Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT

 ,
, 
Abstract
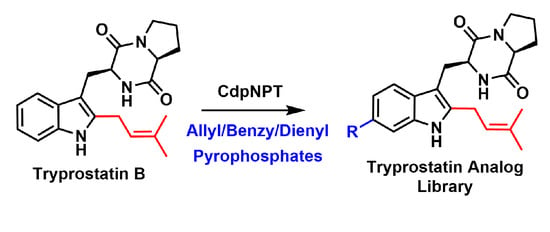
1. Introduction
2. Results and Discussion
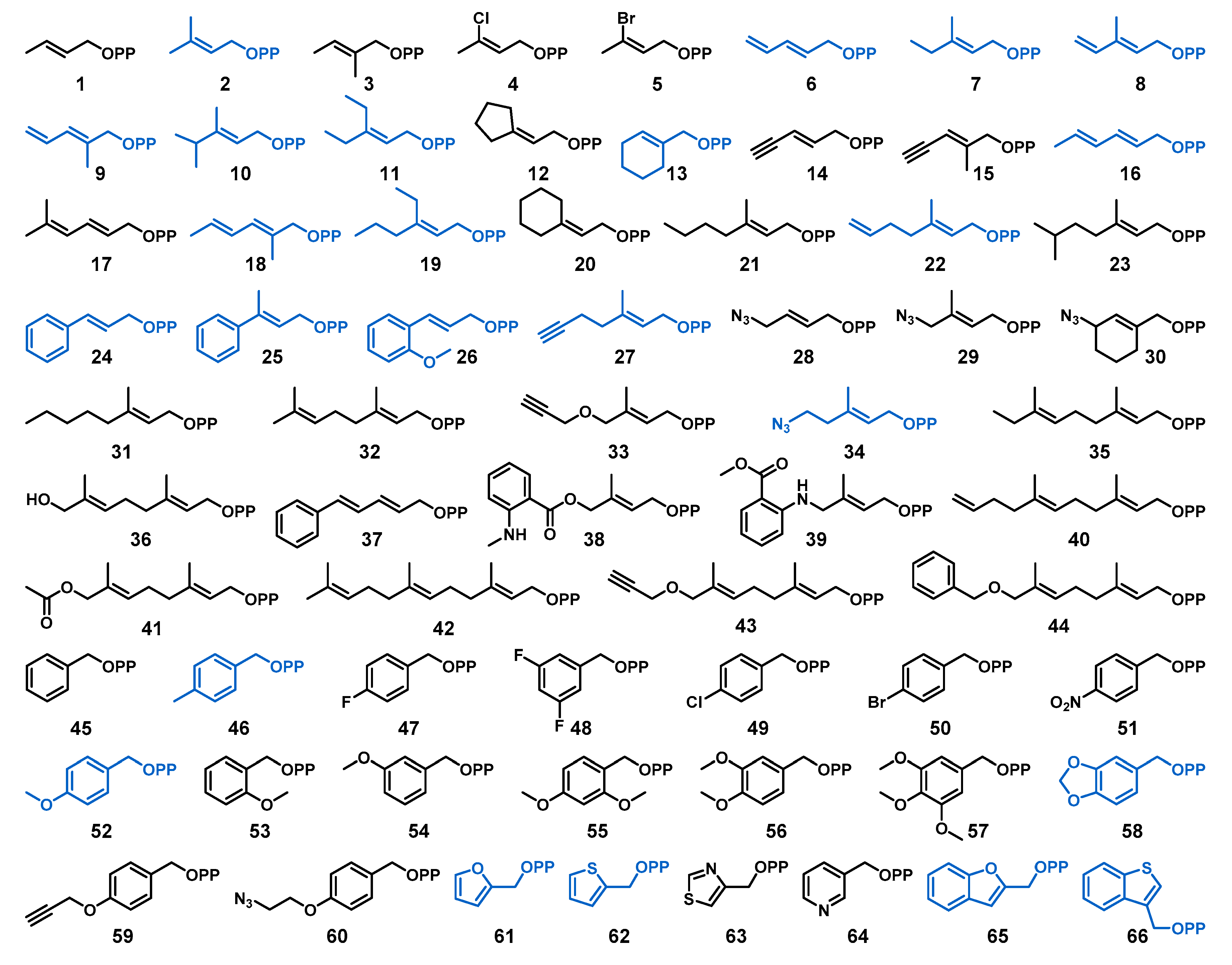
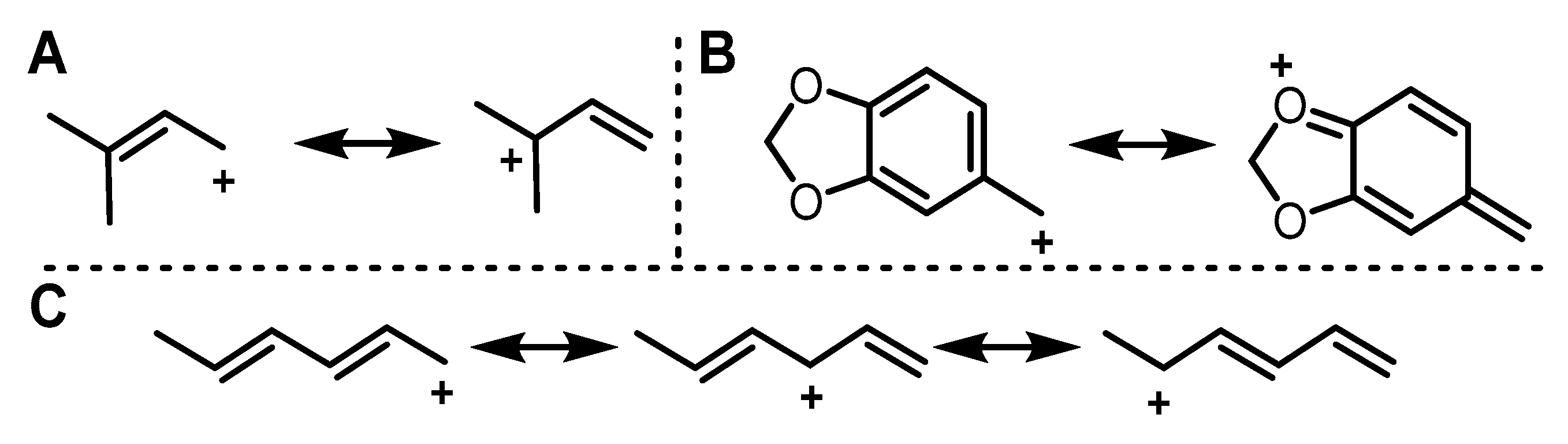
2.1. Design of Alkyl Pyrophosphate Donors
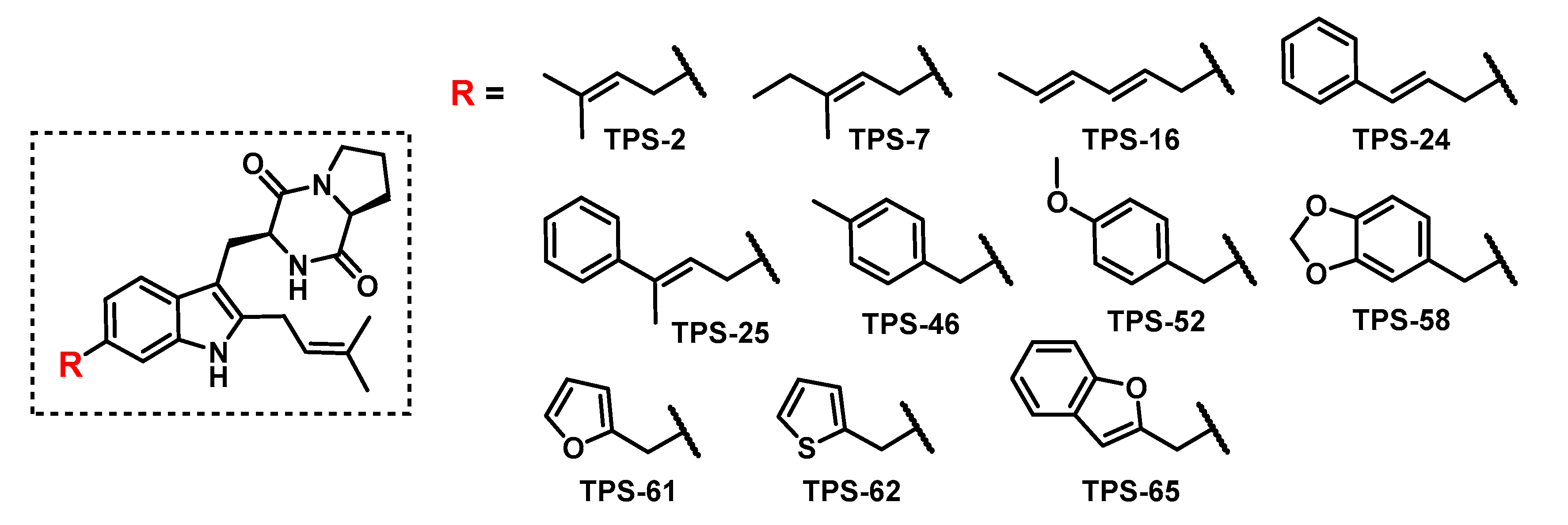
2.2. Donor Library Screening
2.3. Allylic Donors
2.4. Benzylic and Heterocyclic Donors
2.5. Other Functionalized Donors
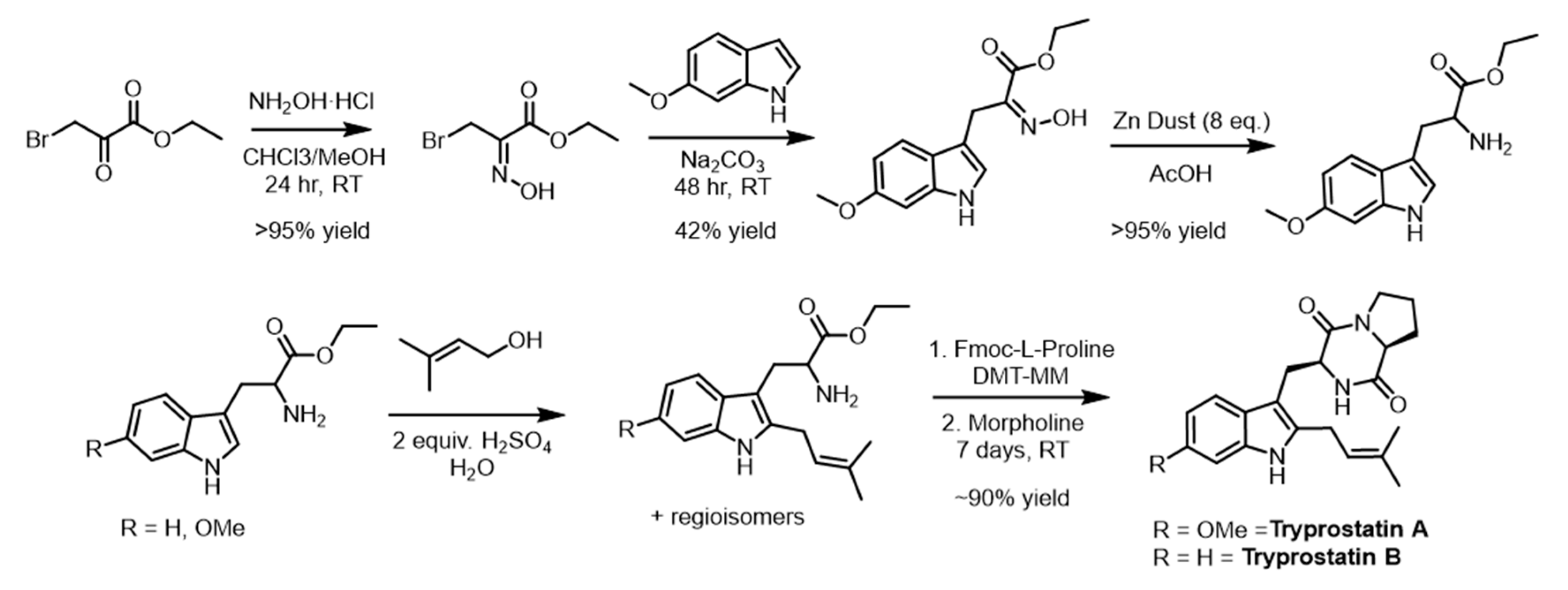
2.6. Scale-Up and Characterization of CdpNPT Reaction Products
2.7. Cytotoxicity Studies
3. Materials and Methods
3.1. General Materials
3.2. General Methods
3.3. In Vitro CdpNPT Assay
3.4. Enzymatic Scale-Up Reactions
3.5. Determination of Structures
3.6. HPLC Method A
3.7. HPLC Method B
3.8. Synthesis Tryprostatin A and B
3.9. Cell Titer-Blue Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
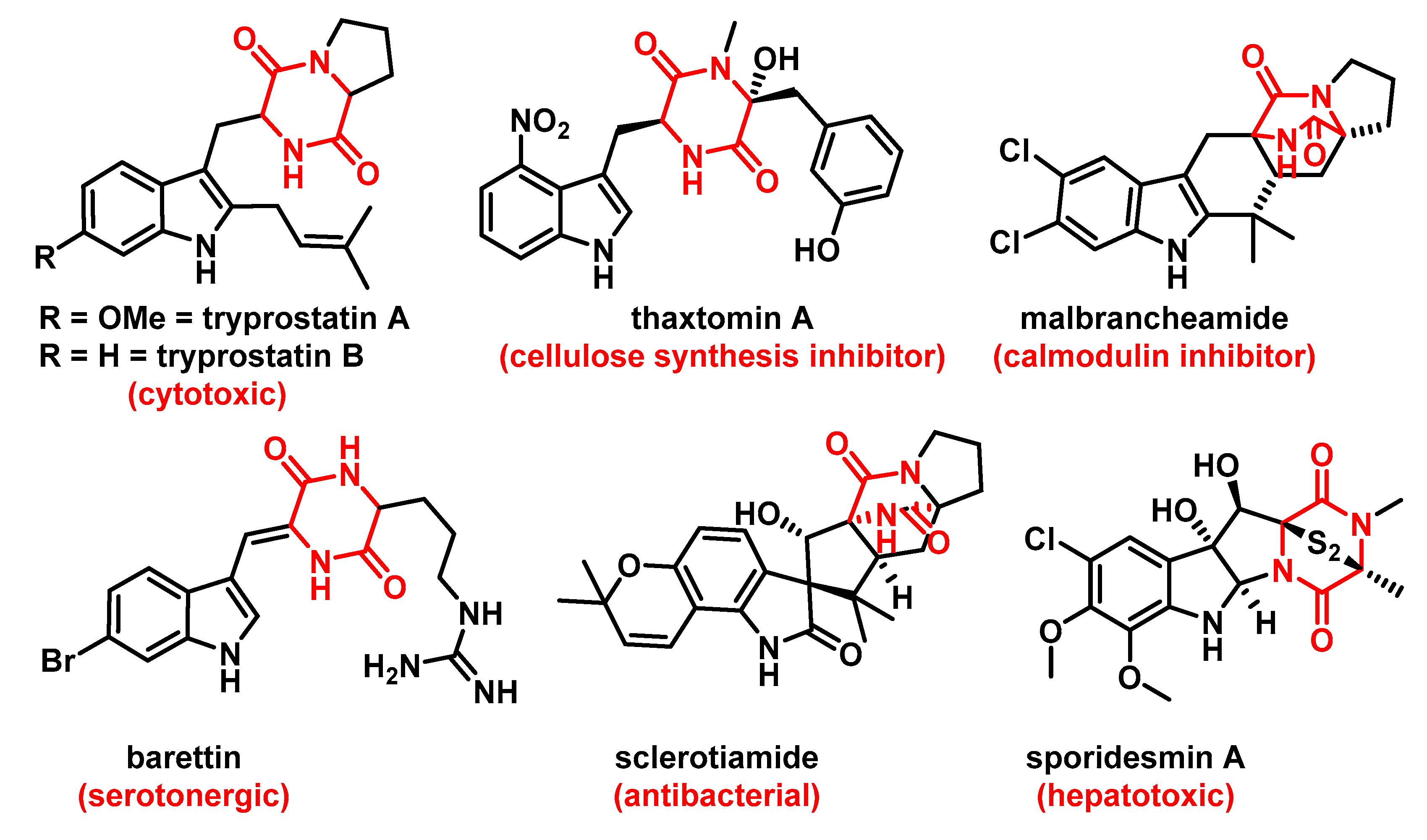
- Evidente, A.; Kornienko, A.; Cimmino, A.; Andolfi, A.; Lefranc, F.; Mathieu, V.; Kiss, R. Fungal metabolites with anticancer activity. Nat. Prod. Rep. 2014, 31, 617–627. [Google Scholar] [CrossRef]
- Ma, Y.-M.; Liang, X.-A.; Kong, Y.; Jia, B. Structural Diversity and Biological Activities of Indole Diketopiperazine Alkaloids from Fungi. J. Agric. Food Chem. 2016, 64, 6659–6671. [Google Scholar] [CrossRef]
