A DFT Study for Catalytic Deoxygenation of Methyl Butyrate on a Lewis Acid Site of ZSM-5 Zeolite


Abstract
:1. Introduction
2. Results and Discussion
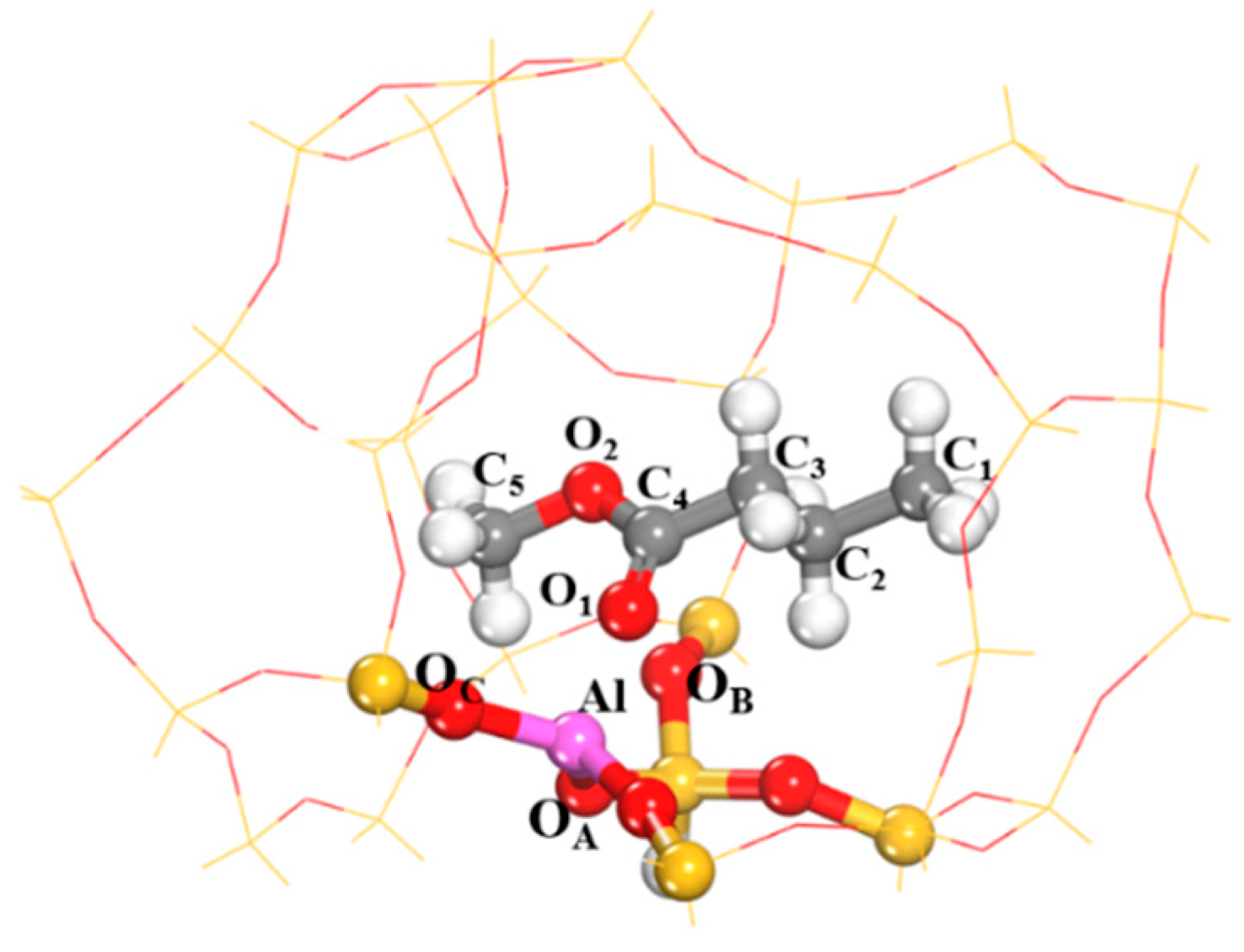
2.1. Adsorption of Methyl Butyrate
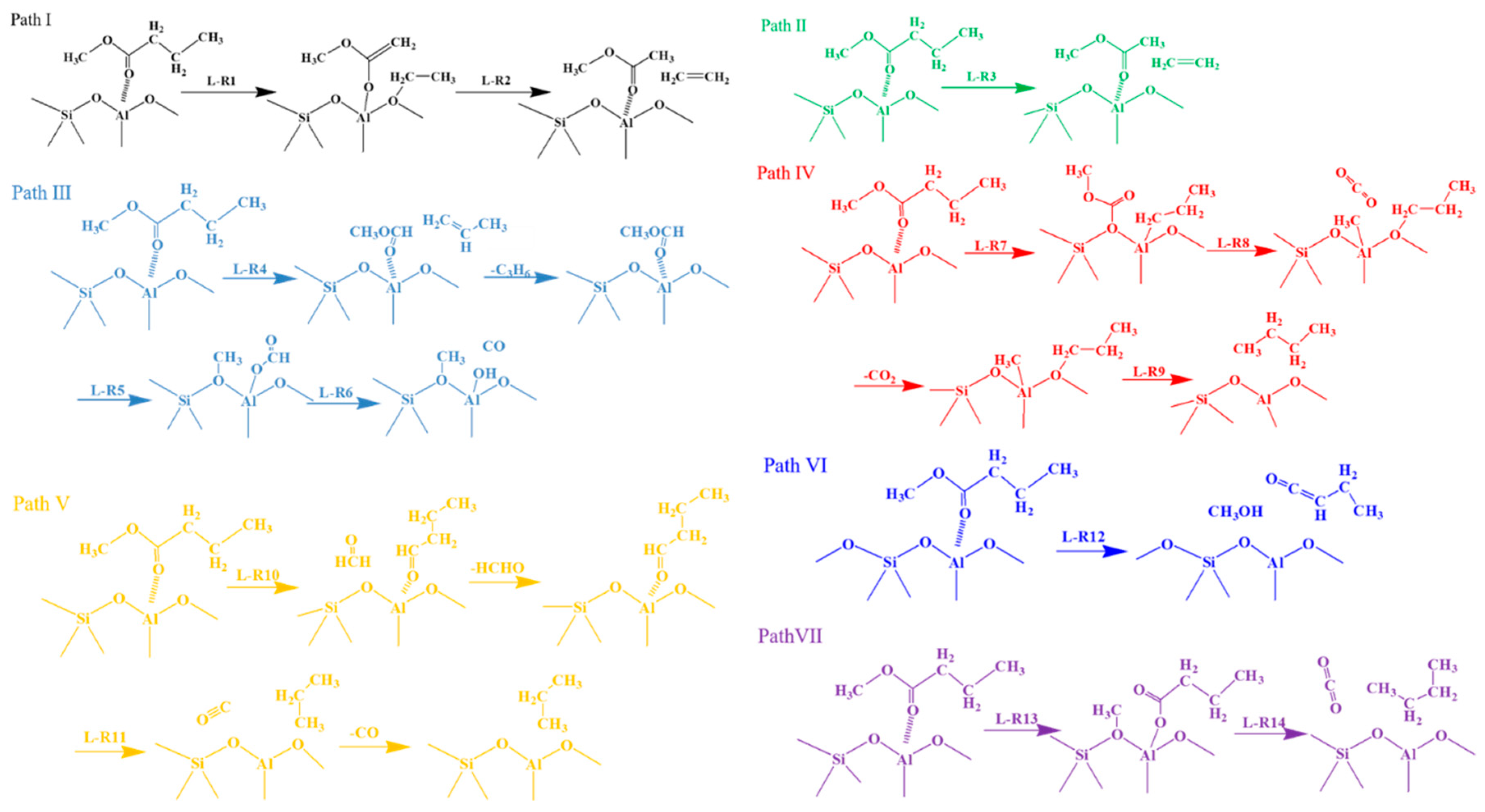
2.2. Deoxygenation Reaction Pathways
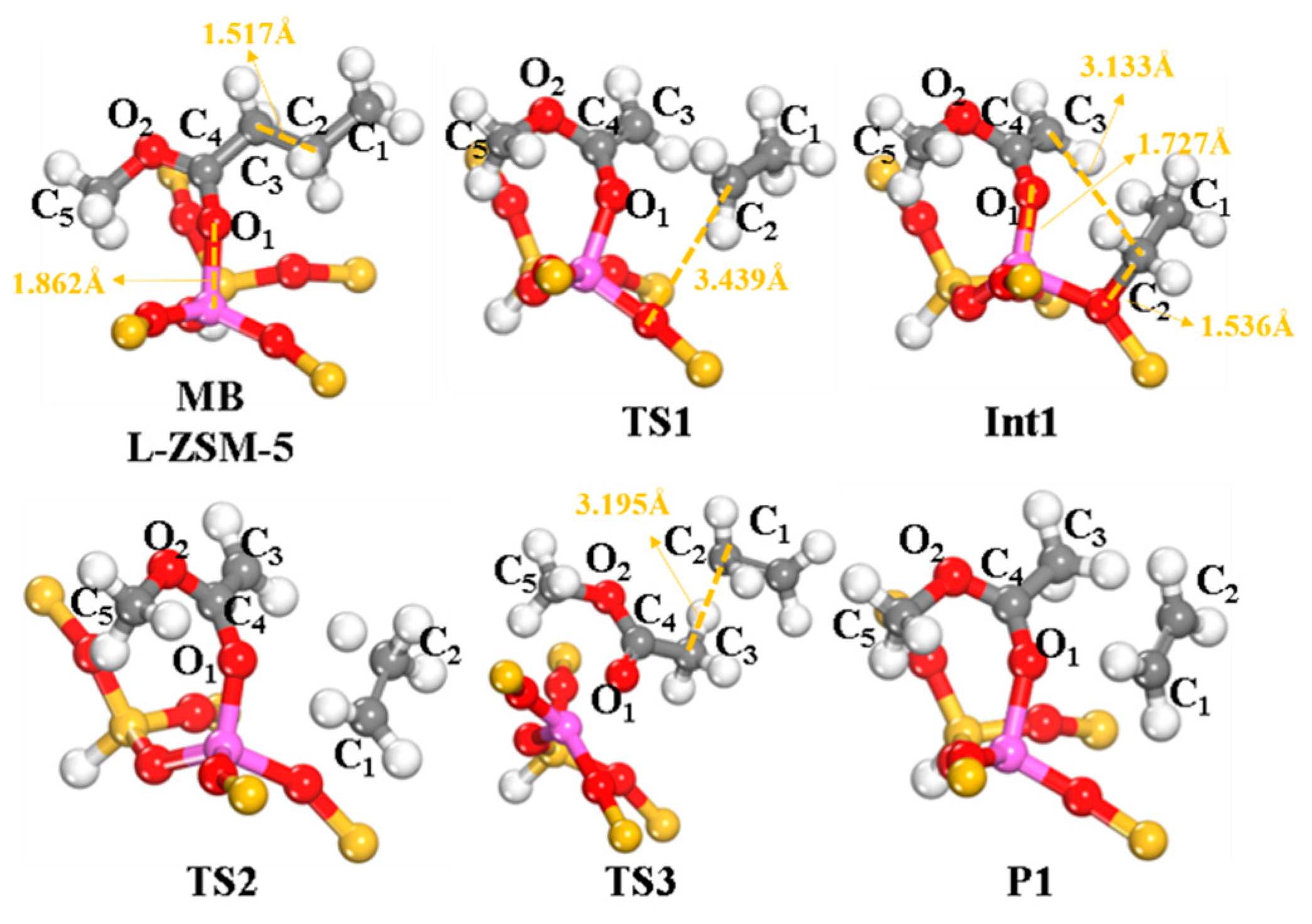
2.2.1. Cracking of β-C–C Bond
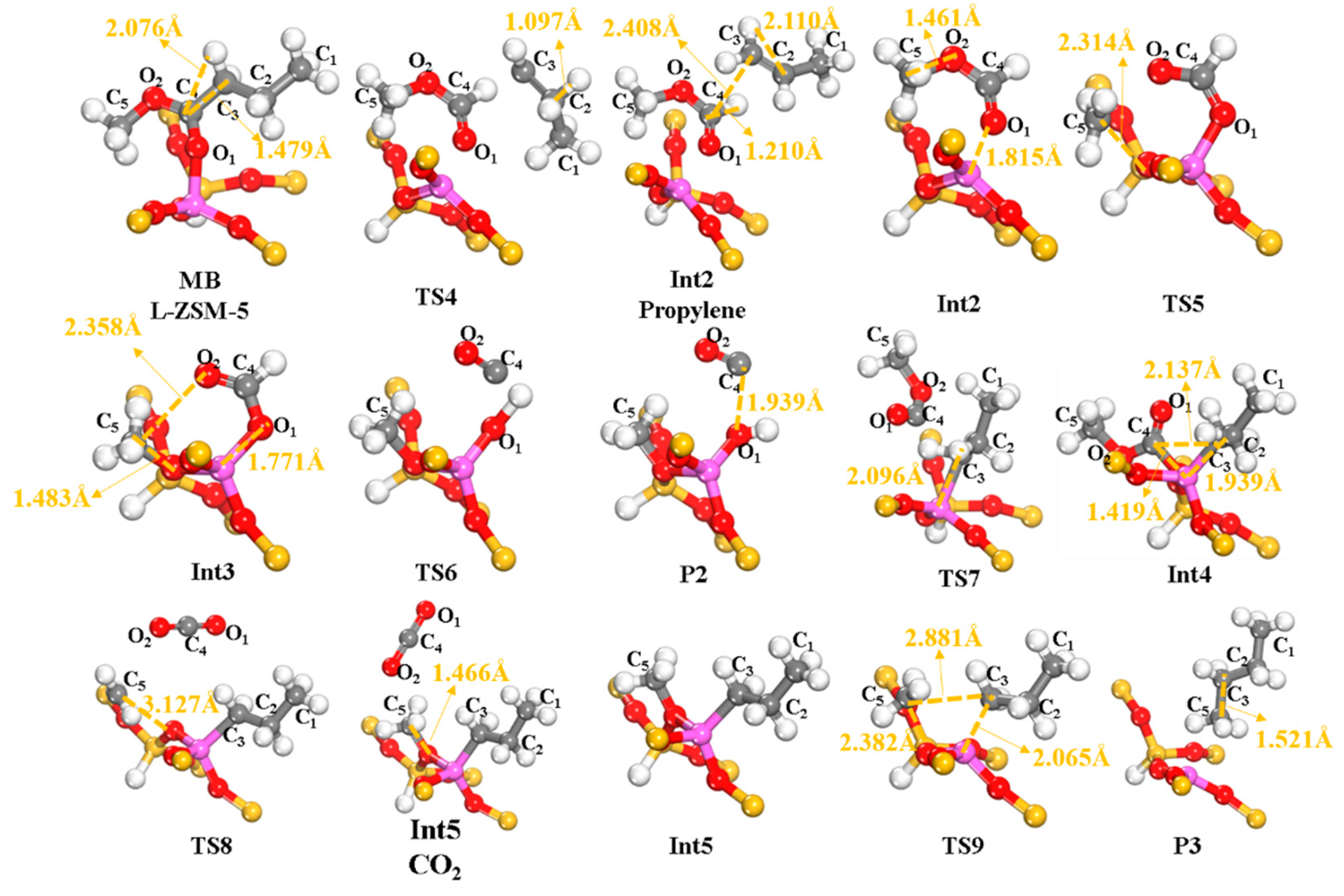
2.2.2. Cracking of α-C–C Bond
2.2.3. Cracking of α-C–O Bond
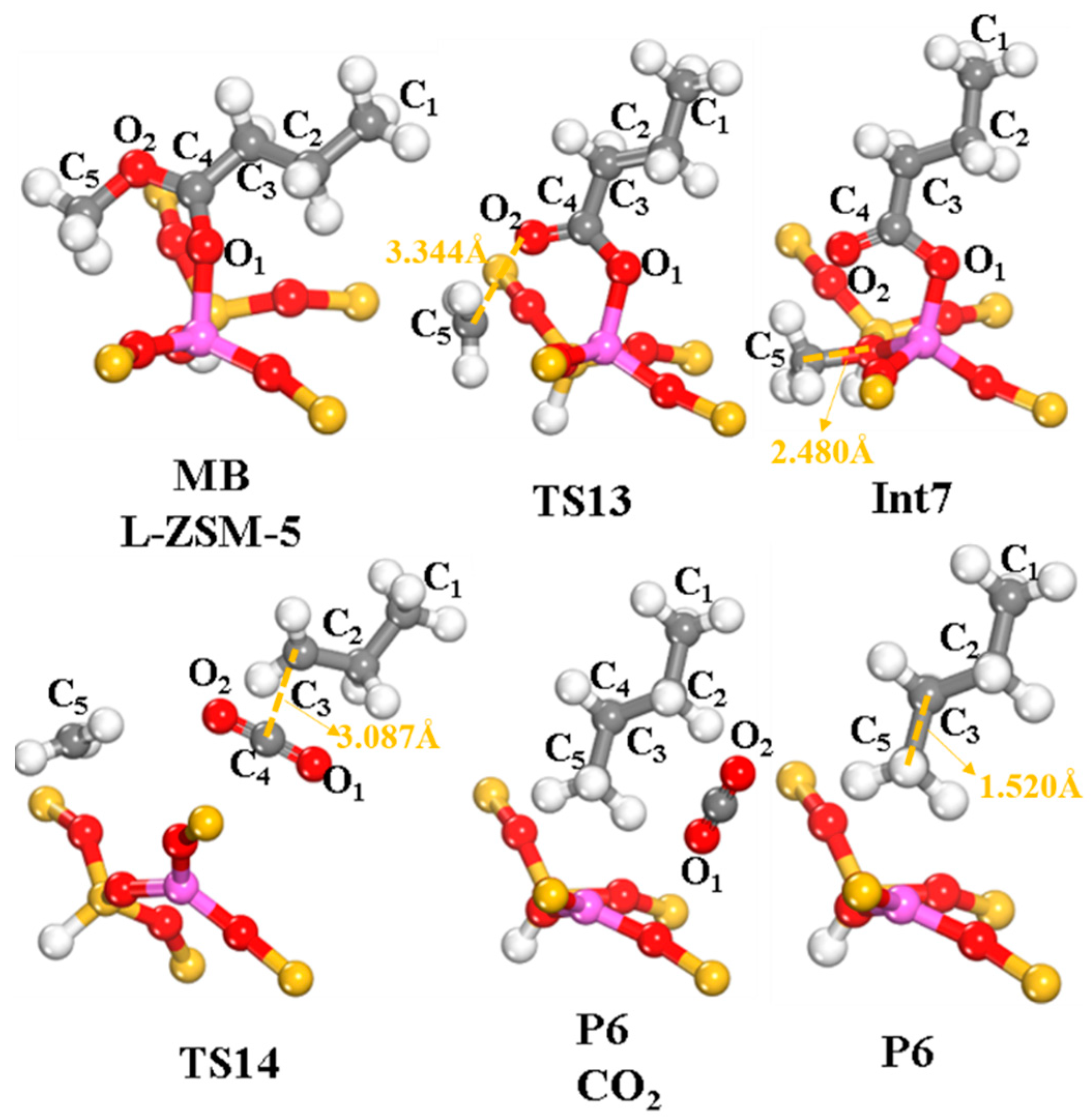
2.2.4. Cracking of β-C–O Bond
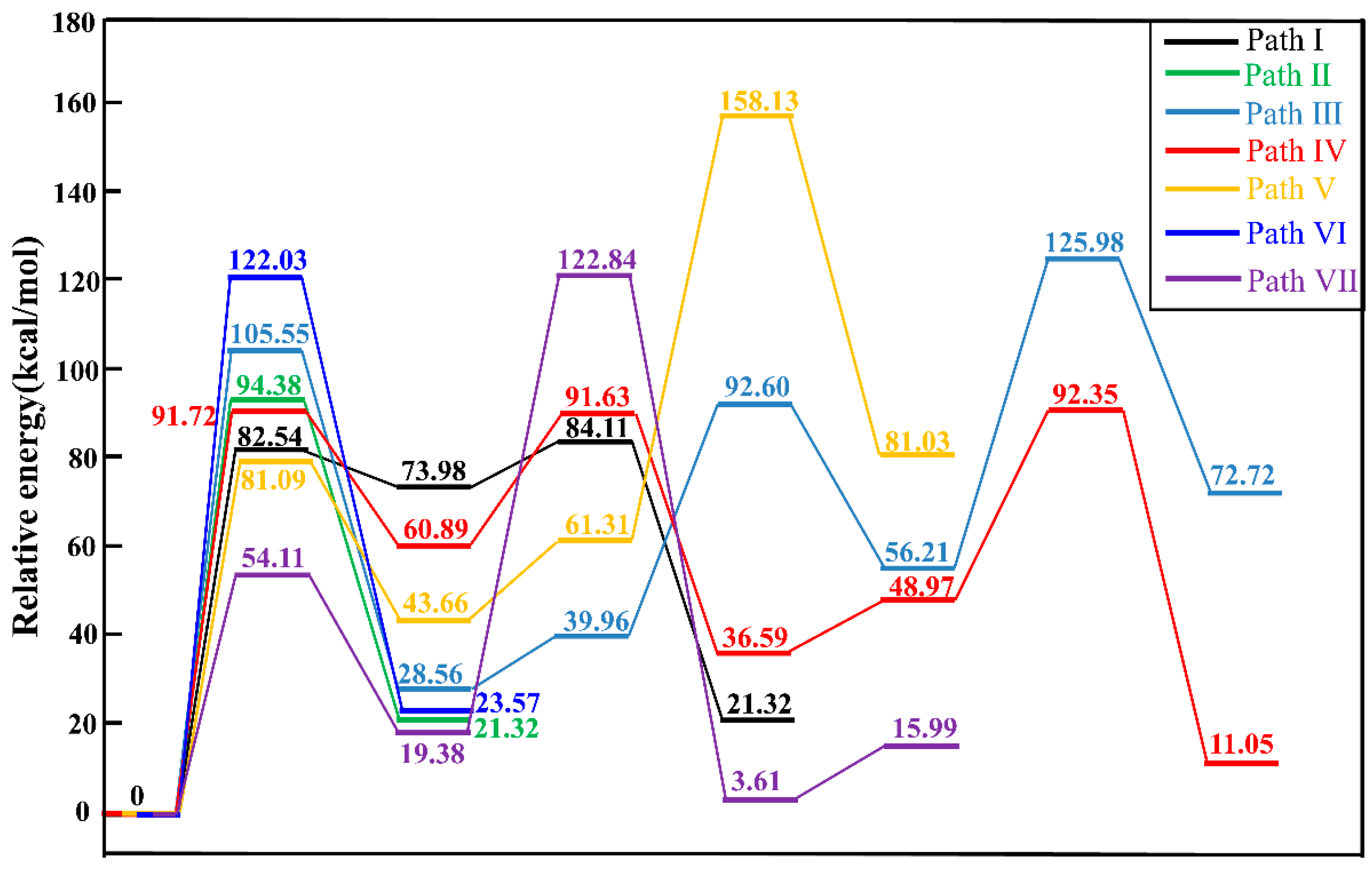
2.3. Activation Energy
2.4. Discussion
3. Model and Methods
3.1. Model
3.2. Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Issariyakul, T.; Dalai, A.K. Biodiesel from vegetable oils. Renew. Sust. Energy Rev. 2014, 31, 446–471. [Google Scholar] [CrossRef]
- Singh, D.; Sharma, D.; Soni, S.L.; Sharma, S.; Sharma, P.K.; Jhalani, A. A review on feedstocks, production processes, and yield for different generations of biodiesel. Fuel 2020, 262, 116553. [Google Scholar] [CrossRef]
- Huber, G.W.; Corma, A. Synergies between bio- and oil refineries for the production of fuels from biomass. Angew. Chem. Int. Ed. 2007, 46, 7184–7201. [Google Scholar] [CrossRef] [PubMed]
- Idem, R.O.; Katikaneni, S.P.; Bakhshi, N.N. Catalytic conversion of canola oil to fuels and chemicals over various cracking catalysts. Fuel Process. Technol. 1997, 51, 101–125. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, Y.; Peng, Y.; Ke, L.; Yang, Q.; Jiang, L.; Dai, L.; Liu, Y.; Ruan, R.; Xia, D.; et al. Microwave-assisted pyrolysis of waste cooking oil for hydrocarbon bio-oil over metal oxides and HZSM-5 catalysts. Energy Conv. Manag. 2020, 220, 113124. [Google Scholar] [CrossRef]
