Abstract
Bicontinuous microemulsions (BCME) were used to synthesize hierarchical superstructures (HSs) of Pt-Co3O4 by reduction/precipitation. BCMEs possess water and oil nanochannels, and therefore, both hydrophilic and lipophilic precursors can be used. Thus, PtAq-CoAq, PtAq-CoOi, PtOi-CoAq and PtOi-CoOi were prepared (where Aq and Oi stand for the precursor present in aqueous or oily phase, respectively). The characterization of the Pt-Co3O4-HS confirmed the formation of metallic Pt and Co3O4 whose composition and morphology are controlled by the initial pH and precursor combination, determining the presence of the reducing/precipitant species in the reaction media. The electrocatalytic activity of the Pt-Co3O4-HSs for oxygen evolution reaction (OER) was investigated using linear sweep voltammetry in 0.1 M KOH and compared with Pt-HS. The lowest onset overpotentials for Pt-Co3O4-Hs were achieved with PtOi-CoOi (1.46 V vs. RHE), while the lowest overpotential at a current density of 10 mA cm−2 (η10) was obtained for the PtAq-CoAq (381 mV). Tafel slopes were 102, 89, 157 and 92 mV dec−1, for PtAq-CoAq, PtAq-CoOi, PtOi-CoAq and PtOi-CoOi, respectively. The Pt-Co3O4-HSs showed a better performance than Pt-HS. Our work shows that the properties and performance of metal–metal oxide HSs obtained in BCMEs depend on the phases in which the precursors are present.
1. Introduction
Electrocatalytic reactions involving HER (hydrogen evolution reaction) and OER (oxygen evolution reaction) are required in the production of hydrogen and oxygen by water electrolysis. These reactions, relevant to renewable energy technologies, require the lowering of onset potential and increasing the reaction kinetics to make the energy balance efficient [1,2,3].
2H2O + 2e− → 2OH− + H2 Hydrogen evolution reaction
4OH− → 2H2O + 4e− + O2 Oxygen evolution reaction
Generally, anodic OER is characterized by a high onset potential, and it exhibits slow kinetics because the reaction requires four-electron transfer and oxygen-oxygen bond formation [4]. These characteristics hamper large-scale water splitting technologies. Therefore, to overcome large onset potential and sluggish anodic OER, the use of an appropriate catalyst is important. RuO2 and IrO2 are widely used as OER catalysts in achieving water electrolysis. However, they are costly, scarce and have unstable catalytic performance in alkaline media [5,6]. These factors have prompted various research efforts geared towards exploring readily available, highly active, cost-effective and efficient catalysts that can lower the onset potential, have good stability and increase the reaction kinetics [7,8,9]. Studies have shown that Co3O4-based nanomaterials are affordable, abundant in nature, resistant to corrosion and possess enhanced catalytic performance towards OER in alkaline media [3,10,11].
Spectroscopic methods such as ex situ electron paramagnetic resonance spectroscopy [12] and in situ Raman spectroscopy have established the formation of Co4+ species in Co3O4-based nanomaterials as the active centers for OER. The EPR spectroscopic studies of Cobalt-phosphate catalyst evidenced the generation and population rise in active Co4+ species during water oxidation. An increase from 3 to 7% in Co4+ population improved the water oxidation by more than 2 orders of magnitude, with a change in the deposition potential from 1.14 to 1.34 V vs. NHE [13]. The in situ Raman spectra of a CoOx monolayers deposited on gold acquired during linear sweep voltammetry from 0 to 1.0 V vs. Hg|HgO in 0.1 M KOH, revealed the oxidation of the Co2+ and Co3+ species to Co4+ by increasing the potential [14]. Therefore, a strategy to increase the population of Co4+ centers on the surface of cobalt oxide will increase its performance as an OER catalyst. The introduction of highly electronegative transition metals by electrochemical or chemical methods, as support in a metal–metal oxide composites has been successful in achieving this.
In recent literature, metal–cobalt oxide nanomaterials have shown better OER performances than either noble metal or Co3O4 nanomaterials alone [10,15,16]. During OER, the presence of noble metals or highly electronegative metal ions promotes the oxidation of Co3+ to Co4+ through electron-withdrawing inductive effect [10,14,17]. The galvanostatic deposition of cobalt oxide on Au support by Yeo and Bell. 2011 resulted in improved OER performances. The turnover frequencies at an overpotential of 351 mV for 75 µC of cobalt oxide deposited on Au, is 1.24 s−1 compared to 0.0071 s−1 for Au without cobalt oxide deposit [14]. The successful synthesis of Au@Co3O4 nanocrystal (NC) catalysts using hydrothermal method has been reported. The draining of electrons from Co3O4 by Au and the stronger binding to oxygen by Co3O4 shell of Au@Co3O4 nanocrystals increases the Co4+ population, resulting in enhanced OER activity. At a fixed potential of 1.58 V vs. RHE, Au@Co3O4 exhibited a current density of 2.84 mA cm−2, which is 7 times as high as Co3O4 having 0.42 mA cm−2 and 55 times that of Au (0.05 mA cm−2) [17]. Qu et al., 2017 demonstrated the ability of Pd-Co3O4 nanostructures to generate high amounts of Co4+ cations. Superior electrochemical performance deemed by the onset potential and Tafel slope for Pd-Co3O4 (1.388 V vs. RHE and 60.7 mV dec−1) in comparison with Co3O4/C (1.481 V vs. RHE and 96.1 mV dec−1) were reported. This was attributed to the synergistic effect between Pd and Co3O4 and the high surface area of the 3D ordered mesoporous structure, which enhanced the charge transfer between the solid/electrolyte interface. High OER activity has been reported for Au-Co3O4/C electrocatalyst compared to Co3O4 nanomaterial. A value of 1.507 V vs. RHE for the onset potential of Au-Co3O4/C was recorded while Au/C and Co3O4 had values of 1.568 V vs. RHE and 1.518 V vs. RHE, respectively. This is suggested, once again, to be due to the charge transfer from the cobalt oxide to electronegative Au, resulting in an increase in the formation of Co4+ cations. The Au nanoparticles with an average diameter of 7 nm were well dispersed in Co3O4 [10]. Recently, the synthesis of noble metal particles (Pt, Pd, Au) embedded within mesoporous Co3O4 resulted in materials exhibiting superior activities and excellent stability in alkaline medium when compared with either the metal catalysts/C (Pt, Pd, Au) or Co3O4/C [16]. An onset potential of 1.481 V vs. RHE was recorded on Co3O4/C electrocatalyst, while 1.582 V, 1.538 V and 1.618 V vs. RHE for Pt/C, Pd/C and Au/C electrodes were reported, respectively. The metal–cobalt oxide electrocatalysts of Pt-Co3O4, Pd-Co3O4 and Au-Co3O4 had onset potentials of 1.383V, 1.388 V and 1.395V vs. RHE, respectively. The OER kinetic measurements showed Tafel slopes of 57.55, 60.70 and 68.01 mV dec−1 for Pt-Co3O4, Pd-Co3O4 and Au-Co3O4 electrocatalyst, respectively. This showed that the introduction of metals into cobalt oxides promotes higher OER activity than that observed on either the metal or cobalt oxide nanomaterial alone.
Furthermore, it has been shown that oxygen vacancies in Co3O4-based nanomaterials would result in a higher Co2+/Co3+ ratio, but conversely provide more active sites and lead to improved performance for OER. Xiao et al., 2020 studied the real active sites during OER process using operando characterization techniques such as EIS (electrochemical impedance spectroscopy), CV (cyclic voltammetry), XPS (X-ray photoelectron spectroscopy) and showed that oxygen vacancies promote the pre-oxidation of low-valence Co (Co2+) as well as the formation of cobalt oxyhydroxide (CoIII-OOH) intermediate species for OER. The authors also showed that a high ratio of Co2+/Co3+ plays a role in the rapid OH ion adsorption and deprotonation of CoOOH [18].
A wide range of noble metal–cobalt oxide nanomaterials used as OER electrocatalysts have been synthesized by the chemical reduction method [6,19], nanocasting using highly ordered mesoporous SiO2 as a hard template [4,16,20,21,22,23], and the hydrothermal method [24]. Likewise, the synthesis of metal nanoparticles (mono and bimetallic), as well as porous materials, could be obtained in one-pot procedures by using microemulsions as confined reaction media and template [25,26,27].