- Cui, C.-B.; Kakeya, H.; Okada, G.; Onose, R.; Osada, H. Novel Mammalian Cell Cycle Inhibitors, Tryprostatins A, B and Other Diketopiperazines Produced by Aspergillus fumigatus. I. Taxonomy, Fermentation, Isolation and Biological Properties. J. Antibiot. 1996, 49, 527–533. [Google Scholar] [CrossRef]
- Bischoff, V.; Cookson, S.J.; Wu, S.; Scheible, W.-R. Thaxtomin A affects CESA-complex density, expression of cell wall genes, cell wall composition, and causes ectopic lignification in Arabidopsis thaliana seedlings. J. Exp. Bot. 2009, 60, 955–965. [Google Scholar] [CrossRef]
- Sjögren, M.; Jonsson, P.R.; Dahlström, M.; Lundälv, T.; Burman, R.; Göransson, U.; Bohlin, L. Two Brominated Cyclic Dipeptides Released by the Coldwater Marine Sponge Geodia barrette Act in Synergy as Chemical Defense. J. Nat. Prod. 2011, 74, 449–454. [Google Scholar] [CrossRef]
- Lavey, N.P.; Coker, J.A.; Ruben, E.A.; Duerfeldt, A.S. Sclerotiamide: The First Non-Peptide-Based Natural Product Activator of Bacterial Caseinolytic Protease P. J. Nat. Prod. 2016, 79, 1193–1197. [Google Scholar] [CrossRef]
- Kuramochi, K.; Ohnishi, K.; Fujieda, S.; Nakajima, M.; Saitoh, Y.; Watanabe, N.; Takeuchi, T.; Nakazaki, A.; Sugawara, F.; Arai, T.; et al. Synthesis and biological activities of neoechinulin A derivatives: New aspects of structure-activity relationships for neoechinulin A. Chem. Pharm. Bull. 2008, 56, 1738–1743. [Google Scholar] [CrossRef]
- Ravikanth, V.; Reddy, V.L.N.; Ramesh, P.; Rao, T.P.; Diwan, P.V.; Khar, A.; Venkateswarlu, Y. An immunosuppressive tryptophan-derived alkaloid from Lepidagathis cristata. Phytochemistry 2001, 58, 1263–1266. [Google Scholar] [CrossRef]