- Atsushi, I.; Takashi, T.; Tadanori, H.; Hiroyuki, N. Catalytic cracking of soybean oil by ZSM-5 zeolite-containing silica-aluminas with three layered micro-meso-meso-structure. Catal. Today 2018, 303, 123–129. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, Z.; Ding, K.; Zhang, B.; Deng, A.; Min, M.; Chen, P.; Ruan, R. Successive desilication and dealumination of HZSM-5 in catalytic conversion of waste cooking oil to produce aromatics. Energy Conv. Manag. 2017, 147, 100–107. [Google Scholar] [CrossRef]
- Phung, T.K.; Casazza, A.A.; Finocchio, E.; Busca, G. Catalytic conversion of ethyl acetate over faujasite zeolites. Appl. Catal. A Gen. 2014, 470, 72–80. [Google Scholar] [CrossRef]
- Cai, W.; Yan, H.; Feng, X.; Liu, Y.; Yang, C. Product distribution in catalytic cracking of fatty acid methyl esters with different carbon chain lengths. CIESC J. 2017, 68, 2057–2065. [Google Scholar] [CrossRef]
- Botas, J.A.; Serrano, D.P.; García, A.; Ramos, R. Catalytic conversion of rapeseed oil for the production of raw chemicals, fuels and carbon nanotubes over Ni-modified nanocrystalline and hierarchical ZSM-5. Appl. Catal. B Environ. 2014, 145, 205–215. [Google Scholar] [CrossRef]
- Li, R.; Yan, H.; Dang, Y.; Liu, Y.; Feng, X.; Chen, X.; Jin, X.; Yang, C. Deoxygenation mechanism of methyl butyrate on HZSM-5: A density functional theory study. Mol. Catal. 2019, 479, 7–26. [Google Scholar] [CrossRef]
