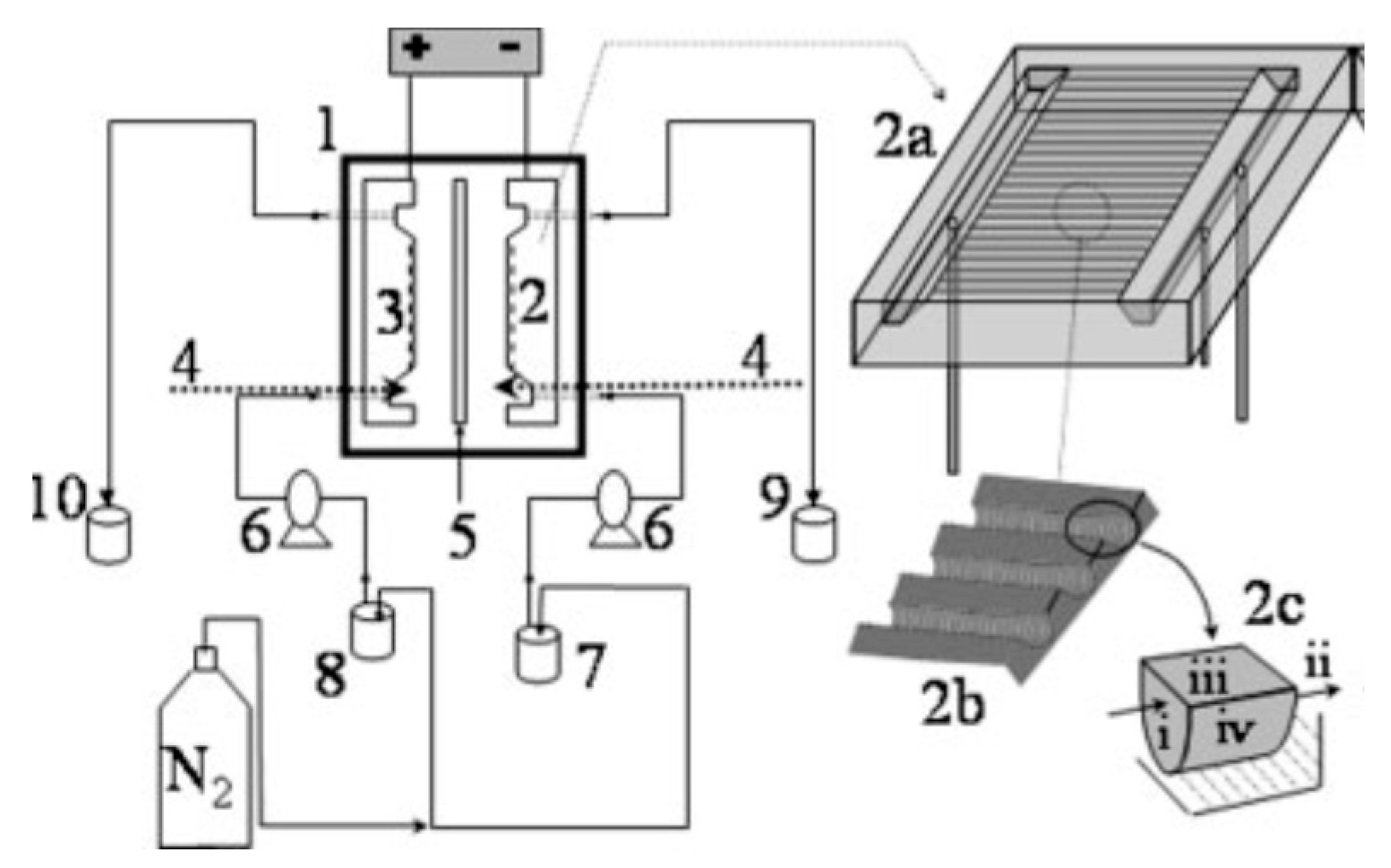
4.3. Electrochemical Reactors
5. Strategies for Improving Bioelectrocatalysts
5.1. High Surface Area Electrodes
5.2. Enzyme Engineering
5.3. Enzyme Cascades
6. Bioelectrosynthesis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jemli, S.; Ayadi-Zouari, D.; Hlima, H.B.; Bejar, S. Biocatalysts: Application and engineering for industrial purposes. Crit. Rev. Biotechnol. 2016, 36, 246–258. [Google Scholar] [CrossRef] [PubMed]
- van Beilen, J.B.; Li, Z. Enzyme technology: An overview. Curr. Opin. Biotechnol. 2002, 13, 338–344. [Google Scholar] [CrossRef]
- Otten, L.G.; Quax, W.J. Directed evolution: Selecting today′s biocatalysts. Biomol. Eng. 2005, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Kostrov, X. Immobilization of enzymes on porous silicas–benefits and challenges. Chem. Soc. Rev. 2013, 42, 6277–6289. [Google Scholar] [CrossRef]
- Schulze, B.; Wubbolts, M.G. Biocatalysis for industrial production of fine chemicals. Curr. Opin. Biotechnol. 1999, 10, 609–615. [Google Scholar] [CrossRef]
- Davis, B.G.; Boyer, V. Biocatalysis and enzymes in organic synthesis. Nat. Prod. Rep. 2001, 18, 618–640. [Google Scholar]
- Martinez, A.T.; Ruiz-Dueñas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv. 2017, 35, 815–831. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, L.M.; Rosenthal, K.; Lütz, S. Enzyme-based electrobiotechnological synthesis. In Bioelectrosynthesis; Springer: Cham, Switzerland, 2017; pp. 87–134. [Google Scholar]
- Giroud, F.; Sawada, K.; Taya, M.; Cosnier, S. 5, 5-Dithiobis (2-nitrobenzoic acid) pyrene derivative-carbon nanotube electrodes for NADH electrooxidation and oriented immobilization of multicopper oxidases for the development of glucose/O2 biofuel cells. Biosens. Bioelectron. 2017, 87, 957–963. [Google Scholar] [CrossRef]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Takagi, K.; Kano, K. High-power formate/dioxygen biofuel cell based on mediated electron transfer type bioelectrocatalysis. ACS Catal. 2017, 7, 5668–5673. [Google Scholar] [CrossRef]
- Kang, Z.; Jiao, K.; Yu, C.; Dong, J.; Peng, R.; Hu, Z.; Jiao, S. Direct electrochemistry and bioelectrocatalysis of glucose oxidase in CS/CNC film and its application in glucose biosensing and biofuel cells. RSC Adv. 2017, 7, 4572–4579. [Google Scholar] [CrossRef] [Green Version]
- Ruff, A.; Conzuelo, F.; Schuhmann, W. Bioelectrocatalysis as the basis for the design of enzyme-based biofuel cells and semi-artificial biophotoelectrodes. Nat. Catal. 2019, 1–11. [Google Scholar] [CrossRef]
- Sekretaryova, A.N.; Eriksson, M.; Turner, A.P. Bioelectrocatalytic systems for health applications. Biotechnol. Adv. 2016, 34, 177–197. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.O.; Maia, L.B.; Loureiro, J.A.; Pereira, M.C.; Delerue-Matos, C.; Moura, I.; Moura, J.J.; Morais, S. Biosensor for direct bioelectrocatalysis detection of nitric oxide using nitric oxide reductase incorporated in carboxylated single-walled carbon nanotubes/lipidic 3 bilayer nanocomposite. Bioelectrochemistry 2019, 127, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Voitechovič, E.; Vektarienė, A.; Vektaris, G.; Jančienė, R.; Razumienė, J.; Gurevičienė, V. 1, 4-Benzoquinone derivatives for enhanced bioelectrocatalysis by fructose dehydrogenase from Gluconobacter japonicus: Towards promising D-fructose biosensor development. Electroanalysis 2020. [Google Scholar] [CrossRef]
- Miyata, M.; Kitazumi, Y.; Shirai, O.; Kataoka, K.; Kano, K. Diffusion-limited biosensing of dissolved oxygen by direct electron transfer-type bioelectrocatalysis of multi-copper oxidases immobilized on porous gold microelectrodes. J. Electroanal. Chem. 2020, 860, 113895. [Google Scholar] [CrossRef]
- Höllrigl, V.; Otto, K.; Schmid, A. Electroenzymatic asymmetric reduction of rac-3-methylcyclohexanone to (1S, 3S)-3-methylcyclohexanol in organic/aqueous media catalyzed by a thermophilic alcohol dehydrogenase. Adv. Synth. Catal. 2007, 349, 1337–1340. [Google Scholar] [CrossRef]
- Kohlmann, C.; Lütz, S. Electroenzymatic synthesis of chiral sulfoxides. Eng. Life Sci. 2006, 6, 170–174. [Google Scholar] [CrossRef]
- Lütz, S.; Steckhan, E.; Liese, A. First asymmetric electroenzymatic oxidation catalyzed by a peroxidase. Electrochem. Commun. 2004, 6, 583–587. [Google Scholar] [CrossRef]
- Atsumi, S.; Hanai, T.; Liao, J.C. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 2008, 451, 86–89. [Google Scholar] [CrossRef]
- Liu, C.; Sakimoto, K.K.; Colón, B.C.; Silver, P.A.; Nocera, D.G. Ambient nitrogen reduction cycle using a hybrid inorganic–biological system. Proc. Natl. Acad. Sci. USA 2017, 114, 6450–6455. [Google Scholar] [CrossRef] [Green Version]
- Cooney, C.L. Bioreactors: Design and operation. Science 1983, 219, 728–733. [Google Scholar] [CrossRef]
- Britton, J.; Majumdar, S.; Weiss, G.A. Continuous flow biocatalysis. Chem. Soc. Rev. 2018, 47, 5891–5918. [Google Scholar] [CrossRef] [PubMed]
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14, 20170253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.V.; Holade, Y.; Minteer, S.D. DNA redox hydrogels: Improving mediated enzymatic bioelectrocatalysis. ACS Catal. 2016, 6, 2603–2607. [Google Scholar] [CrossRef]
- Shiraiwa, S.; So, K.; Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Nishikawa, K.; Higuchi, Y.; Kano, K. Reactivation of standard [NiFe]-hydrogenase and bioelectrochemical catalysis of proton reduction and hydrogen oxidation in a mediated-electron-transfer system. Bioelectrochemistry 2018, 123, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.J.; Chen, X.; Giroud, F.; Travelet, C.; Borsali, R.; Cosnier, S. Redox-active glyconanoparticles as electron shuttles for mediated electron transfer with bilirubin oxidase in solution. J. Am. Chem. Soc. 2017, 139, 16076–16079. [Google Scholar] [CrossRef] [PubMed]