- Usui, T.; Kondoh, M.; Cui, C.-B.; Mayumi, T.; Osada, H. Tryprostatin A, a specific and novel inhibitor of microtubule assembly. Biochem. J. 1998, 333, 543–548. [Google Scholar] [CrossRef]
- Wollinsky, B.; Ludwig, L.; Hamacher, A.; Yu, X.; Kassack, M.U.; Li, S.-M. Prenylation at the indole ring leads to a significant increase of cytotoxicity of tryptophan-containing cyclic dipeptides. Bioorg. Med. Chem. Lett. 2012, 22, 3866–3869. [Google Scholar] [CrossRef]
- Jain, H.D.; Zhang, C.; Zhou, S.; Zhou, H.; Ma, J.; Liu, X.; Liao, X.; Deveau, A.M.; Dieckhaus, C.M.; Johnson, M.A.; et al. Synthesis and structure–activity relationship studies on tryprostatin A, an inhibitor of breast cancer resistance protein. Bioorg. Med. Chem. 2008, 16, 4626–4651. [Google Scholar] [CrossRef][Green Version]
- Woehlecke, H.; Osada, H.; Herrmann, A.; Lage, H. Reversal of breast cancer resistance protein-mediated drug resistance by tryprostatin A. Int. J. Cancer 2003, 107, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.W.; Boebel, T.A.; Hartwig, J.F. Iridium-Catalyzed, Silyl-Directed Borylation of Nitrogen-Containing Heterocycles. J. Am. Chem. Soc. 2010, 132, 4068–4069. [Google Scholar] [CrossRef]
- Song, Z.; Antonchick, A.P. Iridium(iii)-catalyzed regioselective C7-sulfonamidation of indoles. Org. Biomol. Chem. 2016, 14, 4804–4808. [Google Scholar] [CrossRef] [PubMed]