Recently, we reported for the first time the synthesis of hierarchical metallic superstructures using bicontinuous microemulsions (BCME) [28]. A nano-meso-macrostructure of Pt electrocatalyst with a nanocoral morphology was obtained from a soft and facile synthesis approach. Since BCMEs comprise both water and oil interconnected nanochannels, it was possible to obtain platinum hierarchical superstructures (Pt-HSs) using both water-soluble and oil-soluble precursors; it was observed that the resultant morphology depends on the type of Pt precursor. The resultant materials showed an efficient and stable electrocatalytic performance for hydrogen evolution reaction. In the present work, we extend this strategy for the synthesis of a hybrid metal–metal oxide HSs material. To the best of our knowledge, synthesis of Pt-Co3O4 superstructures by bicontinuous microemulsion (BCME) has not been previously investigated. The synthesis of Pt-Co3O4 and Pt hierarchical superstructures with various characteristics and performance is achieved by choosing different combinations of Co and Pt metallic precursors, based on their solubility, and employing bicontinuous microemulsion as a soft template, for achieving unique hierarchical morphologies.
2. Results and Discussion
A one-step chemical reduction/precipitation process was used to synthesize the Pt-Co3O4-HSs. The structures of the nanomaterials obtained reflect the confinement imposed by the bicontinuous microemulsion which acted as an interconnected nanocage network. The SEM images reveal that the Pt-Co3O4-HSs, as well as the reference Pt-HS solid, are made up of hierarchical nanostructures with a unique tailored arrangement. Figure 1A,B show the morphology of the Pt-HS. An interconnected-needle-like structure resembling a nanocoral is observed for Pt-HS. The SEM images for PtAq-CoAq revealed a morphological arrangement similar to that of Pt-HS (Figure 1C,D). This seems to follow from the fact that the precursors for PtAq-CoAq and Pt-HS are dissolved in the same medium in which the reducing agent NaBH4 was dissolved. Therefore, the resulting framework can be attributed to the soft template, morphology-directing nature of the water channels in the BCME and the presence of NaBH4 in the same aqueous phase. Similar observation has been reported by Zhuang et al., where the authors attributed the formation of hierarchical microstructure to the use of CTAB micelles and a strong reducing agent dissolved in the same aqueous solution as the metal precursors [6]. In the reacting systems containing either or both metallic precursors dissolved in the organic phase; (PtAq-CoOi, PtOi-CoAq, and PtOi-CoOi), the SEM images (Figure 1E–J), show a different morphology from that of Pt-HS. In the case of PtAq-CoOi, nanoplatelet-like morphology was observed, and PtOi-CoOi, exhibited a fiber-like morphology, which is attributed to the nanocage property of the oily channels in our BCME and the restricted access of reducing agent through the interface. Also, it is expected that the oil channels to be wider [28] than the water channels, since there is a larger fraction of the oil phase in the BCME, which permits the formation of different shapes when one or both precursors are present in the oil phase. Figure 2A,C present the STEM images of PtAq-CoAq and PtOi-CoAq. Agglomeration of interconnected needle-like nanomaterial was obtained for PtAq-CoAq, while PtOi-CoAq showed small nanoparticle agglomerates without a defined shape. Figure 2B,D show that PtAq-CoOi and PtOi-CoOi are made up of nanoplatelet-like and fiber-like nanomaterials respectively, which can also observed from SEM.


Figure 1.
SEM images of Pt-HS (A,B), Pt-Co3O4(s)-HSs: PtAq-CoAq (C,D), PtAq-CoOi (E,F), PtOi-CoAq (G,H), PtOi-CoOi (I,J).

Figure 2.
STEM images of Pt-Co3O4-HSs: PtAq-CoAq (A), PtAq-CoOi (B), PtOi-CoAq (C), PtOi-CoOi (D).
The Energy-dispersive X-ray spectroscopy (EDS) analysis showed the presence of Pt, Co, and O in all the samples (Figure S1). The ICP-AES analysis results in Table 1 reveal that Pt-Co3O4-HSs synthesized using Pt aqueous precursor yielded higher amounts of Pt than the nominal Pt/Co mole ratios. This observation suggests that a higher Pt reduction was achieved for the aqueous Pt precursor than when Pt organic precursor was used.

Table 1.
Nominal and experimental Pt/Co mole ratios; pH of BCME before and after NaBH4 addition.
In our synthesis, two scenarios should be considered: one in which both metallic precursors are present in the same nanophase and another in which they are individually dissolved in each liquid (water or isooctane). NaBH4 is added to the BCME as a freshly prepared solution in water, and it is expected to incorporate to the aqueous nanochannels initially. It is well known that NaBH4 hydrolysis is a pH-dependent reaction producing basic metaborate ions (which increase the solution pH) and hydrogen gas as products [29]. When NaBH4 is in acidic media, H2 is produced quickly. In contrast, at neutral or alkaline pH, NaBH4 is much more stable, leading to slow H2 release [30]. Therefore, the initial pH in the BCME greatly affects the presence of either the hydrolysis products or reducing BH4− ions as majoritarian species. On the other hand, the catalytic effect of several noble metal nanoparticles and their salts on the NaBH4 hydrolysis reaction has been widely reported, which increases H2 production [31,32,33].
On formation of H2 gas, microbubbles that evolve in situ in the water phase may reach the oil phase due to the low surface tension inside the BCME conferred by the nonionic surfactant [34]. These microbubbles are surrounded by a surfactant monolayer. The hydrocarbon tails orient towards the inside H2 bubble and EO chains towards the aqueous phase. It has been reported that EO chains are capable of preferentially solvate anions [35]; thus, we hypothesize that it is feasible that these H2 microbubbles may be dragging some reducing species (BH4−) adsorbed on its surface, similarly to ion flotation phenomena [36], see Figure S2. Furthermore, H2 itself is a slow reducing agent for platinum salts [37,38,39]. Thus, few nuclei of reduced metal in the oil phase are sufficient to act as seeds for superstructure growth when one of both reagents are present in this phase.
For the preparation of the PtAq-CoAq solid, the presence of H2PtCl6.6H2O as a water-soluble Pt precursor lowers the pH of the BCME prior to NaBH4 addition (pH = 1.57). Under this condition, NaBH4 hydrolysis is promoted; thus, H2 and OH− are produced, as evidenced by the increase of the pH after the recovery of the solids (i.e., 6.36). Both reducing species, as well as OH− ions, become available in the same nanophase, resulting in a one-step reduction (Pt)/precipitation (Co3O4) inside the water nanochannels. In this case, the relatively high standard reduction potentials of the platinum precursor (E0[PtCl6]2−/[PtCl4]2− = 0.68 V; E0[PtCl4]2−/Pt = 0.73 V) explain the higher Pt amount in the sample despite starting from a cobalt-rich mixture. In contrast, when using the Pt oil-soluble precursor (PtCOD) to obtain PtOi-CoAq, the microemulsion had an initial pH of 4.10, closer to BCME without any precursors. In this case, the presence of the reducing agent in the water nanochannels and the platinum precursor in the oil nanochannels suggests that the reduction reaction can only occur at the interface or by contact with the H2 microbubbles dragging BH4− through the oil channels. Consequently, a lower production of metallic Pt is observed, as revealed in the lower Pt/Co ratio compared to the PtAq-CoAq solid.
On the other hand, the use of cobalt 2-ethylhexanoate as Co oil-precursor to obtain either PtAq-CoOi or PtOi-CoOi increases the initial pH up to neutral values due to the presence of 2-ethylhexanoate species at the interface [40]. For the PtAq-CoOi solid, the H+ provided by the acidic platinum precursor neutralizes the unreacted hydroxyl species (produced from the NaBH4 hydrolysis) causing a lower final pH in this system. It is worth noticing that the pH of the BCME barely changes after the synthesis of Pt-HS (initial pH = 0.57; final pH = 0.69). In the case of PtOi-CoOi, the experimental Pt/Co ratio is the lowest of all the samples, since platinum is in the oil phase and the BCME showed the highest initial pH value (Table 1); thus, the production of H2 is lessened and the presence of reducing species passing through the oil phase is not favored.
Regarding the formation of Co species, metallic Co was not obtained. For all the compositions tested, the conditions were not appropriate for the reduction of Co2+ to metallic cobalt; instead, Co3O4 was formed. The formation of this oxide depends on the pH of the BCME; its formation is favored as the pH becomes more alkaline. As observed in Table 1, the final pH of the BCME for the PtAq-CoAq sample was 6.36, leading to a low Co3O4 content and the highest Pt/Co ratio obtained. The relatively low pH did not allow the formation of a large amount of Co3O4, leaving a considerable amount of precursor unreacted. In contrast, PtOi-CoOi, which started at pH = 7.34 and reached pH = 10.30 after NaBH4 addition, allowed the formation of a larger amount of Co3O4. According to Carlo et al., 2015, Co particles were obtained with NaBH4. It seems that in this work the simultaneous presence of Pt and Co precursors implied a competition, in which Pt4+ is reduced to Pt0 and cobalt precipitates due to a consequent increase in pH as shown in Table 1. Actually, Co3O4 is the product obtained when NaOH or Oxalic acid are used [25]. Overall, we observed a significant dependence of initial and final pH values on the combination of precursors used.