- Yan, H.; Feng, X.; Liu, Y.; Yang, C.; Shan, H. Catalytic cracking of acetic acid and its ketene intermediate over HZSM-5 catalyst: A density functional theory study. Mol. Catal. 2017, 437, 11–17. [Google Scholar] [CrossRef]
- Vonghia, E.; Boocock, D.G.B.; Konar, S.K.; Leung, A. Pathways for the deoxygenation of triglycerides to aliphatic hydrocarbons over activated alumina. Energy Fuel 1995, 9, 1090–1096. [Google Scholar] [CrossRef]
- Leung, A.; Boocock, D.G.B.; Konar, S.K. Pathway for the catalytic conversion of carboxylic acids to hydrocarbons over activated alumina. Energy Fuel 1995, 9, 913–920. [Google Scholar] [CrossRef]
- Phung, T.K.; Casazza, A.A.; Aliakbarian, B.; Finocchio, E.; Perego, P.; Busca, G. Catalytic conversion of ethyl acetate and acetic acid on alumina as models of vegetable oils conversion to biofuels. Chem. Eng. J. 2013, 215, 838–848. [Google Scholar] [CrossRef]
- Blaszkowski, S.R.; Nascimento, M.A.C.; van Santen, R.A. Activation of C−H and C−C Bonds by an Acidic Zeolite: A Density Functional Study. J. Phys. Chem. 1996, 100, 3463–3472. [Google Scholar] [CrossRef] [Green Version]
- Kolganov, A.A.; Gabrienko, A.A.; Yashnik, S.A.; Pidko, E.A.; Stepanov, A.G. Nature of the surface intermediates formed from methane on Cu-ZSM-5 zeolite: A combined solid-state nuclear magnetic resonance and density functional theory study. J. Phys. Chem. C 2020, 124, 6242–6252. [Google Scholar] [CrossRef]
- Goncalves, T.J.; Plessow, P.N.; Studt, F. On the accuracy of density functional theory in zeolite catalysis. ChemCatChem 2019, 11, 4368–4376. [Google Scholar] [CrossRef]