- Leitch, J.A.; Bhonoah, Y.; Frost, C.G. Beyond C2 and C3: Transition-Metal-Catalyzed C–H Functionalization of Indole. ACS Catal. 2017, 7, 5618–5627. [Google Scholar] [CrossRef]
- Fukuda, T.; Maeda, R.; Iwao, M. Directed C-7 lithiation of 1-(2,2-diethylbutanoyl)indoles. Tetrahedron 1999, 55, 9151–9162. [Google Scholar] [CrossRef]
- Kona, C.N.; Nishii, Y.; Miura, M. Iridium-Catalyzed Direct C4- and C7-Selective Alkynylation of Indoles Using Sulfur-Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 9856–9860. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qiu, X.; Zhao, Y.; Mu, Y.; Shi, Z. Palladium-Catalyzed C–H Arylation of Indoles at the C7 Position. J. Am. Chem. Soc. 2016, 138, 495–498. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, P.; Zhao, Y.; Shi, Z. Regiocontrolled Direct C−H Arylation of Indoles at the C4 and C5 Positions. Angew. Chem. Int. Ed. 2017, 56, 3966–3971. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, C.; He, Y.; Tan, L.; Ma, D. Rhodium-Catalyzed Regioselective C7-Functionalization of N -Pivaloylindoles. Angew. Chem. Int. Ed. 2016, 55, 321–325. [Google Scholar] [CrossRef]
- Lu, X.; He, S.-J.; Cheng, W.-M.; Shi, J. Transition-metal-catalyzed C H functionalization for late-stage modification of peptides and proteins. Chin. Chem. Lett. 2018, 29, 1001–1008. [Google Scholar] [CrossRef]