Figure 3 shows the X-ray diffraction pattern of the PtAq-CoAq-HS compared to Pt-HS. Reference peaks for fcc Pt and cubic Co3O4 are included. Figure 3a shows the diffraction peaks located at 40.01°, 46.39° and 67.69°, these are assigned to the (111), (200) and (220) planes, respectively and were indexed to face-centered cubic (fcc) Pt (JCPDS file 98-002-1963). In Figure 3b, the broad diffraction peak at Bragg angle of 40.38° corresponds to Pt (111) and a possible contribution from Co3O4 (222) reflection, which is expected in PtAq-CoAq. Likewise, the diffraction peak at around 47.17° is ascribed to fcc crystalline Pt (200), and it closely coincides with cubic crystalline Co3O4 (400), while the peak at 68.39° is ascribed to fcc crystalline Pt (220), coincident with cubic crystalline Co3O4 (440). [Co3O4 JCPDS file number is 04-022-7366 (space group Fd-3m)]. The Pt (111) peak is broadened and shifted to a higher angle in comparison to the reference Pt and Pt-HS peaks. Following the Debye-Scherrer equation, the crystallite size was estimated for PtAq-CoAq to be 3.21 ± 0.45 nm, while the size of Pt-HS was estimated to be 4.81 ± 0.24 nm. The Pt and Co3O4 peaks superimposition, as well as the decrease in the crystallite size could be responsible for the broadening of the XRD pattern of PtAq-CoAq compared to Pt-HS. However, a possible microstrain in the Pt lattice should not be ruled out, due to replacement of some Pt by Co atoms, which have a lower atomic radius (rCo = 1.52 Å vs. rPt = 1.77 Å) [41,42,43,44]. PtAq-CoAq exhibit a diffractogram coinciding to a greater extent with the reference Pt pattern, indicating that crystalline Pt is the main component in this sample, also in agreement with the ICP-AES results which revealed more Pt in the Pt-Co3O4-HSs synthesized with Pt aqueous soluble precursor. The diffractograms for the other Pt-Co3O4-HSs (PtAq-CoOi, PtOi-CoAq, and PtOi-CoOi) present broad, noisy, and undefined peaks (Figure S3).
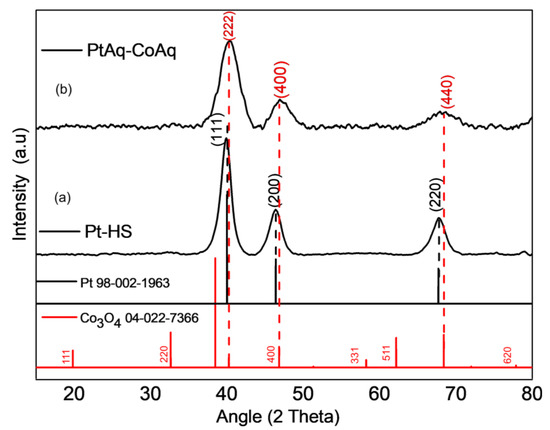
Figure 3.
The XRD patterns obtain for Pt-HS (a) and PtAq-CoAq (b).
Since only one of the Pt-Co3O4-HSs presented a defined XRD pattern (PtAq-CoAq), these samples were analyzed by Raman Spectroscopy in order to confirm the nature of the cobalt phase. Figure 4 shows the Raman spectra of Pt-Co3O4-HSs; all samples presented the characteristic spectra for Co3O4 with a cubic spinel structure. Five Raman bands were observed at about 186, 463, 504, 601, and 662–668 cm−1 for all the samples, which could be ascribed to the , , , , and symmetry of crystalline cubic Co3O4 phase [45,46]. In particular, the Raman band at 186 cm−1 can be assigned to symmetry in tetrahedral sites (CoO4), which can be attributed to vibration of Co2+-O2−; while the band at 662–668 cm−1 can be assigned to species in octahedral sites (CoO6), which can be attributed to vibration of Co3+-O2− [18].
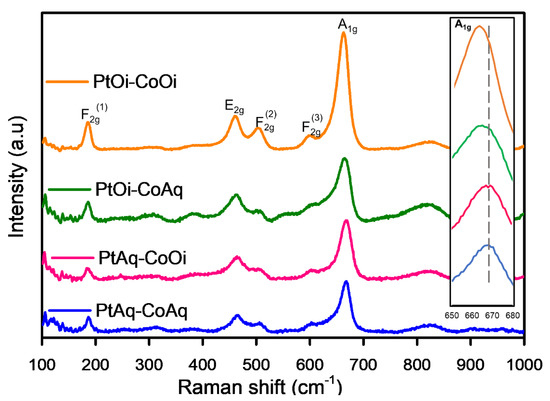
Figure 4.
Raman spectra of Pt-Co3O4-HSs. The inset shows the shift of band towards lower frequency.
Furthermore, as observed in the inset of Figure 4, the position of the band varied consistently from 668 to 662 cm−1 depending on the sample. This shift towards lower frequencies has been associated with the increase in oxygen vacancies [46,47], although other studies attribute this shift to the amorphous state of the samples [48]. In our case, XRD spectra of PtAq-CoOi, PtOi-CoAq and PtOi-CoOi indicate that the samples are rather amorphous (Figure S3), although given the reducing conditions of the reaction, oxygen vacancies cannot be ruled out.
The surface composition and chemical oxidation states of Pt-Co3O4-HSs and Pt-HS was determined by X-ray photoelectron spectroscopy. Figure 5A shows the XPS survey spectra which revealed the expected photoelectron peaks of Pt, C, O and Co for Pt-Co3O4-HSs, while for Pt-HS, as expected, the peak due to Co was not found. Boron or chloride species from the precursors were also disregarded. The highest intensity for the Pt 4f photoelectron peak was found in Pt-HS, while the intensity of the O 1s peak in this sample was much lower as compared to the Pt-Co3O4-HSs solids.
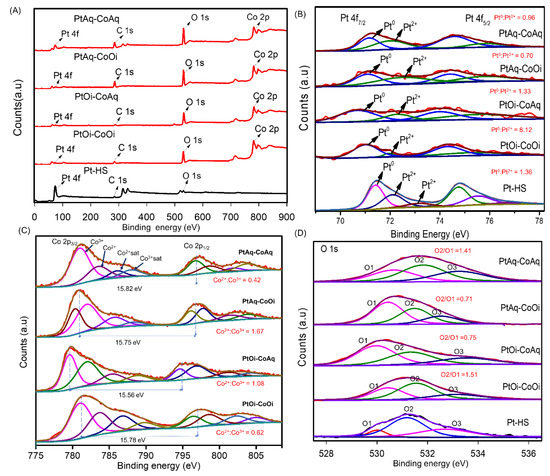
Figure 5.
(A) XPS survey spectrum of Pt-Co3O4-HSs and Pt-HS, high-resolution spectra of (B) Pt 4f, (C) Co 2p, and (D) O 1s.
The Pt 4f high-resolution spectra in Figure 5B show the corresponding spin–orbit coupling (4f7/2 and 4f5/2) centered around 71 and 74 eV, respectively. For the Pt-HS, the deconvolution of the 4f7/2 peak showed the contribution of zerovalent platinum (Pt0), as well as Pt2+ forming Pt-O and Pt-OH type-bonds (71.47, 72.39, and 73.17.4 eV) [49]. The binding energy for metallic Pt0 (71.47 eV) is slightly higher than the value expected for bulk Pt (71.2 eV) [50], as well as the values obtained in the Pt-Co3O4-HS(s) samples. This is explained to be as a result of small cluster-size effect [16,51]. Regarding the platinum species existing in the Pt-Co3O4-HSs nanomaterials, the deconvoluted 4f7/2 peak revealed only two contributions assigned to Pt0 and Pt2+-O centered at 71.16, 72.09 eV for PtAq-CoAq; 71.06, 72.39 eV for PtAq-CoOi; 70.68, 72.31 eV for PtOi-CoAq and 71.00, 71.95 eV for PtOi-CoOi. As observed, the binding energy for Pt0 species are lower in HS prepared using the oil soluble precursor.
On the other hand, the presence of oxidized platinum species, suggests that reduction is not fully completed under these conditions. In the case of the synthesis using chloroplatinic acid the reduction must be done from a Pt4+ oxidation state; while in the PtCOD precursor the Pt exists as Pt2+. The Pt0/Pt2+ ratio was dependent on the combination of precursors used; for Pt-HS, which was prepared using aqueous Pt precursor H2PtCl6 • 6H2O, the Pt0/Pt2+ ratio was 1.36; in comparison, for samples PtAq-CoAq and PtAq-CoOi, which were also prepared with the same precursor, the ratios were 0.96 and 0.7, respectively, indicating that the presence of Co precursor (and the formation of Co3O4) inhibited the reduction of Pt4+ to Pt0. On the contrary, for PtOi-CoAq and PtOi-CoOi that were synthesized using the oil-soluble platinum precursor, the Pt0/Pt2+ ratio is higher. In particular, the sample synthesized with both Pt and Co oil-soluble precursors, presented the highest Pt0/Pt2+ ratio with a value of 8.12. According to these results, it seems easier to reduce the oil-soluble Pt precursor to Pt0, despite the reducing species are less available in the oil phase. However, it should also be considered that in the case of synthesis using the platinum water-soluble precursor, the aqueous environment may favor stabilization of higher Pt oxidation state, in comparison with the oil environment.