- Liu, Y.; Dang, Y.; Feng, X.; Chen, X.; Yang, C. Promoting effect of Ni on the structure and electronic properties of NixMo(1−x)S2 catalyst and benzene adsorption: A periodic DFT study. Appl. Surf. Sci. 2019, 471, 607–614. [Google Scholar] [CrossRef]
- Rangarajan, S.; Mavrikakis, M. DFT insights into the competitive adsorption of sulfur- and nitrogen-containing compounds and hydrocarbons on Co-promoted molybdenum sulfide catalysts. ACS Cat. 2016, 6, 2904–2917. [Google Scholar] [CrossRef]
- Rubes, M.; Trachta, M.; Koudelková, E.; Bulánek, R.; Kasneryk, V.; Bludský, O. Methane adsorption in ADOR zeolites: A combined experimental and DFT/CC study. Phys. Chem. Chem. Phys. 2017, 19, 16533–16540. [Google Scholar] [CrossRef]
- Fischer, M.; Bell, R.G. Modeling CO2 Adsorption in Zeolites Using DFT-Derived Charges: Comparing System-Specific and Generic Models. J. Phys. Chem. C 2013, 117, 24446–24454. [Google Scholar] [CrossRef]
- Hessou, E.P.; Ponce-Vargas, M.; Mensah, J.-B.; Tielens, F.; Santos, J.C.; Badawi, M. Dibenzyl Disulfide Adsorption on Cationic Exchanged Faujasites: A DFT Study. Nanomaterials 2019, 9, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, L.K.; Violi, A. Thermal decomposition of methyl butanoate: Ab initio study of a biodiesel fuel surrogate. J. Org. Chem. 2008, 73, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Violi, A. Reaction pathways for the thermal decomposition of methyl butanoate. J. Org. Chem. 2013, 78, 5898–5908. [Google Scholar] [CrossRef]