- Elouarzaki, K.; Cheng, D.; Fisher, A.C.; Lee, J.-M. Coupling orientation and mediation strategies for efficient electron transfer in hybrid biofuel cells. Nat. Energy 2018, 3, 574–581. [Google Scholar] [CrossRef]
- Sakai, K.; Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Takagi, K.; Kano, K. Direct electron transfer-type bioelectrocatalytic interconversion of carbon dioxide/formate and NAD+/NADH redox couples with tungsten-containing formate dehydrogenase. Electrochim. Acta 2017, 228, 537–544. [Google Scholar] [CrossRef]
- Sakai, K.; Xia, H.-q.; Kitazumi, Y.; Shirai, O.; Kano, K. Assembly of direct-electron-transfer-type bioelectrodes with high performance. Electrochim. Acta 2018, 271, 305–311. [Google Scholar] [CrossRef]
- Kaida, Y.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Ultimate downsizing of D-fructose dehydrogenase for improving the performance of direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2019, 98, 101–105. [Google Scholar] [CrossRef]
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszyńska, D. Immobilization as a strategy for improving enzyme properties-application to oxidoreductases. Molecules 2014, 19, 8995–9018. [Google Scholar] [CrossRef] [Green Version]
- Cooney, M.; Svoboda, V.; Lau, C.; Martin, G.; Minteer, S.D. Enzyme catalysed biofuel cells. Energy Environ. Sci. 2008, 1, 320–337. [Google Scholar] [CrossRef]
- Hickey, D.P.; Milton, R.D.; Rasmussen, M.; Abdellaoui, S.; Nguyen, K.; Minteer, S.D. Fundamentals and applications of bioelectrocatalysis. Electrochemistry 2015, 13, 97. [Google Scholar]
- Chen, H.; Dong, F.; Minteer, S.D. The progress and outlook of bioelectrocatalysis for the production of chemicals, fuels and materials. Nat. Catal. 2020, 3, 1–20. [Google Scholar] [CrossRef]
- Hilt, G.; Lewall, B.; Montero, G.; Utley, J.H.; Steckhan, E. Efficient In-Situ Redox Catalytic NAD (P)+ Regeneration in Enzymatic Synthesis Using Transition-Metal Complexes of 1, 10-Phenanthroline-5, 6-dione and Its N-Monomethylated Derivative as Catalysts. Liebigs Annalen 1997, 1997, 2289–2296. [Google Scholar] [CrossRef]
- Kroutil, W.; Mang, H.; Edegger, K.; Faber, K. Biocatalytic oxidation of primary and secondary alcohols. Adv. Synth. Catal. 2004, 346, 125–142. [Google Scholar] [CrossRef]
- Findrik, Z.; Šimunović, I.; Vasić-Rački, Đ. Coenzyme regeneration catalyzed by NADH oxidase from Lactobacillus brevis in the reaction of L-amino acid oxidation. Biochem. Eng. J. 2008, 39, 319–327. [Google Scholar] [CrossRef]
- Tahar, A.B.; Szymczyk, A.; Tingry, S.; Vadgama, P.; Zelsmanne, M.; Tsujumura, S.; Cinquin, P.; Martin, D.; Zebda, A. One-year stability of glucose dehydrogenase confined in a 3D carbon nanotube electrode with coated poly-methylene green: Application as bioanode for a glucose biofuel cell. J. Electroanal. Chem. 2019, 847, 113069. [Google Scholar] [CrossRef]
- Bonfin, C.S.; Franco, J.H.; de Andrade, A.R. Ethanol bioelectrooxidation in a robust poly (methylene green-pyrrole)-mediated enzymatic biofuel cell. J. Electroanal. Chem. 2019, 844, 43–48. [Google Scholar] [CrossRef]
- Karyakin, A.A.; Karyakina, E.E.; Schuhmann, W.; Schmidt, H.L.; Varfolomeyev, S.D. New amperometric dehydrogenase electrodes based on electrocatalytic NADH-oxidation at poly (methylene blue)-modified electrodes. Electroanalysis 1994, 6, 821–829. [Google Scholar] [CrossRef]
- Yuan, M.; Minteer, S.D. Redox polymers in electrochemical systems: From methods of mediation to energy storage. Curr. Opin. Electrochem. 2019. [Google Scholar] [CrossRef] [Green Version]
- Carucci, C.; Salis, A.; Magner, E. Specific Ion Effects on the Mediated Oxidation of NADH. ChemElectroChem 2017, 4, 3075–3080. [Google Scholar] [CrossRef]
- Abdellaoui, S.; Milton, R.D.; Quah, T.; Minteer, S.D. NAD-dependent dehydrogenase bioelectrocatalysis: The ability of a naphthoquinone redox polymer to regenerate NAD. Chem. Commun. 2016, 52, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hollmann, F. Nonconventional regeneration of redox enzymes–a practical approach for organic synthesis? Chem. Commun. 2018, 54, 7281–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.J.; Yang, H.; Jo, K.; Kwak, J. An electrochemical immunosensor using p-aminophenol redox cycling by NADH on a self-assembled monolayer and ferrocene-modified Au electrodes. Analyst 2008, 133, 1599–1604. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yun, S.-E.; Kang, C. Electrochemical evaluation of the reaction rate between methyl viologen mediator and diaphorase enzyme for the electrocatalytic reduction of NAD+ and digital simulation for its voltammetric responses. J. Electroanal. Chem. 1999, 465, 153–159. [Google Scholar] [CrossRef]
- Kochius, S.; Park, J.B.; Ley, C.; Könst, P.; Hollmann, F.; Schrader, J.; Holtmann, D. Electrochemical regeneration of oxidised nicotinamide cofactors in a scalable reactor. J. Mol. Catal. B Enzym. 2014, 103, 94–99. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I.W.; Buehler, K. Biocatalytic redox reactions for organic synthesis: Nonconventional regeneration methods. ChemCatChem 2010, 2, 762–782. [Google Scholar] [CrossRef]
- Weckbecker, A.; Gröger, H.; Hummel, W. Regeneration of nicotinamide coenzymes: Principles and applications for the synthesis of chiral compounds. In Biosystems Engineering I; Springer: Cham, Switzerland, 2010; pp. 195–242. [Google Scholar]
- Kochius, S.; Magnusson, A.O.; Hollmann, F.; Schrader, J.; Holtmann, D. Immobilized redox mediators for electrochemical NAD (P)+ regeneration. Appl. Microbiol. Biotechnol. 2012, 93, 2251–2264. [Google Scholar] [CrossRef]