The high-resolution spectra of Co 2p are shown in Figure 5C. The Co 2p3/2 and Co 2p1/2 doublet resulted from a contribution of four signals assigned to Co3+, Co2+ species and satellite peaks [11,52]. The full peaks list with the corresponding assignation is shown in Table S1. The spin–orbit splitting of 15.82 eV for PtAq-CoAq; 15.75 eV for PtAq-CoOi; 15.56 eV for PtOi-CoAq; 15.78 eV for PtOi-CoOi are characteristic for Co3O4. The presence of CoO and Co2O3 can be disregarded as splitting values for these oxides are 16.00 and 15.00 eV, respectively [10]. Likewise, the binding energy at 778.00 eV, which indicates the presence of metallic Co0, was not present in the XPS spectra of the synthesized nanomaterials [4,53]. Lower Co2+/Co3+ ratio obtained in the case of the nanostructures prepared with both precursors dissolved in the same phase (Figure 5C) suggests that in these materials the oxidation of Co3+ to Co4+ through an electron-withdrawing inductive effect by Pt should be more favored.
In Figure 5D, each O 1s spectrum can be deconvoluted into three peaks. The peaks labeled as O1 and O2 with binding energies at 530.79 and 531.63 eV for PtAq-CoAq; 530.43 and 531.49 eV for PtAq-CoOi; 529.96 and 531.32 eV for PtOi-CoAq; 530.44 and 531.54 eV for PtOi-CoOi, are attributed to the lattice oxygen in Co-O of Co3O4 and defect sites with low oxygen coordination oxygen-vacancy, respectively. For Pt-HS, these peaks at 530 and 531.19 eV are attributed to the mentioned Pt-O and Pt-OH interaction. The peak labelled as O3 appears at 532.65 eV for PtAq-CoAq, 532.52 eV for PtAq-CoOi, 532.72 eV for Pt-HS is due to the oxygen atoms in -CO bonds, probably arising from the surface contamination with adventitious carbon. However, the contribution of -OH species should not be ruled out [54]. In the case of the peaks at 533.53 eV in PtOi-CoAq and 533.11 eV in PtOi-CoOi solids, it could be ascribed to adsorbed molecular water [6,19,55,56]. A higher O2:O1 ratio, as estimated from the ratio between the area of the peaks, may indicate a larger amount of surface oxygen vacancies [6,18,57,58]. The higher O2:O1 ratio were achieved in those samples prepared incorporating the precursors in the same phase: for PtOi-CoOi this value is 1.51 in agreement with Raman results (higher shift in the band), while for PtAq-CoAq this value is 1.41. This result also correlates with the lower Co2+/Co3+ ratio found in these solids, and it is independent of the Co3O4 content, as the Pt/Co ratio is very different in both samples. The surface oxygen vacancies are useful in OER, and thus, these results would surely reflect in the different performance of the materials prepared. Therefore, having both precursors in the same phase competing for the reducing/precipitating species leads to higher surface defects in Co3O4 and a higher Co3+ population.
In this report, the water splitting capability of Pt-Co3O4-HSs and Pt-HS electrocatalysts was evaluated by investigating the OER activity through linear sweep voltammetry (LSV) in 0.1 M KOH with a sweep rate of 1 mV/s. The total catalysts loading of Pt-Co3O4-HSs and Pt-HS of 200 µg was maintained and drop-casted onto a glassy carbon electrode. As shown in Figure 6A, Pt-HS and Pt-Co3O4-HSs showed different OER performances. In Figure 6A inset, the LSV curve of the Pt-Co3O4-HSs shows anodic peaks at 1.15 V, 1.32 V, 1.19 V and 1.16 V vs. RHE for PtAq-CoAq, PtAq-CoOi, PtOi-CoAq and PtOi-CoOi, respectively, which correspond to Co2+ oxidation to Co3+ (Co2+/Co3+) [6,17,59,60]. This anodic peak precedes the onset of the OER and the sudden increase in the current density. However, Pt-HS did not show a distinct anodic peak for Pt oxidation, this process might be occurring in the same range at which OER happens on the surface of Pt-HS catalyst. The overpotential corresponding to a current density of 10 mA cm−2 (η10) is of high relevance and a primary figure of merit for catalytic activity of oxygen evolution. It is a measure of the current density expected for an integrated solar water-splitting device operating at 10% solar-to-fuels efficiency. This is a common and widely used criterion for assessing OER activities [61,62]. Table 2 shows the overpotential corresponding to the current density at 10 mA cm−2 as well as the onset of oxygen evolution and Tafel slopes for Pt-Co3O4-HSs and Pt-HS. η10 for Pt-HS was calculated by extrapolation of the polarization curve.
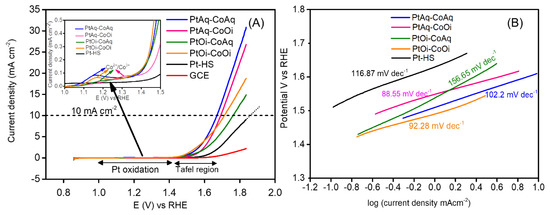
Figure 6.
(A): LSV curves for OER activity of PtAq-CoAq, PtAq-CoOi, PtOi-CoAq, PtOi-CoOi and Pt-HS in 0.1 M KOH at a scan rate of 1 mV/s. (B) Tafel plots for PtAq-CoAq, PtAq-CoOi, PtOi-CoAq, PtOi-CoOi and Pt-HS.
Table 2.
The overpotential at 10 mA cm−2, onset of oxygen evolution and Tafel slopes for the electrocatalysts.
The overpotential was calculated by subtracting water oxidation potential (1.229 V vs. RHE) from the applied potential [63]. In comparison, the Pt-Co3O4-HSs electrocatalysts synthesized in this work showed that PtAq-CoAq had η10 of 381 mV, while PtOi-CoAq had the highest η10 of 513 mV. PtAq-CoOi and PtOi-CoOi had overpotential value of 406 mV and 411 mV respectively. Pt-HS in comparison to the Pt-Co3O4-HSs, exhibited the highest overpotential of 627 mV to achieve a current density of 10 mA cm−2. A good OER electrocatalyst is expected to produce a current density of 10 mA cm−2 at low overpotential. The high surface population of Co3+ species and the interconnected hierarchical structure in PtAq-CoAq could be favoring the Co3+ to Co4+ oxidation driven by the electron-withdrawing inductive effect of Pt, favoring the OER [14]. The potential at which oxygen evolution commences is called the onset potential and it varies between the electrocatalysts. According to the values in Table 2, PtOi-CoOi exhibited the lowest onset potential value of 1.46 V, while Pt-HS showed the onset at 1.56 V. On the other hand, the Tafel slope provides information associated with the rate determining steps and hence, it allows to compare the kinetics of the electrocatalytic reactions occurring on the surface of each material. The potential range at which the Tafel plot was calculated is labeled as the Tafel region in Figure 6A, while The Tafel slope of Pt-Co3O4-HSs and Pt-HS are shown in Figure 6B, these values are also listed in Table 2. PtAq-CoOi exhibited the lowest Tafel slope (89 mV dec−1), which is synonymous with the catalyst having a faster kinetics to evolve oxygen from water as compared to Pt-HS which had a Tafel slope of 117 mV dec−1. Tafel slopes near 120 mV dec−1 are observed when the surface species formed just before the rate-determining step are predominant; under alkaline conditions and assuming a single site mechanism it corresponds to the following equilibrium: M + OH− ↔ MOH + e−. However, when the surface adsorbed species produced in the early stage of the OER remains predominant the Tafel slope decreases [64]. Additionally, the presence and high ratio of Co2+/Co3+ ions in PtAq-CoOi indicate an increase in the pre-oxidation of Co2+, which enhances O2-related electrocatalysis [18,56]. These factors could account for the lower Tafel slope of PtAq-CoOi as compared to Pt-HS.
However, in PtAq-CoAq the lowest Co2+/Co3+ ratio was estimated which suggests a smaller amount of Co2+ (Figure 5C) is available to be effectively bounded by surface oxygen vacancies and oxidized to a higher valence [65]. This is suggested to be responsible for the higher Tafel slope of PtAq-CoAq as compared to PtAq-CoOi. Based on the morphological differences between interconnected needle-like PtAq-CoAq and nanoplatelet-like PtAq-CoOi, it is worthy to note that the electrocatalytic species in the nanoplatelet PtAq-CoOi (Figure 1E,F and Figure 2B) are sufficiently exposed to the electrolyte, thereby improving its surface reaction rate.