- Chukwu, K.C.; Árnadóttir, L. Density functional theory study of decarboxylation and decarbonylation of acetic acid on Pd(111). J. Phys. Chem. C 2020, 124, 13082–13093. [Google Scholar] [CrossRef]
- Banerjee, A.; Mushrif, S.H. Reaction pathways for the deoxygenation of biomass-pyrolysis-derived bio-oil on Ru: A DFT study using furfural as a model compound. ChemCatChem 2017, 9, 2828–2838. [Google Scholar] [CrossRef]
- Baur, W.H.; Fischer, R.X. The floppiness of it all: Bond lengths change with atomic displacement Parameters and the flexibility of various coordination tetrahedra in zeolitic frameworks. An empirical structural study of bond lengths and sngles. Chem. Mater. 2019, 31, 2401–2420. [Google Scholar] [CrossRef]
- Liang, M.; Zhu, X.; Ma, W. The propylene oxide rearrangement catalyzed by the Lewis acid sites of ZSM-5 catalyst with controllable surface acidity. Catal. Lett. 2019, 149, 942–949. [Google Scholar] [CrossRef]
- Boronat, M.; Viruela, P.; Corma, A. Theoretical study on the mechanism of the hydride transfer reaction between alkanes and alkylcarbenium ions. J. Phys. Chem. B 1997, 101, 10069–10074. [Google Scholar] [CrossRef]
- Haw, J.F. Zeolite acid strength and reaction mechanisms in catalysis. Phys. Chem. Chem. Phys. 2002, 4, 5431–5441. [Google Scholar] [CrossRef]
- Stepanov, A.H.; Zamaraev, K.I. 13C solid state NMR evidence for the existence of isobutyl carbenium ion in the reaction of isobutyl alcohol dehydration in H-ZSM-5 zeolite. Catal. Lett. 1993, 19, 153–158. [Google Scholar] [CrossRef]
- Zheng, Z.; Lei, T.; Wang, J.; Wei, Y.; Liu, X.; Yu, F.; Ji, J. Catalytic cracking of soybean oil for biofuel over γ-Al2O3/CaO composite catalyst. J. Braz. Chem. Soc. 2019, 30, 359–370. [Google Scholar] [CrossRef]