- Chen, H.; Cai, R.; Patel, J.; Dong, F.; Chen, H.; Minteer, S.D. Upgraded bioelectrocatalytic N2 fixation: From N2 to chiral amine intermediates. J. Am. Chem. Soc. 2019, 141, 4963–4971. [Google Scholar] [CrossRef]
- Kashiwagi, Y.; Yanagisawa, Y.; Shibayama, N.; Nakahara, K.; Kurashima, F.; Anzai, J.; Osa, T. Preparative, electroenzymatic reduction of ketones on an all components-immobilized graphite felt electrode. Electrochim. Acta 1997, 42, 2267–2270. [Google Scholar] [CrossRef]
- Badalyan, A.; Yang, Z.-Y.; Hu, B.; Luo, J.; Hu, M.; Liu, T.L.; Seefeldt, L.C. An Efficient Viologen-Based Electron Donor to Nitrogenase. Biochemistry 2019, 58, 4590–4595. [Google Scholar] [CrossRef] [PubMed]
- Tosstorff, A.; Kroner, C.; Opperman, D.J.; Hollmann, F.; Holtmann, D. Towards electroenzymatic processes involving old yellow enzymes and mediated cofactor regeneration. Eng. Life Sci. 2017, 17, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Ruff, A. Redox polymers in bioelectrochemistry: Common playgrounds and novel concepts. Curr. Opin. Electrochem. 2017, 5, 66–73. [Google Scholar] [CrossRef]
- Yuan, M.; Kummer, M.J.; Milton, R.D.; Quah, T.; Minteer, S.D. Efficient NADH regeneration by a redox polymer-immobilized enzymatic system. ACS Catal. 2019, 9, 5486–5495. [Google Scholar] [CrossRef]
- Heller, A. Electrical wiring of redox enzymes. Acc. Chem. Res. 1990, 23, 128–134. [Google Scholar] [CrossRef]
- Heller, A. Electrical connection of enzyme redox centers to electrodes. J. Phys. Chem. 1992, 96, 3579–3587. [Google Scholar] [CrossRef]
- Campbell, A.S.; Murata, H.; Carmali, S.; Matyjaszewski, K.; Islam, M.F.; Russell, A.J. Polymer-based protein engineering grown ferrocene-containing redox polymers improve current generation in an enzymatic biofuel cell. Biosens. Bioelectron. 2016, 86, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Tapia, C.; Milton, R.D.; Pankratova, G.; Minteer, S.D.; Åkerlund, H.E.; Leech, D.; De Lacey, A.L.; Pita, M.; Gorton, L. Wiring of photosystem I and hydrogenase on an electrode for photoelectrochemical H2 production by using redox polymers for relatively positive onset potential. ChemElectroChem 2017, 4, 90–95. [Google Scholar] [CrossRef]
- Alkotaini, B.; Abdellaoui, S.; Hasan, K.; Grattieri, M.; Quah, T.; Cai, R.; Yuan, M.; Minteer, S.D. Sustainable bioelectrosynthesis of the bioplastic polyhydroxybutyrate: Overcoming substrate requirement for NADH regeneration. ACS Sustain. Chem. Eng. 2018, 6, 4909–4915. [Google Scholar] [CrossRef]
- Szczesny, J.; Ruff, A.; Oliveira, A.R.; Pita, M.; Pereira, I.A.; De Lacey, A.L.; Schuhmann, W. Electroenzymatic CO2 Fixation Using Redox Polymer/Enzyme-Modified Gas Diffusion Electrodes. ACS Energy Lett. 2020, 5, 321–327. [Google Scholar] [CrossRef]
- Falk, M.; Blum, Z.; Shleev, S. Direct electron transfer based enzymatic fuel cells. Electrochim. Acta 2012, 82, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.D.; Turner, A.P. Home blood glucose biosensors: A commercial perspective. Biosens. Bioelectron. 2005, 20, 2435–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.B.; Larry, R.F. Electrochemical Methods Fundamentals and Applications; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts 2020, 10, 236. [Google Scholar] [CrossRef] [Green Version]
- Shleev, S.; Tkac, J.; Christenson, A.; Ruzgas, T.; Yaropolov, A.I.; Whittaker, J.W.; Gorton, L. Direct electron transfer between copper-containing proteins and electrodes. Biosens. Bioelectron. 2005, 20, 2517–2554. [Google Scholar] [CrossRef]
- Salaj-Kosla, U.; Pöller, S.; Schuhmann, W.; Shleev, S.; Magner, E. Direct electron transfer of Trametes hirsuta laccase adsorbed at unmodified nanoporous gold electrodes. Bioelectrochemistry 2013, 91, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Baek, S.; Lee, H.; Reginald, S.S.; Kim, Y.; Kang, H.; Choi, I.-G.; Chang, I.S. Construction of Uniform Monolayer-and Orientation-Tunable Enzyme Electrode by a Synthetic Glucose Dehydrogenase without Electron-Transfer Subunit via Optimized Site-Specific Gold-Binding Peptide Capable of Direct Electron Transfer. ACS Appl. Mater. Interfaces 2018, 10, 28615–28626. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.-J.; Zhao, G.; Xu, J.-J.; Chen, H.-Y. Direct electrochemistry and electrocatalysis of heme proteins immobilized on gold nanoparticles stabilized by chitosan. Anal. Biochem. 2005, 342, 280–286. [Google Scholar] [CrossRef]
- Kuk, S.K.; Gopinath, K.; Singh, R.K.; Kim, T.-D.; Lee, Y.; Choi, W.S.; Lee, J.-K.; Park, C.B. NADH-Free Electroenzymatic Reduction of CO2 by Conductive Hydrogel-Conjugated Formate Dehydrogenase. ACS Catal. 2019, 9, 5584–5589. [Google Scholar] [CrossRef]
- Hickey, D.P.; Lim, K.; Cai, R.; Patterson, A.R.; Yuan, M.; Sahin, S.; Abdellaoui, S.; Minteer, S.D. Pyrene hydrogel for promoting direct bioelectrochemistry: ATP-independent electroenzymatic reduction of N 2. Chem. Sci. 2018, 9, 5172–5177. [Google Scholar] [CrossRef] [Green Version]
- Bollella, P.; Gorton, L.; Antiochia, R. Direct electron transfer of dehydrogenases for development of 3rd generation biosensors and enzymatic fuel cells. Sensors 2018, 18, 1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Takagi, K.; Kano, K. Direct electron transfer-type four-way bioelectrocatalysis of CO2/formate and NAD+/NADH redox couples by tungsten-containing formate dehydrogenase adsorbed on gold nanoparticle-embedded mesoporous carbon electrodes modified with 4-mercaptopyridine. Electrochem. Commun. 2017, 84, 75–79. [Google Scholar] [CrossRef]
- Yates, N.D.; Fascione, M.A.; Parkin, A. Methodologies for “wiring” redox proteins/enzymes to electrode surfaces. Chem. Eur. J. 2018, 24, 12164–12182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepenkoetter, T.; Salaj-Kosla, U.; Xiao, X.; Conghaile, P.Ó.; Pita, M.; Ludwig, R.; Magner, E. Immobilization of redox enzymes on nanoporous gold electrodes: Applications in biofuel cells. ChemPlusChem 2017, 82, 553–560. [Google Scholar] [CrossRef]