- Shah, T.A.; De, P.B.; Pradhan, S.; Punniyamurthy, T. Transition-metal-catalyzed site-selective C7-functionalization of indoles: Advancement and future prospects. Chem. Commun. 2019, 55, 572–587. [Google Scholar] [CrossRef]
- Demopoulos, V.J.; Nicolaou, I. Electrophilic Substitution of Indole on the Benzene Moiety: A Synthesis of 5-Acyl- and 5-Aroylindoles. Synthesis 1998, 1998, 1519–1522. [Google Scholar] [CrossRef]
- Feng, Y.; Holte, D.; Zoller, J.; Umemiya, S.; Simke, L.R.; Baran, P.S. Total Synthesis of Verruculogen and Fumitremorgin A Enabled by Ligand-Controlled C–H Borylation. J. Am. Chem. Soc. 2015, 137, 10160–10163. [Google Scholar] [CrossRef]
- Bandari, C.; Scull, E.M.; Bavineni, T.; Nimmo, S.L.; Gardner, E.D.; Bensen, R.C.; Burgett, A.W.; Singh, S. FgaPT2, a biocatalytic tool for alkyl-diversification of indole natural products. MedChemComm 2019, 10, 1465–1475. [Google Scholar] [CrossRef]
- Johnson, B.P.; Scull, E.M.; Dimas, D.A.; Bavineni, T.; Bandari, C.; Batchev, A.L.; Gardner, E.D.; Nimmo, S.L.; Singh, S. Acceptor substrate determines donor specificity of an aromatic prenyltransferase: Expanding the biocatalytic potential of NphB. Appl. Microbiol. Biotechnol. 2020, 104, 4383–4395. [Google Scholar] [CrossRef]
- Bandari, C.; Scull, E.M.; Masterson, J.M.; Tran, R.H.Q.; Foster, S.B.; Nicholas, K.M.; Singh, S. Determination of Alkyl-Donor Promiscuity of Tyrosine-O -Prenyltransferase SirD from Leptosphaeria maculans. ChemBioChem 2017, 18, 2323–2327. [Google Scholar] [CrossRef] [PubMed]