- Senol, O.I.; Viljava, T.R.; Krause, A.O.I. Hydrodeoxygenation of methyl esters on sulphided NiMo/γ-Al2O3 and CoMo/γ-Al2O3 catalysts. Catal. Today 2005, 100, 331–335. [Google Scholar] [CrossRef]
- Katikaneni, S.P.R.; Adjaye, J.D.; Bakhshi, N.N. Performance of Aluminophosphate Molecular Sieve Catalysts for the Production of Hydrocarbons from Wood-Derived and Vegetable Oils. Energy Fuel 1995, 9, 1065–1078. [Google Scholar] [CrossRef]
- Katikaneni, S.P.R.; Adjaye, J.D.; Bakhshi, N.N. Studies on the Catalytic Conversion of Canola Oil to Hydrocarbons: Influence of Hybrid Catalysts and Steam. Energy Fuels 1995, 9, 599–609. [Google Scholar] [CrossRef]
- Katikaneni, S.P.R.; Adjaye, J.D.; Bakhshi, N.N. Catalytic conversion of canola oil to fuels and chemicals over various cracking catalysts. Can. J. Chem. Eng. 2010, 73, 484–497. [Google Scholar] [CrossRef]
- Tian, H.; Chunyi, L.; Yang, C.; Shan, H. Alternative Processing Technology for Converting Vegetable Oils and Animal Fats to Clean Fuels and Light Olefins. Chin. J. Chem. Eng. 2008, 16, 394–400. [Google Scholar] [CrossRef]
- Mota, C.J.; Bhering, D.L.; Rosenbach, N., Jr. A DFT study of the acidity of ultrastable Y zeolite: Where is the Bronsted/Lewis acid synergism? Angew. Chem. Int. Ed. 2004, 43, 3050–3053. [Google Scholar] [CrossRef]
- Bucko, T.; Hafner, J.; Benco, L. Adsorption and vibrational spectroscopy of ammonia at mordenite: Ab initio study. J. Chem. Phys. 2004, 120, 10263–10277. [Google Scholar] [CrossRef]