- Besic, S.; Minteer, S.D. Micellar Polymer Encapsulation of Enzymes. In Enzyme Stabilization and Immobilization; Springer: Cham, Swtzerland, 2017; pp. 93–108. [Google Scholar]
- Xiao, X.; Xia, H.-Q.; Wu, R.; Bai, L.; Yan, L.; Magner, E.; Cosnier, S.; Lojou, E.; Zhu, Z.; Liu, A. Tackling the challenges of enzymatic (bio) fuel cells. Chem. Rev. 2019, 119, 9509–9558. [Google Scholar] [CrossRef]
- Leech, D.; Kavanagh, P.; Schuhmann, W. Enzymatic fuel cells: Recent progress. Electrochim. Acta 2012, 84, 223–234. [Google Scholar] [CrossRef]
- Milton, R.D.; Hickey, D.P.; Abdellaoui, S.; Lim, K.; Wu, F.; Tan, B.; Minteer, S.D. Rational design of quinones for high power density biofuel cells. Chem. Sci. 2015, 6, 4867–4875. [Google Scholar] [CrossRef] [Green Version]
- Prévoteau, A.; Mano, N. Oxygen reduction on redox mediators may affect glucose biosensors based on “wired” enzymes. Electrochim. Acta 2012, 68, 128–133. [Google Scholar] [CrossRef]
- Pankratov, D.; Conzuelo, F.; Pinyou, P.; Alsaoub, S.; Schuhmann, W.; Shleev, S. A Nernstian biosupercapacitor. Angew. Chem. Int. Ed. 2016, 55, 15434–15438. [Google Scholar] [CrossRef]
- Ruff, A.; Szczesny, J.; Zacarias, S.N.; Pereira, I.S.A.; Plumeré, N.; Schuhmann, W. Protection and reactivation of the [NiFeSe] hydrogenase from Desulfovibrio vulgaris Hildenborough under oxidative conditions. ACS Energy Lett. 2017, 2, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Alsaoub, S.; Ruff, A.; Conzuelo, F.; Ventosa, E.; Ludwig, R.; Shleev, S.; Schuhmann, W. An Intrinsic Self-Charging Biosupercapacitor Comprised of a High-Potential Bioanode and a Low-Potential Biocathode. ChemPlusChem 2017, 82, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Spahn, C.; Minteer, S.D. Enzyme immobilization in biotechnology. Recent Pat. Eng. 2008, 2, 195–200. [Google Scholar] [CrossRef]
- Jankowska, K.; Bachosz, K.; Zdarta, J.; Jesionowski, T. Application of Enzymatic-Based Bioreactors. In Seminar on Practical Aspects of Chemical Engineering; Springer: Cham, Switzerland, 2019; pp. 110–121. [Google Scholar]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow bioreactors as complementary tools for biocatalytic process intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef]
- Märkle, W.; Lütz, S. Electroenzymatic strategies for deracemization, stereoinversion and asymmetric synthesis of amino acids. Electrochim. Acta 2008, 53, 3175–3180. [Google Scholar] [CrossRef]
- Mazurenko, I.; Ghach, W.; Kohring, G.-W.; Despas, C.; Walcarius, A.; Etienne, M. Immobilization of membrane-bounded (S)-mandelate dehydrogenase in sol–gel matrix for electroenzymatic synthesis. Bioelectrochemistry 2015, 104, 65–70. [Google Scholar] [CrossRef]
- Ali, I.; Gill, A.; Omanovic, S. Direct electrochemical regeneration of the enzymatic cofactor 1, 4-NADH employing nano-patterned glassy carbon/Pt and glassy carbon/Ni electrodes. Chem. Eng. J. 2012, 188, 173–180. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, Q.; Shao, L.; Jia, Y.; Zhang, X. Microfluidic immobilized enzyme reactors for continuous biocatalysis. React. Chem. Eng. 2020, 5, 9–32. [Google Scholar] [CrossRef]
- Kundu, S.; Bhangale, A.S.; Wallace, W.E.; Flynn, K.M.; Guttman, C.M.; Gross, R.A.; Beers, K.L. Continuous flow enzyme-catalyzed polymerization in a microreactor. J. Am. Chem. Soc. 2011, 133, 6006–6011. [Google Scholar] [CrossRef]
- Aguillón, A.R.; Avelar, M.N.; Gotardo, L.E.; de Souza, S.P.; Leão, R.A.; Itabaiana Jr, I.; Miranda, L.S.; de Souza, R.O. Immobilized lipase screening towards continuous-flow kinetic resolution of (±)-1, 2-propanediol. Mol. Catal. 2019, 467, 128–134. [Google Scholar] [CrossRef]
- Cosgrove, S.C.; Mattey, A.P.; Riese, M.; Chapman, M.R.; Birmingham, W.R.; Blacker, A.J.; Kapur, N.; Turner, N.J.; Flitsch, S.L. Biocatalytic oxidation in continuous flow for the generation of carbohydrate dialdehydes. ACS Catal. 2019, 9, 11658–11662. [Google Scholar] [CrossRef]
- Yue, J. Multiphase flow processing in microreactors combined with heterogeneous catalysis for efficient and sustainable chemical synthesis. Catal. Today 2018, 308, 3–19. [Google Scholar] [CrossRef]
- Zhang, P.; Russell, M.G.; Jamison, T.F. Continuous flow total synthesis of rufinamide. Org. Process Res. Dev. 2014, 18, 1567–1570. [Google Scholar] [CrossRef]
- Cantillo, D.; Kappe, C.O. Halogenation of organic compounds using continuous flow and microreactor technology. React. Chem. Eng. 2017, 2, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Jo, C.; Groombridge, A.S.; De La Verpilliere, J.; Lee, J.T.; Son, Y.; Liang, H.-L.; Boies, A.M.; De Volder, M. Continuous Flow Synthesis of Carbon Coated Silicon/Iron Silicide Secondary Particles for Li-Ion Batteries. ACS Nano 2019, 14, 698–707. [Google Scholar] [CrossRef]
- Rubio-Martinez, M.; Hadley, T.D.; Batten, M.P.; Constanti-Carey, K.; Barton, T.; Marley, D.; Mönch, A.; Lim, K.S.; Hill, M.R. Scalability of Continuous Flow Production of Metal–Organic Frameworks. ChemSusChem 2016, 9, 938–941. [Google Scholar] [CrossRef]
- Pastre, J.C.; Browne, D.L.; Ley, S.V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 2013, 42, 8849–8869. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [Green Version]
- Valikhani, D.; Bolivar, J.M.; Nidetzky, B. Enzyme Immobilization in Wall-Coated Flow Microreactors. In Immobilization of Enzymes and Cells; Springer: Cham, Switzerland, 2020; pp. 243–257. [Google Scholar]
- Thompson, M.P.; Peñafiel, I.; Cosgrove, S.C.; Turner, N.J. Biocatalysis using immobilized enzymes in continuous flow for the synthesis of fine chemicals. Org. Process Res. Dev. 2018, 23, 9–18. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Nidetzky, B. Smart enzyme immobilization in microstructured reactors. Chim Oggi 2013, 31, 50–54. [Google Scholar]