- Scull, E.M.; Bandari, C.; Johnson, B.P.; Gardner, E.D.; Tonelli, M.; You, J.; Cichewicz, R.H.; Singh, S. Chemoenzymatic synthesis of daptomycin analogs active against daptomycin-resistant strains. Appl. Microbiol. Biotechnol. 2020, 104, 7853–7865. [Google Scholar] [CrossRef]
- Chen, R.; Gao, B.; Liu, X.; Ruan, F.; Zhang, Y.; Lou, Y.Z.J.; Feng, K.; Wunsch, C.; Li, S.-M.; Dai, J.; et al. Molecular insights into the enzyme promiscuity of an aromatic prenyltransferase. Nat. Chem. Biol. 2016, 13, 226–234. [Google Scholar] [CrossRef]
- Tarcz, S.; Xie, X.; Li, S.-M. Substrate and catalytic promiscuity of secondary metabolite enzymes: O-prenylation of hydroxyxanthones with different prenyl donors by a bisindolyl benzoquinone C- and N-prenyltransferase. RSC Adv. 2014, 4, 17986–17992. [Google Scholar] [CrossRef]
- Zou, H.; Zheng, X.; Li, S.-M. Substrate Promiscuity of the Cyclic Dipeptide Prenyltransferases fromAspergillus fumigatus. J. Nat. Prod. 2009, 72, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Elshahawi, S.I.; Cao, H.; Shaaban, K.A.; Ponomareva, L.V.; Subramanian, T.; Farman, M.L.; Spielmann, H.P.; Phillips, G.N., Jr.; Thorson, J.S.; Singh, S. Structure and specificity of a permissive bacterial C-prenyltransferase. Nat. Chem. Biol. 2017, 13, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.E. Mechanistic studies on the indole prenyltransferases. Nat. Prod. Rep. 2015, 32, 88–101. [Google Scholar] [CrossRef]