- Bucko, T.; Hafner, J.; Benco, L. Adsorption and vibrational spectroscopy of CO on mordenite: Ab initio density-functional study. J. Phys. Chem. B 2005, 109, 7345–7357. [Google Scholar] [CrossRef]
- Sokol, A.A.; Catlow, C.R.A.; Garcés, J.M.; Kuperman, A. Computational investigation into the origins of Lewis acidity in zeolites. Adv. Mater. 2000, 12, 1801–1805. [Google Scholar] [CrossRef]
- van Bokhoven, J.A.; van der Eerden, A.M.; Koningsberger, D.C. Three-coordinate aluminum in zeolites observed with in situ x-ray absorption near-edge spectroscopy at the Al K-edge: Flexibility of aluminum coordinations in zeolites. J. Am. Chem. Soc. 2003, 125, 7435–7442. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Tamargo, C.E.; Roldan, A.; de Leeuw, N.H. Density functional theory study of the zeolite-mediated tautomerization of phenol and catechol. Mol. Catal. 2017, 433, 334–345. [Google Scholar] [CrossRef]
- Benco, L.; Bucko, T.; Hafner, J.; Toulhoat, H. Ab initio simulation of Lewis sites in mordenite and comparative study of the strength of active sites via CO adsorption. J. Phys. Chem. B 2004, 108, 13656–13666. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Applications and validations of the Minnesota density functionals. Chem. Phys. Lett. 2011, 502, 1–13. [Google Scholar] [CrossRef]
- Zhao, Y.; Ng, H.T.; Hanson, E. Benchmark data for noncovalent interactions in HCOOH···benzene complexes and their use for validation of density functionals. J. Chem. Theory Comput. 2009, 5, 2726–2733. [Google Scholar] [CrossRef]