On the other hand, the atomic ratio of Co2+/Co3+ and O2/O1 for PtOi-CoOi is 0.62 and 1.51, with Tafel slopes of 92 mV dec−1; whereas PtOi-CoAq had a Co2+/Co3+ ratio of 1.08 and O2/O1 of 0.75, with a Tafel slope of 157 mV dec−1. In these two HSs, we identified that their morphology and arrangement (Figure 1G–J and Figure 2C,D), higher Co/Pt ratio (Table 1 ICP values), high surface oxygen vacancies (Raman) as well as atomic Co2+/Co3+ ratio from XPS, had significant effect on the electrocatalytic activity. These favored the early OER onset potential, comparable overpotential for 10 mA cm−2 (Table 2) and lower Tafel slope for PtOi-CoOi when compared with PtOi-CoAq (Figure 6B). The fibrous morphological structure of PtOi-CoOi permits rapid electron transfer through the OER catalytic active sites [6,63].
From the LSV plots for the OER process, (Figure 6A,B) Pt-HS had the highest onset potential value, a Tafel slope higher than all the Pt-Co3O4(s) evaluated except PtOi-CoAq, whereas the potential corresponding to a current density of 10 mA cm−2 for Pt-HS was obtained by extrapolation. This shows that the catalytic performance of Pt-HS is significantly low without the presence of the mixed metal oxide. This highlights that the formation of interconnected structures in which Pt atom and mixed valence Co3O4 exist together enhances the performance of nanomaterials as electrocatalyst.
In general, the presence of Co3O4 in the nanomaterial using either oil soluble or aqueous soluble cobalt precursor showed an enhancement in the oxygen evolution reaction electrocatalytic activity of platinum. Therefore, for practical applications, the use of PtAq-CoAq is preferred not only because it has a unique interconnected needle-like morphology but because the overpotential to achieve the primary figure of merit η10 is low, earlier onset potential and a comparable Tafel slope (Table 3). In addition, the use of bicontinuous microemulsion as confined reaction media resulted in a good control of the morphology, as well as platinum and cobalt oxide incorporation into the nanomaterial, which are key for electrocatalytic applications.
Table 3.
Comparison of the Pt-Co3O4-HSs and Pt-HS electrocatalysts synthesized in this work versus reported literature.
The catalytic performance of the BCME synthesized Pt-Co3O4-HSs is comparable to reported literature for metal–metal oxides, and performed better than most metal oxides catalysts. This suggests that the Pt-Co3O4-HSs can serve as efficient electrocatalysts.
Chronoamperometry is a useful technique to evaluate the stability and durability of electrocatalysts [16]. Figure 7 shows the stability test of Pt-Co3O4-HSs and Pt-HS conducted at a potential of 1.50 V vs. RHE in 0.1 M KOH using chronoamperometry method for 4000 s. This potential was used because it is close to onset of OER as observed from the LSV (Figure 6A). The reaction is performed at constant potential with oxygen bubbles steadily generated on the surface of the electrocatalyst. The generated bubbles attach to the surface of the electrode, which resulted in decreased current density. The steady oxygen bubbles grow bigger with time and leave the catalyst surface. This increases current density immediately after the bubbles leave the active sites (Figure 7 inset). This process repeats throughout time under which chronoamperometry is performed. This gives a fluctuating current density-time curve. The decrease in the current density was 25%, 10.7%, 2.8%, 11.6% for PtAq-CoAq, PtAq-CoOi, PtOi-CoAq, PtOi-CoOi and 20.5% for Pt-HS after a long time of polarization. However, this change shows that the electrocatalyst are stable and can be used for long duration without substantial changes.
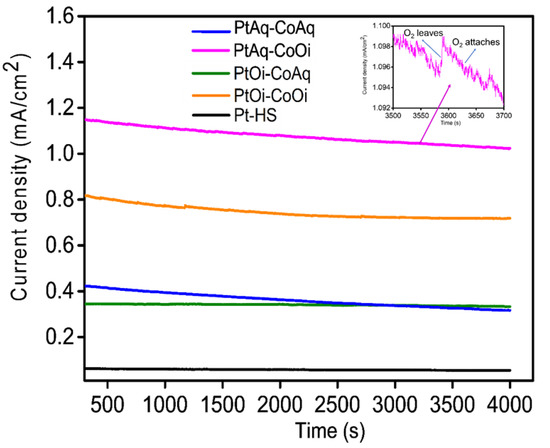
Figure 7.
The current density-time curves for PtAq-CoAq, PtAq-CoOi, PtOi-CoAq, PtOi-CoOi and Pt-HS in 0.1 M KOH at 1.50 vs. RHE.
The mechanism of the OER using noble metal–cobalt oxide as electrocatalyst has been investigated [4,6,16,49]. It is reported that in metal–cobalt oxide catalysts, highly electronegative metals such as Pt facilitate the oxidation of Co3+ to produce Co4+ cations. This is majorly the rate limiting step, which highlights the importance of the noble metal. The electronegative metal allows for the easier formation of Co4+, which is the active center for OER. Higher number of active centers on the electrode correspond to increase in the OER performance. Co4+ is more electrophilic, and will form intermediates by nucleophilic reactions with O species and OH− at the reaction interface [16,70].
Co3+(s) → Co4+(s) + e−
Co4+(s) + OH− → Co4+(OH)ad + e−
Co4+(OH) ad + OH− → Co4+(s) + 1/2O2 + H2O + e−
The OER activity of Pt-Co3O4-HSs is better when compared to Pt-HS because multielectron transfer steps involved are promoted by the synergistic effect of the electrophilic Pt, the oxidation of Co3+ to Co4+ cation and surface oxygen vacancies. Additionally, the hierarchical structure of these materials made the active sites accessible for a higher electrocatalytic activity.
3. Materials and Methods
3.1. Chemicals
Cobalt (II) 2-ethylhexanoate (65 wt.% in mineral spirits, CoC16H30O4, referred to as Co organic precursor), Cobalt (II) nitrate hexahydrate 99% (Co(NO3)2.6H2O, referred to as Co aqueous precursor), (1,5-cyclooctadiene) dimethylplatinum (II) 97% (C10H16Pt, referred to as Pt organic precursor), Chloroplatinic acid hexahydrate 37.68% (H2PtCl6.6H2O, referred to as Pt aqueous precursor), Isooctane 99% (C8H18), Sodium borohydride 99.99% (NaBH4), KOH 90%, Nafion 5 wt.%, were purchased from Sigma Aldrich (Darmstadt, Germany). SynperonicTM 91/5 (C19O6H40) was purchased from CRODA. Isopropyl alcohol 99.9% (C3H8O) was supplied by Fisher Scientific (Hampton, NH, USA). Sulphuric acid 93–98% H2SO4 was purchased from Fermont.
3.2. Synthesis of Pt-Co3O4-HSs and Pt-HS
BCME was prepared using 52% of Isooctane, 27.36% of SynperonicTM 91/5 and 20.64% of Milli-Q water. More details on this microemulsion system and its characterization can be found elsewhere [28]. Pt-Co3O4-HSs were produced by the reduction/precipitation of the aqueous or organic soluble precursors present in the BCME as follows:
- I.
- Pt and Co aqueous precursors both present in the aqueous phase is referred as PtAq-CoAq
- II.
- Pt and Co organic precursors both present in the organic phase is referred to as PtOi-CoOi
- III.
- Pt aqueous precursor and Co organic precursor is referred to as PtAq-CoOi
- IV.
- Pt organic precursor and Co aqueous precursor is referred to as PtOi-CoAq
The preparation of BCME was carried out in 20 mL glass vials. To prepare PtAq-CoAq, proper amounts of Pt and Co aqueous precursors were dissolved in Milli-Q water to give a final aqueous mixture containing 1 wt.% of Pt and 1 wt.% of Co. This was followed by the addition of SynperonicTM 91/5 and Isooctane to obtain a final composition of 20.64% aqueous phase, 27.36% surfactant, and 52% oil phase. The mixture was vortexed for few seconds, resulting in a transparent, fluid, optically isotropic liquid phase and kept at 27 °C. A 100 µL of NaBH4 solution with concentration equivalent to twice the total sum of Co and Pt moles, was prepared and added to the BCME within 3 s, and vortexed during 5 s. Then the reaction mixture was left to stand unstirred at 27 °C during 48 h prior to recovery and isolation of the nanomaterials. Detailed descriptions of the synthesis method for the other Pt-Co3O4-HSs are found in the Supplementary document. In brief, PtOi-CoOi, PtAq-CoOi and PtOi-CoAq are prepared by dissolving each precursor in the appropriate solvent, with the final mixture containing 1 wt.% of Pt and 1 wt.% of Co. The pH of BCMEs containing Pt and Co precursors before the addition of the NaBH4 and after the recovery of the solids were measured using a Thermo Scientific Orion Star A211 pH Benchtop Meter.