- Liebhold, M.; Li, S.-M. Regiospecific Benzylation of Tryptophan and Derivatives Catalyzed by a Fungal Dimethylallyl Transferase. Org. Lett. 2013, 15, 5834–5837. [Google Scholar] [CrossRef]
- Liebhold, M.; Xie, X.; Li, S.-M. Breaking Cyclic Dipeptide Prenyltransferase Regioselectivity by Unnatural Alkyl Donors. Org. Lett. 2013, 15, 3062–3065. [Google Scholar] [CrossRef] [PubMed]
- Liebhold, M.; Xie, X.; Li, S.M. Expansion of enzymatic Friedel-Crafts alkylation on indoles: Acceptance of unnatural beta-unsaturated allyl diphospates by dimethylallyl-tryptophan synthases. Org. Lett. 2012, 14, 4882–4885. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Liang, S.; Wu, X.-L.; Xiong, J.; Zong, M.-H.; Lou, W.-Y. Metal-organic frameworks as novel matrices for efficient enzyme immobilization: An update review. Coord. Chem. Rev. 2020, 406, 213149. [Google Scholar] [CrossRef]
- Bhushan, I.; Parshad, R.; Qazi, G.N.; Gupta, V.K. Immobilization of Lipase by Entrapment in Ca-alginate Beads. J. Bioact. Compat. Polym. 2008, 23, 552–562. [Google Scholar] [CrossRef]
- Kim, J.; Grate, J.W.; Wang, P. Nanostructures for enzyme stabilization. Chem. Eng. Sci. 2006, 61, 1017–1026. [Google Scholar] [CrossRef]
- Kumar, K.; Wang, P.; Sanchez, R.; Swartz, E.A.; Stewart, A.F.; DeVita, R.J. Development of Kinase-Selective, Harmine-Based DYRK1A Inhibitors that Induce Pancreatic Human β-Cell Proliferation. J. Med. Chem. 2018, 61, 7687–7699. [Google Scholar] [CrossRef]