- Xiao, X.; Siepenkoetter, T.; Whelan, R.; Salaj-Kosla, U.; Magner, E. A continuous fluidic bioreactor utilising electrodeposited silica for lipase immobilisation onto nanoporous gold. J. Electroanal. Chem. 2018, 812, 180–185. [Google Scholar] [CrossRef]
- Svec, F. Less common applications of monoliths: I. Microscale protein mapping with proteolytic enzymes immobilized on monolithic supports. Electrophoresis 2006, 27, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, R.; Plazl, I.; Žnidaršič-Plazl, P.; Gernaey, K.V.; Woodley, J.M. Microscale technology and biocatalytic processes: Opportunities and challenges for synthesis. Trends Biotechnol. 2015, 33, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, K.; Pietrowska, M.; Kocurek, J.; Maresz, K.; Koreniuk, A.; Mrowiec-Białoń, J.; Widłak, P.; Magner, E.; Jarzębski, A. Low back-pressure hierarchically structured multichannel microfluidic bioreactors for rapid protein digestion–Proof of concept. Chem. Eng. J. 2016, 287, 148–154. [Google Scholar] [CrossRef]
- Hong, J.; Tsao, G.; Wankat, P. Membrane reactor for enzymatic hydrolysis of cellobiose. Biotechnol. Bioeng. 1981, 23, 1501–1516. [Google Scholar] [CrossRef]
- Rios, G.; Belleville, M.; Paolucci, D.; Sanchez, J. Progress in enzymatic membrane reactors–A review. J. Membr. Sci. 2004, 242, 189–196. [Google Scholar] [CrossRef]
- Lopez, C.; Mielgo, I.; Moreira, M.; Feijoo, G.; Lema, J. Enzymatic membrane reactors for biodegradation of recalcitrant compounds. Application to dye decolourisation. J. Biotechnol. 2002, 99, 249–257. [Google Scholar] [CrossRef]
- Prazeres, D.; Cabral, J. Enzymatic membrane bioreactors and their applications. Enzyme Microb. Technol. 1994, 16, 738–750. [Google Scholar] [CrossRef]
- Tanimu, A.; Jaenicke, S.; Alhooshani, K. Heterogeneous catalysis in continuous flow microreactors: A review of methods and applications. Chem. Eng. J. 2017, 327, 792–821. [Google Scholar] [CrossRef]
- Dall’Oglio, F.; Contente, M.L.; Conti, P.; Molinari, F.; Monfredi, D.; Pinto, A.; Romano, D.; Ubiali, D.; Tamborini, L.; Serra, I. Flow-based stereoselective reduction of ketones using an immobilized ketoreductase/glucose dehydrogenase mixed bed system. Catal. Commun. 2017, 93, 29–32. [Google Scholar] [CrossRef]
- Döbber, J.; Pohl, M.; Ley, S.; Musio, B. Rapid, selective and stable HaloTag-Lb ADH immobilization directly from crude cell extract for the continuous biocatalytic production of chiral alcohols and epoxides. Reaction Chem. Eng. 2018, 3, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Döbber, J.; Gerlach, T.; Offermann, H.; Rother, D.; Pohl, M. Closing the gap for efficient immobilization of biocatalysts in continuous processes: HaloTag™ fusion enzymes for a continuous enzymatic cascade towards a vicinal chiral diol. Green Chem. 2018, 20, 544–552. [Google Scholar] [CrossRef]
- Peschke, T.; Bitterwolf, P.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Oxidoreductases for Flow Biocatalysis in Miniaturized Packed-Bed Reactors. Chem. Eng. Technol. 2019, 42, 2009–2017. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Wiesbauer, J.; Nidetzky, B. Biotransformations in microstructured reactors: More than flowing with the stream? Trends Biotechnol. 2011, 29, 333–342. [Google Scholar] [CrossRef]
- Russell, M.G.; Veryser, C.; Hunter, J.F.; Beingessner, R.L.; Jamison, T.F. Monolithic Silica Support for Immobilized Catalysis in Continuous Flow. Adv. Synth. Catal. 2020, 362, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhu, X.; Camara, M.A.; Qu, Q.; Shan, Y.; Yang, L. Surface modification with highly-homogeneous porous silica layer for enzyme immobilization in capillary enzyme microreactors. Talanta 2019, 197, 539–547. [Google Scholar] [CrossRef]
- Zhao, X.; Fan, P.-R.; Mo, C.-E.; Huang, Y.-P.; Liu, Z.-S. Green synthesis of monolithic enzyme microreactor based on thiol-ene click reaction for enzymatic hydrolysis of protein. J. Chromatogr. A 2020, 1611, 460618. [Google Scholar] [CrossRef]
- Meller, K.; Pomastowski, P.; Szumski, M.; Buszewski, B. Preparation of an improved hydrophilic monolith to make trypsin-immobilized microreactors. J. Chromatogr. B 2017, 1043, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Van den Biggelaar, L.; Soumillion, P.; Debecker, D.P. Enantioselective transamination in continuous flow mode with transaminase immobilized in a macrocellular silica monolith. Catalysts 2017, 7, 54. [Google Scholar] [CrossRef]
- Szymańska, K.; Odrozek, K.; Zniszczoł, A.; Torrelo, G.; Resch, V.; Hanefeld, U.; Jarzębski, A.B. MsAcT in siliceous monolithic microreactors enables quantitative ester synthesis in water. Catal. Sci. Technol. 2016, 6, 4882–4888. [Google Scholar] [CrossRef] [Green Version]
- Sandig, B.; Buchmeiser, M.R. Highly productive and enantioselective enzyme catalysis under continuous supported liquid–liquid conditions using a hybrid monolithic bioreactor. ChemSusChem 2016, 9, 2917–2921. [Google Scholar] [CrossRef] [PubMed]
- Logan, T.C.; Clark, D.S.; Stachowiak, T.B.; Svec, F.; Frechet, J.M. Photopatterning enzymes on polymer monoliths in microfluidic devices for steady-state kinetic analysis and spatially separated multi-enzyme reactions. Anal. Chem. 2007, 79, 6592–6598. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Qi, L.; Mu, X.; Chen, Y. Monolith and coating enzymatic microreactors of L-asparaginase: Kinetics study by MCE–LIF for potential application in acute lymphoblastic leukemia (ALL) treatment. Analyst 2011, 136, 2077–2083. [Google Scholar] [CrossRef]