3.3. Recovery and Isolation of Pt-Co3O4-HSs and Pt-HS
Isopropanol was added to the BCME containing the Pt-Co3O4-HSs in order to break the microemulsion and separate the solid; this was followed by washings with isopropanol, Milli-Q water and finally Isopropanol. For Pt-HS, a solution containing HNO3: H2O at 50:50 wt. ratio was used as additional washing solvent. Milli-Q water was then used to wash Pt-HS, until a neutral pH was recorded. The Pt-Co3O4-HSs and Pt-HS were then dried at room temperature, recovered and used for further characterizations.
3.4. Characterization of Pt-Co3O4-HSs and Pt-HS
The structure and morphologies of the synthesized Pt-Co3O4-HSs and Pt-HS were analyzed by scanning electron microscopy (SEM) and Scanning transmission electron microscopy (STEM) with a Field Emission Model 200 Nova NanoSEM at working voltages of 15–18 kV and equipped with an energy dispersive X-ray detector. The crystalline structure was characterized using a X-ray diffractometer PANalytical Empyrean equipped with a CuKα cathode using a continuous scan mode from 10 to 80° (2θ) with 0.001 step size. The surface analysis was carried out using X-ray photoelectron spectroscopy (XPS) with a Thermo Fisher Scientific Escalab 250 Xi with an Al-Kα (1486.7 eV) X-ray source. Raman spectra were recorded with a Labram HR Evolution from Horiba, equipped with a 532 nm Nd: Yag laser (50 mW); the measurements were run with 10 s acquisition time, 50 accumulations. The metal content was measured using Inductively coupled plasma atomic emission spectroscopy (ICP-AES) with a Thermo Jarrell Ash iCAP 6000 equipment; appropriate sample treatment and dilution was carried out prior to ICP-AES measurement by open-beaker digestion with aqua regia.
3.5. Electrocatalytic Characteristics of Pt-Co3O4-HSs and Pt-HS
The electrocatalytic activity was evaluated using CH Instruments 440C Potentiostat/Galvanostat. The experiments were conducted at room temperature. A glassy carbon electrode (GCE) was used as the working electrode. It was polished using alumina powder suspensions of different particle sizes (1 µm, 0.3 µm and 0.1 µm). A three-electrode cell was assembled with a platinum mesh counter electrode and a Mercury/Mercurous oxide Reference Electrode (Hg|HgO, 20% KOH; 0.098 V vs. RHE). The oxygen evolution reaction was investigated by linear sweep voltammetry (LSV) in a range of 0 to 1.05 V vs. Hg|HgO in 0.1 M KOH at a scan rate of 1 mV/s.
The Pt-Co3O4-HSs and Pt-HS were dispersed in a 2:1 (Isopropanol: water) mixture, followed by a vortex mixing after ultrasonication. The concentration of the Pt-Co3O4-HSs and Pt-HS suspension prepared were 10 mg mL−1. 20 µL of the solution containing the HSs was drop-casted onto the GCE electrode with an exposed cross section diameter of 7.9 mm. The HSs were allowed to dry at room temperature, after which 5 µL of a 0.05 wt.% Nafion solution was added to fix the HSs to the surface of the GCE. The stability of the HSs was investigated by chronoamperometry using a potential step of 650 mV vs. Hg|HgO in 0.1 M KOH for 4000 s. IR correction (Ecorr = ERHE − iRu) was done on all the potentials, where Ecorr is the iRu-compensated potential, ERHE is the potential obtained from experimentally measured potential and Ru is the resistance of the electrolyte determined by the iR compensation function of the CH Instruments 440 C electrochemical workstation.
4. Conclusions
The synthesis of Pt-Co3O4-HSs and Pt-HS using bicontinuous microemulsion was successfully carried out by a facile, fast, reproducible soft chemical method under ambient temperature and without further treatments. The characteristics, properties and performance of the obtained materials were dependent on the type of Pt and Co precursor (hydrophilic or lipophilic) and their combination; the best material was the one synthesized using both Pt and Co hydrophilic precursors. Oxygen evolution reaction performances of Pt-Co3O4-HSs were shown to be significantly faster and higher than on Pt-HS. The overpotential required to achieve 10 mA cm−2 when PtAq-CoAq catalyst was used was 381 mV, compared to 627 mV obtained when Pt-HS was used as catalyst. The onset potential of oxygen evolution on Pt-HS at 1.56 V vs. RHE was much higher than all the Pt-Co3O4-HSs (Eonset of PtAq-CoAq, PtAq-CoOi, PtOi-CoAq and PtOi-CoOi were 70, 60, 80, and 100 mV, respectively, more negative than that of Pt-HS). Additionally, the Tafel slopes of Pt-Co3O4-HSs are lower and the kinetics are faster than Pt-HS. The improvement in OER activity is due to the synergistic effect of electronegative Pt on Co3O4, which allows the formation of Co4+ species. This current research provides an opportunity to explore bicontinuous microemulsion as a structure-directing template for metal–metal oxide catalyst that are efficient in water electrolysis.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/11/1311/s1, Figure S1: EDS images of Pt-Co3O4(s): PtAq-CoAq (A), PtAq-CoOi (B), PtOi-CoAq (C), PtOi-CoOi (D), Pt HS, Figure S2: Schematic representation of the H2 microbubbles evolved in situ at the water channels, surrounded by surfactant molecules and dragging reducing species to the oil channels, Figure S3: The XRD pattern of Pt-Co3O4-HSs: PtOi-CoOi (a), PtAq-CoOi (b), PtOi-CoAq (c), Table S1: Table showing the peaks derive from the deconvolution of the Co 2p3/2 peak. The signals were assigned to Co3+ and Co2+ species with the corresponding satellite signals (shake-up).
Author Contributions
E.T.A.: methodology, validation, formal analysis, investigation and writing original draft, writing-review & editing; E.G.-V.: methodology and investigation; K.M.F.: methodology, investigation, writing-review & editing; M.V.: conceptualization, resources, writing-review & editing, supervision; M.S.-D.: conceptualization, resources, writing-review & editing, supervision, project administration and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research and the APC were funded by CONACYT (Fronteras de la Ciencia) grant number FC 2016/1700.
Acknowledgments
E.T. Adesuji and K.M. Fuentes acknowledge financial support from CONACYT (Ph.D. CONACYT Grant and postdoctoral grant from FC 2016/1700, respectively). M. Videa thanks the School of Engineering and Sciences at Tecnológico de Monterrey for partial funding provided through the Research Chair of Photonics and Quantum Systems. The authors acknowledge Francisco E. Longoria, J. Alejandro Arizpe, Nayely Pineda and L. Gerardo Silva (CIMAV Monterrey) for their help with XRD, HRTEM-STEM and Raman, SEM, and XPS measurements, respectively. We also thank Victor A. Luna Flores and Miguel A. Esneider Alcala for their help in cutting the electrodes.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ozoemena, K.I. Nanostructured platinum-free electrocatalysts in alkaline direct alcohol fuel cells: Catalyst design, principles and applications. RSC Adv. 2016, 6, 89523–89550. [Google Scholar] [CrossRef]
- Lim, T.; Sung, M.; Kim, J. Oxygen evolution reaction at microporous pt layers: Differentiated electrochemical activity between acidic and basic media. Sci. Rep. 2017, 7, 15382. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.U.; Kim, B.J.; Chen, Z. One-pot synthesis of a mesoporous nico2o4 nanoplatelet and graphene hybrid and its oxygen reduction and evolution activities as an efficient bi-functional electrocatalyst. J. Mater. Chem. A 2013, 1, 4754–4762. [Google Scholar] [CrossRef]