- Tanaka, S.; Shiomi, S.; Ishikawa, H. Bioinspired Indole Prenylation Reactions in Water. J. Nat. Prod. 2017, 80, 2371–2378. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | K562 GI50 (µM) |
|---|---|
| TPS-A | 97 ± 21 |
| TPS-B | 78 ± 31 |
| TPS-2 | 54 ± 15 |
| TPS-24 | 50 ± 29 |
| TPS-25 | 56 ± 19 |
| TPS-46 | 37 ± 8.8 |
| TPS-52 | 77 ± 22 |
| TPS-58 | 44 ± 18 |
| TPS-61 | 89 ± 29 |
| TPS-62 | 100 ± 33 |
| TPS-65 | 53 ± 19 |
| Taxol | 0.0058 ± 0.0039 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardner, E.D.; Dimas, D.A.; Finneran, M.C.; Brown, S.M.; Burgett, A.W.; Singh, S. Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT. Catalysts 2020, 10, 1247. https://doi.org/10.3390/catal10111247
Gardner ED, Dimas DA, Finneran MC, Brown SM, Burgett AW, Singh S. Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT. Catalysts. 2020; 10(11):1247. https://doi.org/10.3390/catal10111247
Chicago/Turabian StyleGardner, Eric D., Dustin A. Dimas, Matthew C. Finneran, Sara M. Brown, Anthony W. Burgett, and Shanteri Singh. 2020. "Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT" Catalysts 10, no. 11: 1247. https://doi.org/10.3390/catal10111247
APA StyleGardner, E. D., Dimas, D. A., Finneran, M. C., Brown, S. M., Burgett, A. W., & Singh, S. (2020). Indole C6 Functionalization of Tryprostatin B Using Prenyltransferase CdpNPT. Catalysts, 10(11), 1247. https://doi.org/10.3390/catal10111247