- Irfan, M.; Glasnov, T.N.; Kappe, C.O. Heterogeneous catalytic hydrogenation reactions in continuous-flow reactors. ChemSusChem 2011, 4, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Schilke, K.F.; Wilson, K.L.; Cantrell, T.; Corti, G.; McIlroy, D.N.; Kelly, C. A novel enzymatic microreactor with Aspergillus oryzae β-galactosidase immobilized on silicon dioxide nanosprings. Biotechnol. Progr. 2010, 26, 1597–1605. [Google Scholar] [CrossRef]
- Li, X.; Yin, Z.; Cui, X.; Yang, L. Capillary electrophoresis-integrated immobilized enzyme microreactor with graphene oxide as support: Immobilization of negatively charged l-lactate dehydrogenase via hydrophobic interactions. Electrophoresis 2020, 41, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhang, B.; Zhu, X.; Chen, R.; Liao, Q.; Ye, D.-d.; Liu, J.; Liu, M.; Chen, G. Multilayered Pd nanocatalysts with nano-bulge structure in a microreactor for multiphase catalytic reaction. Chem. Eng. Res. Des. 2018, 138, 190–199. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, X.; Liao, Q.; Chen, R.; Ye, D.; Feng, H.; Liu, M.; Chen, G. Layer-by-layer self-assembly of palladium nanocatalysts with polyelectrolytes grafted on the polydopamine functionalized gas-liquid-solid microreactor. Chem. Eng. J. 2018, 332, 174–182. [Google Scholar] [CrossRef]
- Munirathinam, R.; Huskens, J.; Verboom, W. Supported catalysis in continuous-flow microreactors. Adv. Synth. Catal. 2015, 357, 1093–1123. [Google Scholar] [CrossRef]
- Bi, Y.; Zhou, H.; Jia, H.; Wei, P. A flow-through enzymatic microreactor immobilizing lipase based on layer-by-layer method for biosynthetic process: Catalyzing the transesterification of soybean oil for fatty acid methyl ester production. Process Biochem. 2017, 54, 73–80. [Google Scholar] [CrossRef]
- Valikhani, D.; Bolivar, J.M.; Viefhues, M.; McIlroy, D.N.; Vrouwe, E.X.; Nidetzky, B. A spring in performance: Silica nanosprings boost enzyme immobilization in microfluidic channels. ACS Appl. Mater. Interfaces 2017, 9, 34641–34649. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Zhou, H.; Jia, H.; Wei, P. Polydopamine-mediated preparation of an enzyme-immobilized microreactor for the rapid production of wax ester. RSC Adv. 2017, 7, 12283–12291. [Google Scholar] [CrossRef] [Green Version]
- Britton, J.; Dyer, R.P.; Majumdar, S.; Raston, C.L.; Weiss, G.A. Ten-Minute Protein Purification and Surface Tethering for Continuous-Flow Biocatalysis. Angew. Chem. 2017, 129, 2336–2341. [Google Scholar] [CrossRef] [Green Version]
- Valikhani, D.; Bolivar, J.M.; Pfeiffer, M.; Nidetzky, B. Multivalency effects on the immobilization of sucrose phosphorylase in flow microchannels and their use in the development of a high-performance biocatalytic microreactor. ChemCatChem 2017, 9, 161–166. [Google Scholar] [CrossRef]
- Dixon, M.; Webb, E. Enzyme techniques. In Enzymes, 3rd ed.; Academic Press: New York, NY, USA, 1979; pp. 7–11. [Google Scholar]
- Yoon, S.K.; Choban, E.R.; Kane, C.; Tzedakis, T.; Kenis, P.J. Laminar flow-based electrochemical microreactor for efficient regeneration of nicotinamide cofactors for biocatalysis. J. Am. Chem. Soc. 2005, 127, 10466–10467. [Google Scholar] [CrossRef]
- Cheikhou, K.; Tzédakis, T. Electrochemical microreactor for chiral syntheses using the cofactor NADH. AlChE J. 2008, 54, 1365–1376. [Google Scholar] [CrossRef]
- Ruinatscha, R.; Buehler, K.; Schmid, A. Development of a high performance electrochemical cofactor regeneration module and its application to the continuous reduction of FAD. J. Mol. Catal. B Enzym. 2014, 103, 100–105. [Google Scholar] [CrossRef]
- Rodríguez-Hinestroza, R.A.; López, C.; López-Santín, J.; Kane, C.; Benaiges, M.D.; Tzedakis, T. HLADH-catalyzed synthesis of β-amino acids, assisted by continuous electrochemical regeneration of NAD+ in a filter press microreactor. Chem. Eng. Sci. 2017, 158, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Fisher, K.; Mohr, S.; Mansell, D.; Goddard, N.J.; Fielden, P.R.; Scrutton, N.S. Electro-enzymatic viologen-mediated substrate reduction using pentaerythritol tetranitrate reductase and a parallel, segmented fluid flow system. Catal. Sci. Technol. 2013, 3, 1505–1511. [Google Scholar] [CrossRef]
- Mano, N. Recent advances in high surface area electrodes for bioelectrochemical applications. Curr. Opin. Electrochem. 2020, 19, 8–13. [Google Scholar] [CrossRef]
- Xiao, X.; Si, P.; Magner, E. An overview of dealloyed nanoporous gold in bioelectrochemistry. Bioelectrochemistry 2016, 109, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Salaj-Kosla, U.; Scanlon, M.D.; Baumeister, T.; Zahma, K.; Ludwig, R.; Conghaile, P.Ó.; MacAodha, D.; Leech, D.; Magner, E. Mediated electron transfer of cellobiose dehydrogenase and glucose oxidase at osmium polymer-modified nanoporous gold electrodes. Anal. Bioanal. Chem. 2013, 405, 3823–3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The immobilization of fructose dehydrogenase on nanoporous gold electrodes for the detection of fructose. ChemElectroChem 2017, 4, 905–912. [Google Scholar] [CrossRef]
- Xiao, X.; Siepenkoetter, T.; Conghaile, P.O.; Leech, D.n.; Magner, E. Nanoporous gold-based biofuel cells on contact lenses. ACS Appl. Mater. Interfaces 2018, 10, 7107–7116. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, J.K.; Neupane, D.; Nepal, B.; Mikhaylov, V.; Demchenko, A.V.; Stine, K.J. Preparation, modification, characterization, and biosensing application of nanoporous gold using electrochemical techniques. Nanomaterials 2018, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Falk, M.; Ortiz, R.; Matsumura, H.; Bobacka, J.; Ludwig, R.; Bergelin, M.; Gorton, L.; Shleev, S. Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes. Biosens. Bioelectron. 2012, 31, 219–225. [Google Scholar] [CrossRef]