- Halgren, T.A.; Lipscomb, W.N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MB | Distance (Å) | Angle (°) | Charge (e) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| O1–C4 | O1–Al | ∠OA–Al–OB | ∠OB–Al–OC | ∠OA–Al–OC | OA | OB | OC | Al | O1 | |
| Before adsorption | 1.207 | - | 119.926 | 120.46 | 119.478 | −0.588 | −0.612 | −0.587 | 0.998 | −0.920 |
| After adsorption | 1.236 | 1.862 | 114.435 | 114.761 | 116.883 | −1.041 | −1.022 | -1.031 | 1.727 | −0.663 |
| Model | C-O Distance (Å) | X-Y a Distance (Å) | ||
|---|---|---|---|---|
| -CO | -OC | -CO | -OC | |
| 32 T cluster in this paper | 1.121 | 1.137 | 2.183 | 2.209 |
| Model in literature [37] | 1.135 | 1.146 | 2.173 | 2.200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Li, R.; Yan, H.; Liu, Y.; Yang, C. A DFT Study for Catalytic Deoxygenation of Methyl Butyrate on a Lewis Acid Site of ZSM-5 Zeolite. Catalysts 2020, 10, 1233. https://doi.org/10.3390/catal10111233
Chen X, Li R, Yan H, Liu Y, Yang C. A DFT Study for Catalytic Deoxygenation of Methyl Butyrate on a Lewis Acid Site of ZSM-5 Zeolite. Catalysts. 2020; 10(11):1233. https://doi.org/10.3390/catal10111233
Chicago/Turabian StyleChen, Xiaobo, Ruiying Li, Hao Yan, Yibin Liu, and Chaohe Yang. 2020. "A DFT Study for Catalytic Deoxygenation of Methyl Butyrate on a Lewis Acid Site of ZSM-5 Zeolite" Catalysts 10, no. 11: 1233. https://doi.org/10.3390/catal10111233
APA StyleChen, X., Li, R., Yan, H., Liu, Y., & Yang, C. (2020). A DFT Study for Catalytic Deoxygenation of Methyl Butyrate on a Lewis Acid Site of ZSM-5 Zeolite. Catalysts, 10(11), 1233. https://doi.org/10.3390/catal10111233