- Qu, Q.; Zhang, J.H.; Wang, J.; Li, Q.Y.; Xu, C.W.; Lu, X. Three-dimensional ordered mesoporous co3o4 enhanced by pd for oxygen evolution reaction. Sci. Rep. 2017, 7, 41542. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Sheng, W.; Zhuang, Z.; Fang, Q.; Gu, S.; Jiang, J.; Yan, Y. Efficient water oxidation using nanostructured alpha-nickel-hydroxide as an electrocatalyst. J. Am. Chem. Soc. 2014, 136, 7077–7084. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Ge, L.; Yang, Y.; Li, M.; Jia, Y.; Yao, X.; Zhu, Z. Ultrathin iron-cobalt oxide nanosheets with abundant oxygen vacancies for the oxygen evolution reaction. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, C.W.; Zhang, L.; Lee, K.; Liu, H.; Marques, A.L.; Marques, E.P.; Wang, H.; Zhang, J. A review of fe–n/c and co–n/c catalysts for the oxygen reduction reaction. Electrochim. Acta 2008, 53, 4937–4951. [Google Scholar] [CrossRef]
- Wu, L.; Liu, Z.; Xu, M.; Zhang, J.; Yang, X.; Huang, Y.; Lin, J.; Sun, D.; Xu, L.; Tang, Y. Facile synthesis of ultrathin pd–pt alloy nanowires as highly active and durable catalysts for oxygen reduction reaction. Int. J. Hydrogen Energy 2016, 41, 6805–6813. [Google Scholar] [CrossRef]
- Yuan, N.; Jiang, Q.; Li, J.; Tang, J. A review on non-noble metal based electrocatalysis for the oxygen evolution reaction. Arab. J. Chem. 2020, 13, 4294–4309. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Ye, K.-H.; Zhong, Q.-S.; Zhang, C.-J.; Shi, S.-T.; Xu, C.-W. Au-co3o4/c as an efficient electrocatalyst for the oxygen evolution reaction. ChemPlusChem 2014, 79, 1569–1572. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, Q.; Xiao, Z.; Li, X.; Huo, J.; Wang, S.; Dai, L. Plasma-engraved co3 o4 nanosheets with oxygen vacancies and high surface area for the oxygen evolution reaction. Angew. Chem. Int. Ed. Engl. 2016, 55, 5277–5281. [Google Scholar] [CrossRef]
- Ceyssens, F.; Sree, S.P.; Martens, J.; Puers, R. Fabrication of nanostructured platinum with multilevel porosity for low impedance biomedical recording and stimulation electrodes. Procedia Eng. 2015, 120, 355–359. [Google Scholar] [CrossRef][Green Version]
- McAlpin, J.G.; Surendranath, Y.; Dinca, M.; Stich, T.A.; Stoian, S.A.; Casey, W.H.; Nocera, D.G.; Britt, R.D. Epr evidence for co (iv) species produced during water oxidation at neutral ph. J. Am. Chem. Soc. 2010, 132, 6882–6883. [Google Scholar] [CrossRef]
- Yeo, B.S.; Bell, A.T. Enhanced activity of gold-supported cobalt oxide for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 2011, 133, 5587–5593. [Google Scholar] [CrossRef]
- Hao, Y.; Xu, Y.; Liu, J.; Sun, X. Nickel–cobalt oxides supported on co/n decorated graphene as an excellent bifunctional oxygen catalyst. J. Mater. Chem. A 2017, 5, 5594–5600. [Google Scholar] [CrossRef]
- Qu, Q.; Pan, G.-L.; Lin, Y.-T.; Xu, C.-W. Boosting the electrocatalytic performance of pt, pd and au embedded within mesoporous cobalt oxide for oxygen evolution reaction. Int. J. Hydrogen Energy 2018, 43, 14252–14264. [Google Scholar] [CrossRef]
- Zhuang, Z.; Sheng, W.; Yan, Y. Synthesis of monodispere au@co3o4 core-shell nanocrystals and their enhanced catalytic activity for oxygen evolution reaction. Adv. Mater. 2014, 26, 3950–3955. [Google Scholar] [CrossRef]
- Xiao, Z.; Huang, Y.-C.; Dong, C.-L.; Xie, C.; Liu, Z.; Du, S.; Chen, W.; Yan, D.; Tao, L.; Shu, Z.; et al. Operando identification of the dynamic behavior of oxygen vacancy-rich co3o4 for oxygen evolution reaction. J. Am. Chem. Soc. 2020, 142, 12087–12095. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, L.; Min, Y.; Zhang, Y. Facile synthesis of noble metal (au,pt)/co3o4 nanocomposite microspheres and their catalytic activity. Mater. Chem. Phys. 2010, 124, 1166–1171. [Google Scholar] [CrossRef]
- Grewe, T.; Deng, X.; Tüysüz, H. Influence of fe doping on structure and water oxidation activity of nanocast co3o4. Chem. Mater. 2014, 26, 3162–3168. [Google Scholar] [CrossRef]
- Lu, X.; Ng, Y.H.; Zhao, C. Gold nanoparticles embedded within mesoporous cobalt oxide enhance electrochemical oxygen evolution. ChemSusChem 2014, 7, 82–86. [Google Scholar] [CrossRef]
- Rosen, J.; Hutchings, G.S.; Jiao, F. Ordered mesoporous cobalt oxide as highly efficient oxygen evolution catalyst. J. Am. Chem. Soc. 2013, 135, 4516–4521. [Google Scholar] [CrossRef]
- Deng, X.; Ozturk, S.; Weidenthaler, C.; Tuysuz, H. Iron-induced activation of ordered mesoporous nickel cobalt oxide electrocatalyst for the oxygen evolution reaction. ACS Appl. Mater. Interfaces 2017, 9, 21225–21233. [Google Scholar] [CrossRef]
- Zhan, T.; Lu, S.; Liu, X.; Teng, H.; Hou, W. Alginate derived co3o4/co nanoparticles decorated in n-doped porous carbon as an efficient bifunctional catalyst for oxygen evolution and reduction reactions. Electrochim. Acta 2018, 265, 681–689. [Google Scholar] [CrossRef]
- Carlo, G.D.; Lualdi, M.; Venezia, A.M.; Boutonnet, M.; Sanchez-Dominguez, M. Design of cobalt nanoparticles with tailored structural and morphological properties via o/w and w/o microemulsions and their deposition onto silica. Catalysts 2015, 5, 442–459. [Google Scholar] [CrossRef]
- König, R.Y.; Schwarze, M.; Schomäcker, R.; Stubenrauch, C. Catalytic activity of mono-and bi-metallic nanoparticles synthesized via microemulsions. Catalysts 2014, 4, 256–275. [Google Scholar] [CrossRef]
- Serrà, A.; Vallés, E. Microemulsion-based one-step electrochemical fabrication of mesoporous catalysts. Catalysts 2018, 8, 395. [Google Scholar] [CrossRef]
- Adesuji, E.T.; Khalil-Cruz, L.E.; Videa, M.; Sánchez-Domínguez, M. From nano to macro: Hierarchical platinum superstructures synthesized using bicontinuous microemulsion for hydrogen evolution reaction. Electrochim. Acta 2020, 354, 136608. [Google Scholar] [CrossRef]
- Schlesinger, H.I.; Brown, H.C.; Finholt, A.E.; Gilbreath, J.R.; Hoekstra, H.R.; Hyde, E.K. Sodium borohydride, its hydrolysis and its use as a reducing agent and in the generation of hydrogen1. J. Am. Chem. Soc. 1953, 75, 215–219. [Google Scholar] [CrossRef]
- Liu, B.H.; Li, Z.P. A review: Hydrogen generation from borohydride hydrolysis reaction. J. Power Sources 2009, 187, 527–534. [Google Scholar] [CrossRef]
- Brack, P.; Dann, S.E.; Wijayantha, K.G.U. Heterogeneous and homogenous catalysts for hydrogen generation by hydrolysis of aqueous sodium borohydride (nabh4) solutions. Energy Sci. Eng. 2015, 3, 174–188. [Google Scholar] [CrossRef]
- Demirci, U.B.; Miele, P. Reaction mechanisms of the hydrolysis of sodium borohydride: A discussion focusing on cobalt-based catalysts. Comptes Rendus Chim. 2014, 17, 707–716. [Google Scholar] [CrossRef]
- Kaufman, C.M.; Sen, B. Hydrogen generation by hydrolysis of sodium tetrahydroborate: Effects of acids and transition metals and their salts. J. Chem. Soc. Dalton Trans. 1985, 307–313. [Google Scholar] [CrossRef]
- Folgueiras-Amador, A.A.; Jolley, K.E.; Birkin, P.R.; Brown, R.C.D.; Pletcher, D.; Pickering, S.; Sharabi, M.; de Frutos, O.; Mateos, C.; Rincón, J.A. The influence of non-ionic surfactants on electrosynthesis in extended channel, narrow gap electrolysis cells. Electrochem. Commun. 2019, 100, 6–10. [Google Scholar] [CrossRef]
- Ohki, T.; Harada, M.; Okada, T. Solvation of ions in hydrophilic layer of polyoxyethylated nonionic micelle. Cooperative approach by electrophoresis and ion-transfer voltammetry. J. Phys. Chem. B 2006, 110, 15486–15492. [Google Scholar] [CrossRef]
- Chang, L.; Cao, Y.; Fan, G.; Li, C.; Peng, W. A review of the applications of ion floatation: Wastewater treatment, mineral beneficiation and hydrometallurgy. RSC Adv. 2019, 9, 20226–20239. [Google Scholar] [CrossRef]