- Kizling, M.; Dzwonek, M.; Nowak, A.; Tymecki, Ł.; Stolarczyk, K.; Więckowska, A.; Bilewicz, R. Multi-Substrate Biofuel Cell Utilizing Glucose, Fructose and Sucrose as the Anode Fuels. Nanomaterials 2020, 10, 1534. [Google Scholar] [CrossRef]
- Kumar, S.A.; Chen, S.-M. Electroanalysis of NADH using conducting and redox active polymer/carbon nanotubes modified electrodes—A review. Sensors 2008, 8, 739–766. [Google Scholar] [CrossRef] [Green Version]
- Gooding, J.J. Nanostructuring electrodes with carbon nanotubes: A review on electrochemistry and applications for sensing. Electrochim. Acta 2005, 50, 3049–3060. [Google Scholar] [CrossRef]
- Wang, J. Carbon-nanotube based electrochemical biosensors: A review. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2005, 17, 7–14. [Google Scholar] [CrossRef]
- Zhu, Z.; Garcia-Gancedo, L.; Flewitt, A.J.; Xie, H.; Moussy, F.; Milne, W.I. A critical review of glucose biosensors based on carbon nanomaterials: Carbon nanotubes and graphene. Sensors 2012, 12, 5996–6022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. Enhanced direct electron transfer of fructose dehydrogenase rationally immobilized on a 2-aminoanthracene diazonium cation grafted single-walled carbon nanotube based electrode. ACS Catal. 2018, 8, 10279–10289. [Google Scholar] [CrossRef]
- Feizabadi, M.; Ajloo, D.; Soleymanpour, A.; Faridnouri, H. Study of electron transport in the functionalized nanotubes and their impact on the electron transfer in the active site of horseradish peroxidase. J. Phys. Chem. Solids 2018, 116, 313–323. [Google Scholar] [CrossRef]
- Gross, A.J.; Chen, X.; Giroud, F.; Abreu, C.; Le Goff, A.; Holzinger, M.; Cosnier, S. A high power buckypaper biofuel cell: Exploiting 1, 10-phenanthroline-5, 6-dione with FAD-dependent dehydrogenase for catalytically-powerful glucose oxidation. ACS Catal. 2017, 7, 4408–4416. [Google Scholar] [CrossRef]
- Sim, H.J.; Lee, D.Y.; Kim, H.; Choi, Y.-B.; Kim, H.-H.; Baughman, R.H.; Kim, S.J. Stretchable fiber biofuel cell by rewrapping multiwalled carbon nanotube sheets. Nano Lett. 2018, 18, 5272–5278. [Google Scholar] [CrossRef] [PubMed]
- Jourdin, L.; Freguia, S.; Donose, B.C.; Chen, J.; Wallace, G.G.; Keller, J.; Flexer, V. A novel carbon nanotube modified scaffold as an efficient biocathode material for improved microbial electrosynthesis. J. Mater. Chem. A 2014, 2, 13093–13102. [Google Scholar] [CrossRef] [Green Version]
- Barin, R.; Biria, D.; Rashid-Nadimi, S.; Asadollahi, M.A. Enzymatic CO2 reduction to formate by formate dehydrogenase from Candida boidinii coupling with direct electrochemical regeneration of NADH. J. CO2 Util. 2018, 28, 117–125. [Google Scholar] [CrossRef]
- Bulutoglu, B.; Macazo, F.C.; Bale, J.; King, N.; Baker, D.; Minteer, S.D.; Banta, S. Multimerization of an Alcohol Dehydrogenase by Fusion to a Designed Self-Assembling Protein Results in Enhanced Bioelectrocatalytic Operational Stability. ACS Appl. Mater. Interfaces 2019, 11, 20022–20028. [Google Scholar] [CrossRef]
- Gross, A.; Holzinger, M.; Cosnier, S. Buckypaper bioelectrodes: Emerging materials for implantable and wearable biofuel cells. Energy Environ. Sci. 2018, 11, 1670–1687. [Google Scholar] [CrossRef]
- Choi, D.S.; Lee, H.; Tieves, F.; Lee, Y.W.; Son, E.J.; Zhang, W.; Shin, B.; Hollmann, F.; Park, C.B. Bias-Free In Situ H2O2 Generation in a Photovoltaic-Photoelectrochemical Tandem Cell for Biocatalytic Oxyfunctionalization. ACS Catal. 2019, 9, 10562–10566. [Google Scholar] [CrossRef]
- Seelajaroen, H.; Bakandritsos, A.; Otyepka, M.; Zbořil, R.; Sariciftci, N.S. Immobilized Enzymes on Graphene as Nanobiocatalyst. ACS Appl. Mater. Interfaces 2019, 12, 250–259. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Engineering new catalytic activities in enzymes. Nat. Catal. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Wong, T.S.; Schwaneberg, U. Protein engineering in bioelectrocatalysis. Curr. Opin. Biotechnol. 2003, 14, 590–596. [Google Scholar] [CrossRef]
- Yin, L.L.; Yuan, H.; Liu, C.; He, B.; Gao, S.-Q.; Wen, G.-B.; Tan, X.; Lin, Y.-W. A rationally designed myoglobin exhibits a catalytic dehalogenation efficiency more than 1000-fold that of a native dehaloperoxidase. ACS Catal. 2018, 8, 9619–9624. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Engineering cytochrome P450s for enantioselective cyclopropenation of internal alkynes. J. Am. Chem. Soc. 2020, 142, 6891–6895. [Google Scholar] [CrossRef] [Green Version]
- Brandenberg, O.F.; Miller, D.C.; Markel, U.; Ouald Chaib, A.; Arnold, F.H. Engineering Chemoselectivity in Hemoprotein-Catalyzed Indole Amidation. ACS Catal. 2019, 9, 8271–8275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateljak, I.; Monza, E.; Lucas, M.F.; Guallar, V.; Aleksejeva, O.; Ludwig, R.; Leech, D.; Shleev, S.; Alcalde, M. Increasing redox potential, redox mediator activity, and stability in a fungal laccase by computer-guided mutagenesis and directed evolution. ACS Catal. 2019, 9, 4561–4572. [Google Scholar] [CrossRef] [Green Version]
- Aleksejeva, O.; Mateljak, I.; Ludwig, R.; Alcalde, M.; Shleev, S. Electrochemistry of a high redox potential laccase obtained by computer-guided mutagenesis combined with directed evolution. Electrochem. Commun. 2019, 106, 106511. [Google Scholar] [CrossRef]
- Vidal, L.S.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD (P) H-dependent oxidoreductases: Properties, engineering and application. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef]
- Liu, X.; Bastian, S.; Snow, C.D.; Brustad, E.M.; Saleski, T.E.; Xu, J.-H.; Meinhold, P.; Arnold, F.H. Structure-guided engineering of Lactococcus lactis alcohol dehydrogenase LlAdhA for improved conversion of isobutyraldehyde to isobutanol. J. Biotechnol. 2013, 164, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chen, J.; Li, Y. Enhanced activity of yqhD oxidoreductase in synthesis of 1, 3-propanediol by error-prone PCR. Prog. Natl. Sci. 2008, 18, 1519–1524. [Google Scholar] [CrossRef]