- Boutonnet, M.; Kizling, J.; Stenius, P.; Maire, G. The preparation of monodisperse colloidal metal particles from microemulsions. Colloids Surf. 1982, 5, 209–225. [Google Scholar] [CrossRef]
- Henglein, A.; Giersig, M. Reduction of pt(ii) by h2: Effects of citrate and naoh and reaction mechanism. J. Phys. Chem. B 2000, 104, 6767–6772. [Google Scholar] [CrossRef]
- Zeng, J. A simple eco-friendly solution phase reduction method for the synthesis of polyhedra platinum nanoparticles with high catalytic activity for methanol electrooxidation. J. Mater. Chem. 2012, 22, 3170–3176. [Google Scholar] [CrossRef]
- Sanchez-Dominguez, M.; Pemartin, K.; Boutonnet, M. Preparation of inorganic nanoparticles in oil-in-water microemulsions: A soft and versatile approach. Curr. Opin. Colloid Interface Sci. 2012, 17, 297–305. [Google Scholar] [CrossRef]
- Huang, H.; Hu, X.; Zhang, J.; Su, N.; Cheng, J. Facile fabrication of platinum-cobalt alloy nanoparticles with enhanced electrocatalytic activity for a methanol oxidation reaction. Sci. Rep. 2017, 7, 45555. [Google Scholar] [CrossRef]
- Chattot, R.; Asset, T.; Bordet, P.; Drnec, J.; Dubau, L.; Maillard, F. Beyond strain and ligand effects: Microstrain-induced enhancement of the oxygen reduction reaction kinetics on various ptni/c nanostructures. ACS Catal. 2017, 7, 398–408. [Google Scholar] [CrossRef]
- Maniammal, K.; Madhu, G.; Biju, V. X-ray diffraction line profile analysis of nanostructured nickel oxide: Shape factor and convolution of crystallite size and microstrain contributions. Phys. E Low Dimens. Syst. Nanostruct. 2017, 85, 214–222. [Google Scholar] [CrossRef]
- Clementi, E.; Raimondi, D.L.; Reinhardt, W.P. Atomic screening constants from scf functions. Ii. Atoms with 37 to 86 electrons. J. Chem. Phys. 1967, 47, 1300–1307. [Google Scholar] [CrossRef]
- Wang, J.; Gao, R.; Zhou, D.; Chen, Z.; Wu, Z.; Schumacher, G.; Hu, Z.; Liu, X. Boosting the electrocatalytic activity of co3o4 nanosheets for a li-o2 battery through modulating inner oxygen vacancy and exterior CO3+/CO2+ ratio. ACS Catal. 2017, 7, 6533–6541. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Mu, J.; Fan, S.; Chen, X.; Wang, L.; Yin, Z.; Tadé, M.; Liu, S. Oxygen vacancy-rich porous co3o4 nanosheets toward boosted no reduction by co and co oxidation: Insights into the structure–activity relationship and performance enhancement mechanism. ACS Appl. Mater. Interfaces 2019, 11, 41988–41999. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Fu, L.; Jiang, D.; Ouyang, J.; Hu, Y.; Yang, H.; Xi, Y. Nanoclay-modulated oxygen vacancies of metal oxide. Commun. Chem. 2019, 2, 11. [Google Scholar] [CrossRef]
- Khajehbashi, S.M.B.; Li, J.; Wang, M.; Xu, L.; Zhao, K.; Wei, Q.; Shi, C.; Tang, C.; Huang, L.; Wang, Z.; et al. A crystalline/amorphous cobalt(ii,iii) oxide hybrid electrocatalyst for lithium-air batteries. Energy Technol. 2017, 5, 568–579. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Shi, S.-T.; Zhong, Q.-S.; Zhang, C.-J.; Xu, C.-W. Pt-mn 3 o 4/c as efficient electrocatalyst for oxygen evolution reaction in water electrolysis. Electrochim. Acta 2014, 146, 119–124. [Google Scholar] [CrossRef]
- Peuckert, M.; Bonzel, H.P. Characterization of oxidized platinum surfaces by X-ray photoelectron spectroscopy. Surf. Sci. 1984, 145, 239–259. [Google Scholar] [CrossRef]
- Eberhardt, W.; Fayet, P.; Cox, D.M.; Fu, Z.; Kaldor, A.; Sherwood, R.; Sondericker, D. Photoemission from mass-selected monodispersed pt clusters. Phys. Rev. Lett. 1990, 64, 780–783. [Google Scholar] [CrossRef]
- Alexander, V.N.; Anna, K.; Stephen, W.G.; Cedric, J.P. Nist X-ray photoelectron spectroscopy database. In NIST Standard Reference Database 20, Version 4.1; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2012. [Google Scholar]
- Li, Z.-Y.; Liu, Z.-L.; Liang, J.-C.; Xu, C.-W.; Lu, X. Facile synthesis of pd–mn3o4/c as high-efficient electrocatalyst for oxygen evolution reaction. J. Mater. Chem. A 2014, 2, 18236–18240. [Google Scholar] [CrossRef]
- Payne, B.P.; Biesinger, M.C.; McIntyre, N.S. X-ray photoelectron spectroscopy studies of reactions on chromium metal and chromium oxide surfaces. J. Electron. Spectrosc. Relat. Phenom. 2011, 184, 29–37. [Google Scholar] [CrossRef]
- Aricò, A.S.; Shukla, A.K.; Kim, H.; Park, S.; Min, M.; Antonucci, V. An xps study on oxidation states of pt and its alloys with co and cr and its relevance to electroreduction of oxygen. Appl. Surf. Sci. 2001, 172, 33–40. [Google Scholar] [CrossRef]
- Xin, W.-L.; Lu, K.-K.; Zhu, D.-R.; Zeng, H.-B.; Zhang, X.-J.; Marks, R.-S.; Shan, D. Highly reactive n,n’-carbonyldiimidazole-tailored bifunctional electrocatalyst for oxygen reduction and oxygen evolution. Electrochim. Acta 2019, 307, 375–384. [Google Scholar] [CrossRef]
- Kuznetsov, D.A.; Naeem, M.A.; Kumar, P.V.; Abdala, P.M.; Fedorov, A.; Müller, C.R. Tailoring lattice oxygen binding in ruthenium pyrochlores to enhance oxygen evolution activity. J. Am. Chem. Soc. 2020, 142, 7883–7888. [Google Scholar] [CrossRef]
- Ramos-Moore, E.; Ferrari, P.; Diaz-Droguett, D.E.; Lederman, D.; Evans, J.T. Raman and X-ray photoelectron spectroscopy study of ferroelectric switching in pb(nb,zr,ti)o3 thin films. J. Appl. Phys. 2012, 111, 014108. [Google Scholar] [CrossRef]
- Bajdich, M.; Garcia-Mota, M.; Vojvodic, A.; Norskov, J.K.; Bell, A.T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 2013, 135, 13521–13530. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, T.; Jiang, K.; Da, P.; Peng, Z.; Tang, J.; Kong, B.; Cai, W.-B.; Yang, Z.; Zheng, G. Reduced mesoporous co3o4nanowires as efficient water oxidation electrocatalysts and supercapacitor electrodes. Adv. Energy Mater. 2014, 4. [Google Scholar] [CrossRef]
- Kunhiraman, A.K. Hydrogen evolution reaction catalyzed by platinum nanoislands decorated on three-dimensional nanocarbon hybrid. Ionics 2019, 25, 3787–3797. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef]
- Gong, H.; Zhang, W.; Li, F.; Yang, R. Enhanced electrocatalytic performance of self-supported aucuco for oxygen reduction and evolution reactions. Electrochim. Acta 2017, 252, 261–267. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Zhang, L.; Jiang, D. Surface oxygen vacancies on co3o4 mediated catalytic formaldehyde oxidation at room temperature. Catal. Sci. Technol. 2016, 6, 3845–3853. [Google Scholar] [CrossRef]
- Han, X.; Cheng, F.; Zhang, T.; Yang, J.; Hu, Y.; Chen, J. Hydrogenated uniform pt clusters supported on porous camno(3) as a bifunctional electrocatalyst for enhanced oxygen reduction and evolution. Adv. Mater. 2014, 26, 2047–2051. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Ji, Y.; Xie, M.; You, C.; Yang, L.; Liu, Z.; Asiri, A.M.; Sun, X. Mno2-cop3 nanowires array: An efficient electrocatalyst for alkaline oxygen evolution reaction with enhanced activity. Electrochem. Commun. 2018, 86, 161–165. [Google Scholar] [CrossRef]
- Zhong, L.; Bao, Y.; Feng, L. Fe-doping effect on cote catalyst with greatly boosted intrinsic activity for electrochemical oxygen evolution reaction. Electrochim. Acta 2019, 321. [Google Scholar] [CrossRef]
- Su, Y.; Zhu, Y.; Jiang, H.; Shen, J.; Yang, X.; Zou, W.; Chen, J.; Li, C. Cobalt nanoparticles embedded in n-doped carbon as an efficient bifunctional electrocatalyst for oxygen reduction and evolution reactions. Nanoscale 2014, 6, 15080–15089. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jiang, R.; Chen, X.; Chen, Y.; Wang, L. 3d nickel-cobalt diselenide nanonetwork for highly efficient oxygen evolution. Sci. Bull. 2017, 62, 1373–1379. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).