- Guterl, J.K.; Sieber, V. Biosynthesis “debugged”: Novel bioproduction strategies. Eng. Life Sci. 2013, 13, 4–18. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.l.; Kroutil, W. Artificial biocatalytic linear cascades for preparation of organic molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, F.; Chen, H.; Malapit, C.A.; Prater, M.B.; Li, M.; Yuan, M.; Lim, K.; Minteer, S.D. Biphasic bioelectrocatalytic synthesis of chiral β-hydroxy nitriles. J. Am. Chem. Soc. 2020, 142, 8374–8382. [Google Scholar] [CrossRef]
- Abdellaoui, S.; Macazo, F.C.; Cai, R.; De Lacey, A.L.; Pita, M.; Minteer, S.D. Enzymatic electrosynthesis of alkanes by bioelectrocatalytic decarbonylation of fatty aldehydes. Angew. Chem. Int. Ed. 2018, 57, 2404–2408. [Google Scholar] [CrossRef]
- Kuk, S.K.; Singh, R.K.; Nam, D.H.; Singh, R.; Lee, J.K.; Park, C.B. Photoelectrochemical reduction of carbon dioxide to methanol through a highly efficient enzyme cascade. Angew. Chem. Int. Ed. 2017, 56, 3827–3832. [Google Scholar] [CrossRef]
- Hickey, D.P.; Gaffney, E.M.; Minteer, S.D. Electrometabolic pathways: Recent developments in bioelectrocatalytic cascades. In Electrocatalysis; Springer: Cham, Switzerland, 2020; pp. 149–165. [Google Scholar]
- Duca, M.; Weeks, J.R.; Fedor, J.G.; Weiner, J.H.; Vincent, K.A. Combining noble metals and enzymes for relay cascade electrocatalysis of nitrate reduction to ammonia at neutral pH. ChemElectroChem 2015, 2, 1086–1089. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, Y.; Zhao, W.; Zhu, C.; Zhu, B. Cloning, expression, and characterization of a novel (S)-specific alcohol dehydrogenase from Lactobacillus kefir. Appl. Biochem. Biotechnol. 2010, 160, 19. [Google Scholar] [CrossRef]
- Vedha-Peters, K.; Gunawardana, M.; Rozzell, J.D.; Novick, S.J. Creation of a broad-range and highly stereoselective D-amino acid dehydrogenase for the one-step synthesis of D-amino acids. J. Am. Chem. Soc. 2006, 128, 10923–10929. [Google Scholar] [CrossRef] [Green Version]
- Richter, N.; Neumann, M.; Liese, A.; Wohlgemuth, R.; Weckbecker, A.; Eggert, T.; Hummel, W. Characterization of a whole-cell catalyst co-expressing glycerol dehydrogenase and glucose dehydrogenase and its application in the synthesis of l-glyceraldehyde. Biotechnol. Bioeng. 2010, 106, 541–552. [Google Scholar] [CrossRef]
- Wu, R.; Zhu, Z. Self-powered enzymatic electrosynthesis of l-3, 4-Dihydroxyphenylalanine in a hybrid bioelectrochemical system. ACS Sustain. Chem. Eng. 2018, 6, 12593–12597. [Google Scholar] [CrossRef]
- Chen, H.; Prater, M.B.; Cai, R.; Dong, F.; Chen, H.; Minteer, S.D. Bioelectrocatalytic Conversion from N2 to Chiral Amino Acids in a H2/α-keto Acid Enzymatic Fuel Cell. J. Am. Chem. Soc. 2020, 142, 4028–4036. [Google Scholar] [CrossRef]
- Cai, R.; Milton, R.D.; Abdellaoui, S.; Park, T.; Patel, J.; Alkotaini, B.; Minteer, S.D. Electroenzymatic C–C bond formation from CO2. J. Am. Chem. Soc. 2018, 140, 5041–5044. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Sahin, S.; Cai, R.; Abdellaoui, S.; Hickey, D.P.; Minteer, S.D.; Milton, R.D. Creating a Low-Potential Redox Polymer for Efficient Electroenzymatic CO2 Reduction. Angew. Chem. 2018, 130, 6692–6696. [Google Scholar] [CrossRef]
- Schlager, S.; Dumitru, L.M.; Haberbauer, M.; Fuchsbauer, A.; Neugebauer, H.; Hiemetsberger, D.; Wagner, A.; Portenkirchner, E.; Sariciftci, N.S. Electrochemical reduction of carbon dioxide to methanol by direct injection of electrons into immobilized enzymes on a modified electrode. ChemSusChem 2016, 9, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Sciarria, T.P.; Batlle-Vilanova, P.; Colombo, B.; Scaglia, B.; Balaguer, M.; Colprim, J.; Puig, S.; Adani, F. Bio-electrorecycling of carbon dioxide into bioplastics. Green Chem. 2018, 20, 4058–4066. [Google Scholar] [CrossRef]
- Cadoux, C.M.; Milton, R.D. Recent enzymatic electrochemistry for reductive reactions. ChemElectroChem 2020. [Google Scholar] [CrossRef]
- Lee, Y.S.; Ruff, A.; Cai, R.; Lim, K.; Schuhmann, W.; Minteer, S.D. Electroenzymatic Nitrogen Fixation Using an Organic Redox Polymer-Immobilized MoFe Protein System. Angew. Chem. 2020. [Google Scholar] [CrossRef]
- Milton, R.D.; Abdellaoui, S.; Khadka, N.; Dean, D.R.; Leech, D.; Seefeldt, L.C.; Minteer, S.D. Nitrogenase bioelectrocatalysis: Heterogeneous ammonia and hydrogen production by MoFe protein. Energy Environ. Sci. 2016, 9, 2550–2554. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Yuan, M.; Cai, R.; Lim, K.; Minteer, S.D. Nitrogenase Bioelectrocatalysis: ATP-Independent Ammonia Production Using a Redox Polymer/MoFe Protein System. ACS Catal. 2020, 10, 6854–6861. [Google Scholar] [CrossRef]
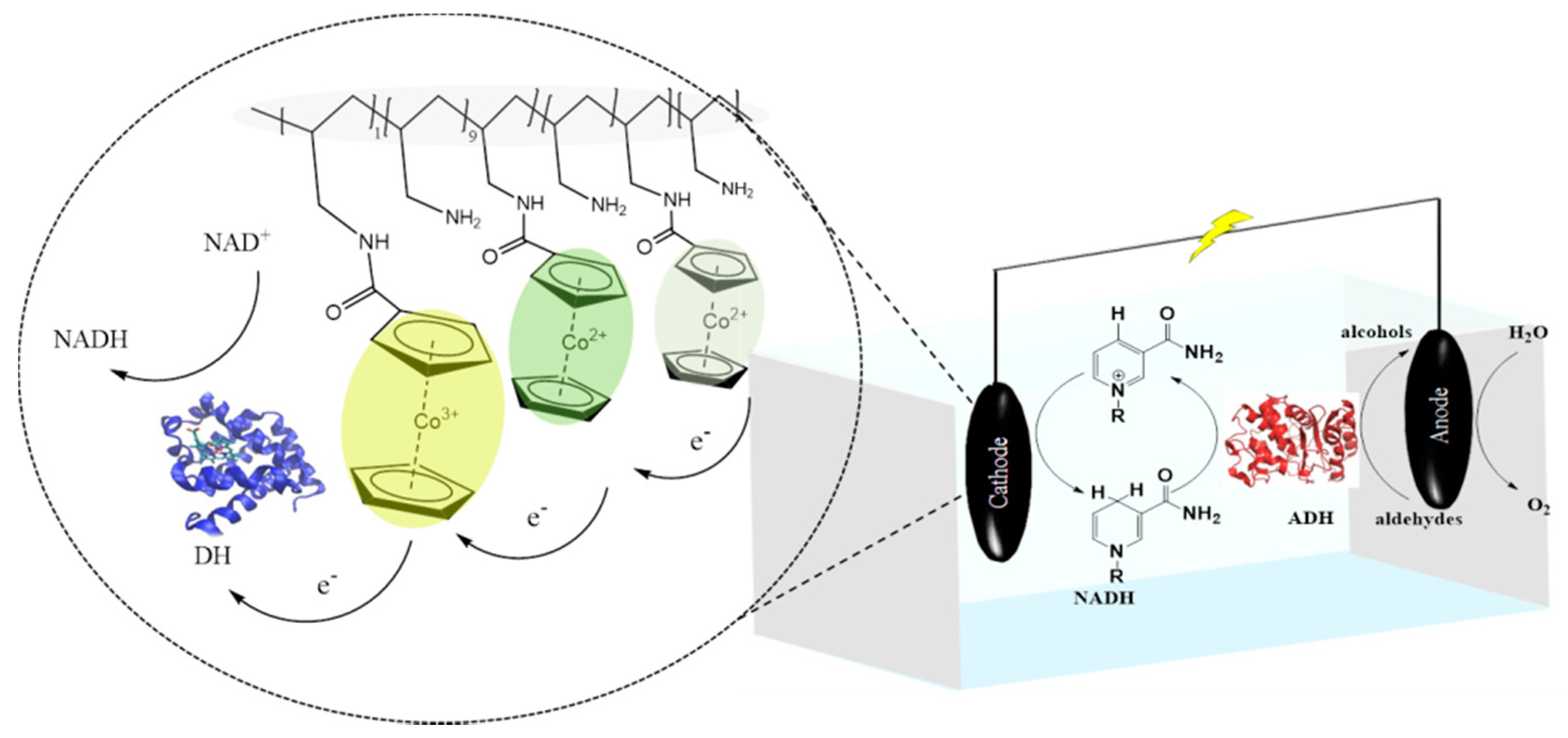




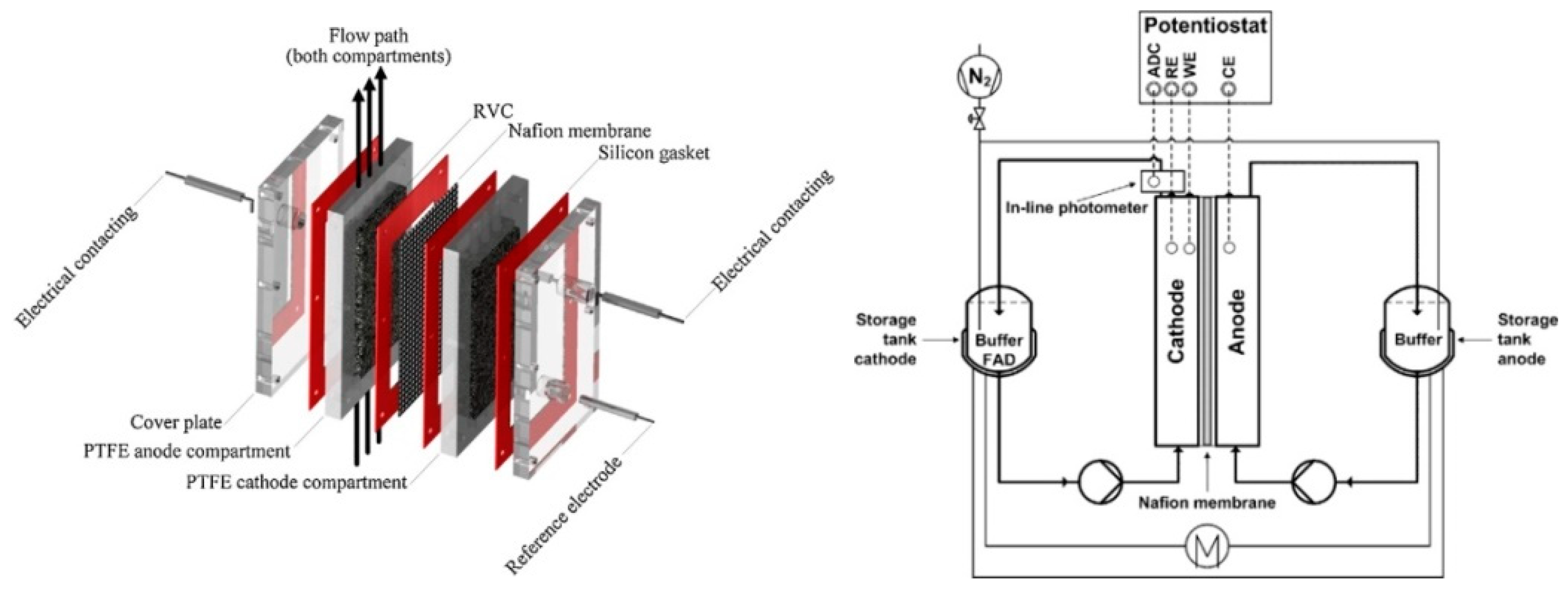
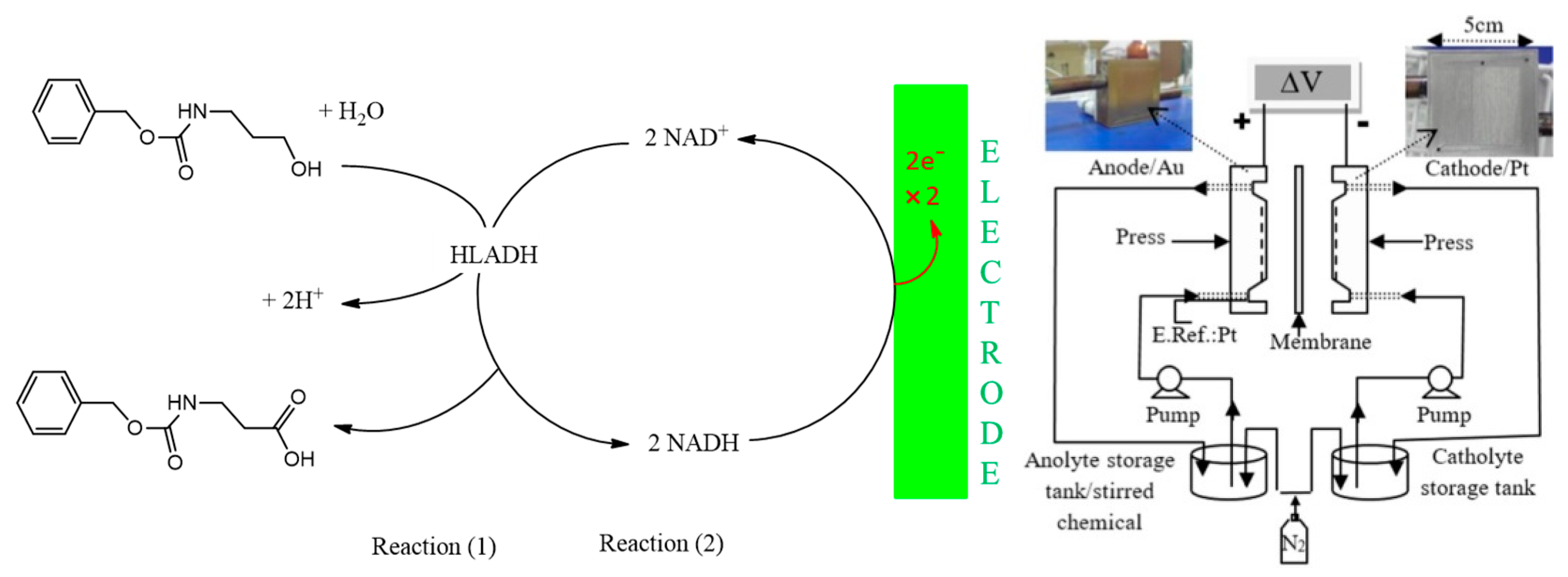
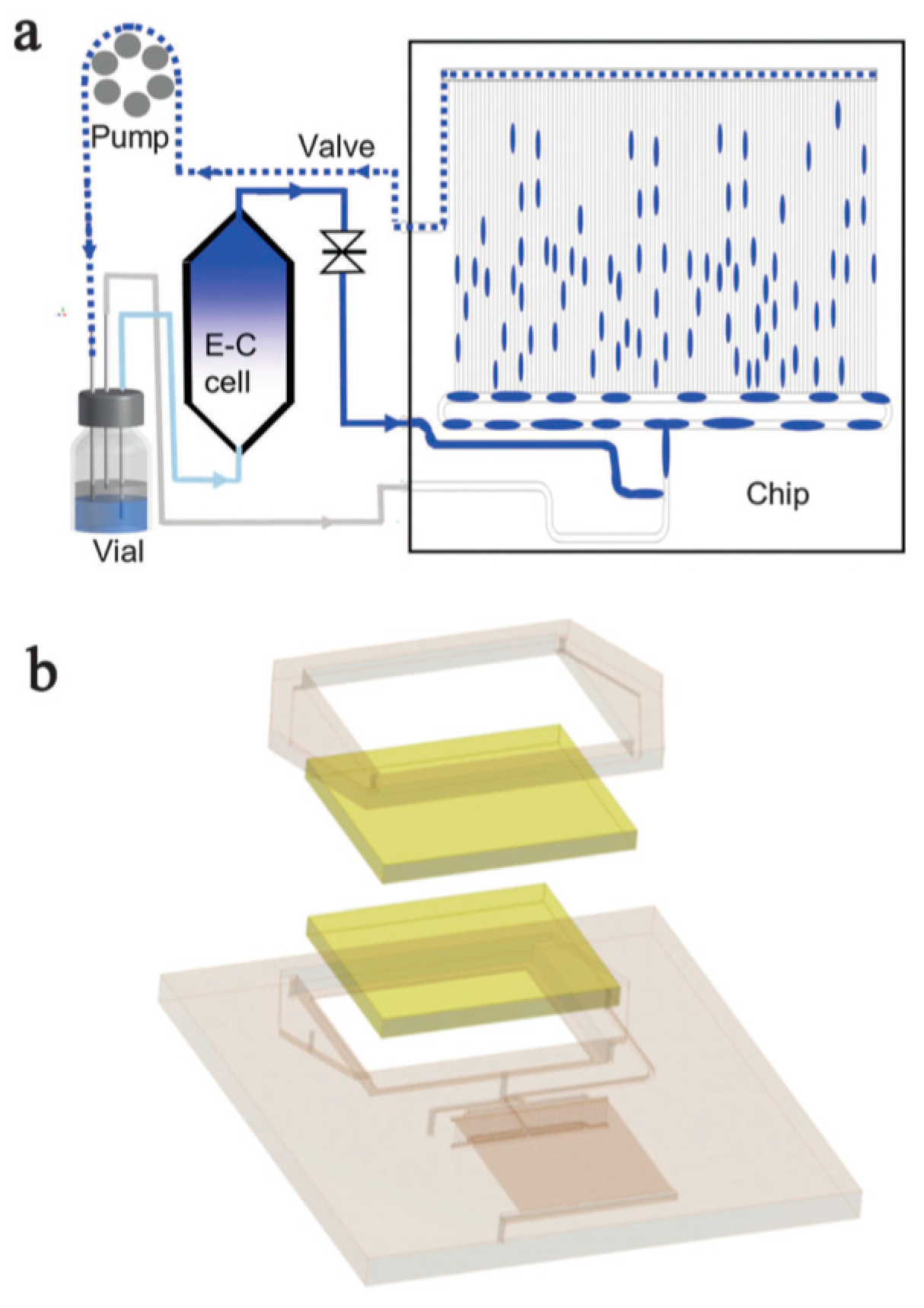


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arshi, S.; Nozari-Asbemarz, M.; Magner, E. Enzymatic Bioreactors: An Electrochemical Perspective. Catalysts 2020, 10, 1232. https://doi.org/10.3390/catal10111232
Arshi S, Nozari-Asbemarz M, Magner E. Enzymatic Bioreactors: An Electrochemical Perspective. Catalysts. 2020; 10(11):1232. https://doi.org/10.3390/catal10111232
Chicago/Turabian StyleArshi, Simin, Mehran Nozari-Asbemarz, and Edmond Magner. 2020. "Enzymatic Bioreactors: An Electrochemical Perspective" Catalysts 10, no. 11: 1232. https://doi.org/10.3390/catal10111232
APA StyleArshi, S., Nozari-Asbemarz, M., & Magner, E. (2020). Enzymatic Bioreactors: An Electrochemical Perspective. Catalysts, 10(11), 1232. https://doi.org/10.3390/catal10111232