Upgrading of Oils from Biomass and Waste: Catalytic Hydrodeoxygenation



Abstract
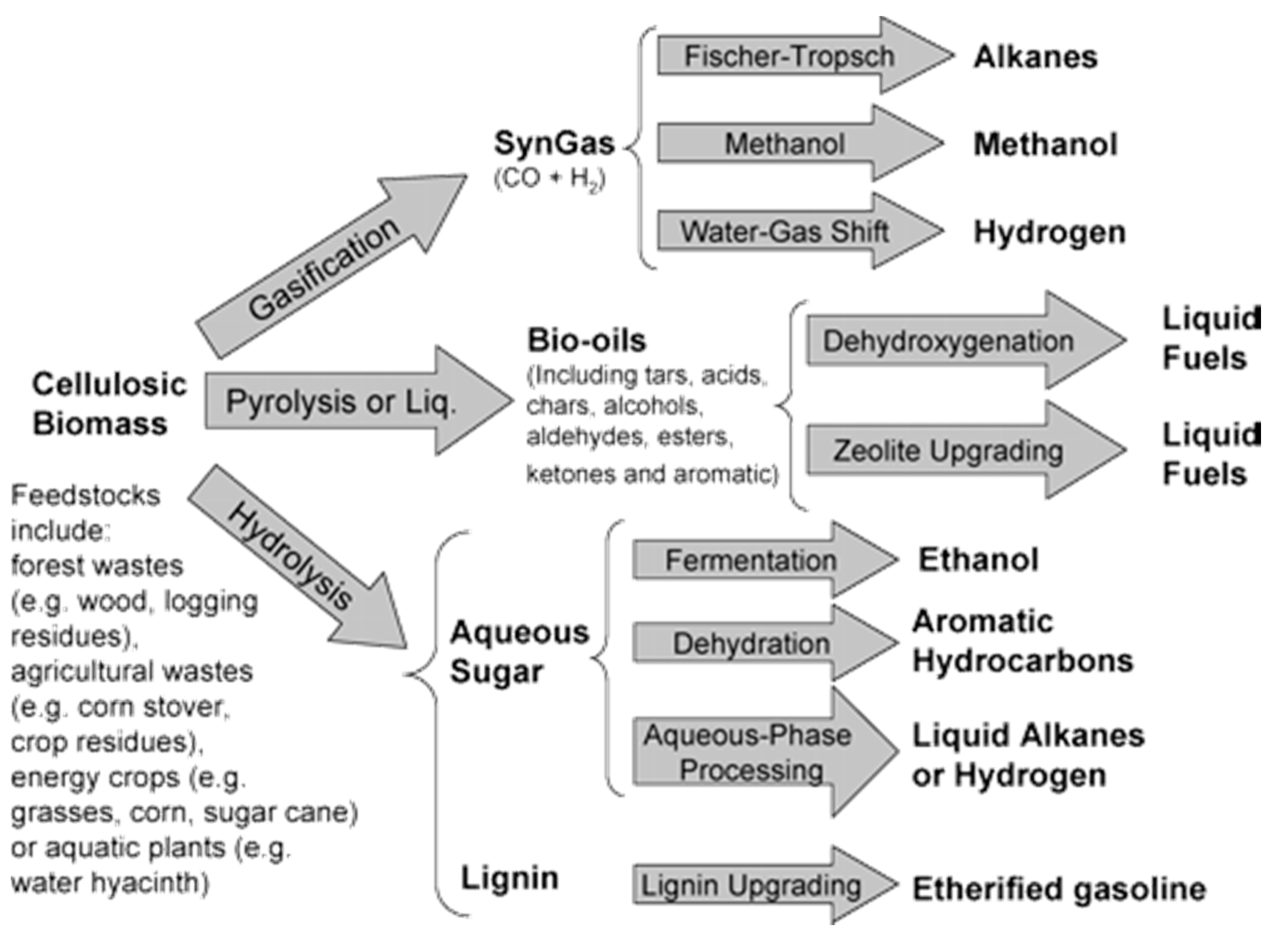
:1. Introduction
- Conventional Pyrolysis: Pyrolysis at the slow heating rate, which permits the formation of the solid, liquid and gaseous products in significant portions.
- Fast Pyrolysis: If the procedure aims to produce a liquid or gaseous product, fast pyrolysis is employed. In this case, the products are considered marginal with a low yield. The operating conditions are declared: High operating temperature, short residence time and very fine particles.
- Flash Pyrolysis: In this method, the feedstock is directly fed into the high-temperature zone of the reactor, which subsequently undergoes flash pyrolysis where the final products are in the gaseous phase with a short contact time inside the reactor.
1.1. Oxygenated Compounds Exist in Biomass Pyrolysis Oil
1.2. Issues Related to the Presence of Oxygen in the Feedstock or Products
2. Different Deoxygenation Approaches for Bio-Oil
2.1. Physical Upgrading Technologies
2.1.1. Solvent Addition
2.1.2. Emulsion
2.2. Chemical Upgrading Technologies
2.2.1. Esterification
2.2.2. Supercritical Fluid
2.2.3. Steam Reforming
2.2.4. Hydrocracking
2.2.5. Hydrotreatment
3. Hydrodeoxygenation
Catalytic Hydrodeoxygenation
4. HDO Catalysts
4.1. Sulphide Catalysts
4.2. Oxide Catalysts
4.3. Transition Metal Catalysts
Phosphide, Carbide, and Nitride Catalysts
5. HDO of Different Biomass-Derived Oxygenates
5.1. HDO of Lignin-Derived Oxygenates
5.1.1. Phenol and Alkylated Phenol
5.1.2. Guaiacol
5.1.3. Phenolic Dimers
5.2. HDO of Carbohydrates-Derived Oxygenates
5.2.1. Levulinic Acid
5.2.2. Acrylic Acid
5.2.3. Furfurals
6. The Issue of Catalytic Deactivation in an Application and How It Can Be Solved
6.1. Coke Formation
6.2. Catalyst Deactivation
7. Techno-Economic Analysis of HDO
8. Applying the Microwave in the Hydrodeoxygenation Process
8.1. Microwave Heating
8.2. Applying the Microwave in the Case of the HDO Processes
Author Contributions
Funding
Conflicts of Interest
References
- Vasilevich, A.V.; Baklanova, O.N.; Lavrenov, A.V. Hydrodeoxygenation of Guaiacol with Molybdenum-Carbide-Based Carbon Catalysts. ChemistrySelect 2020, 5, 4575–4579. [Google Scholar] [CrossRef]
- Isahak, W.N.R.W.; Hisham, M.W.M.; Yarmo, M.A.; Hin, T.-Y.Y. A review on bio-oil production from biomass by using pyrolysis method. Renew. Sustain. Energy Rev. 2012, 16, 5910–5923. [Google Scholar] [CrossRef]
- Rubin, E.M. Genomics of cellulosic biofuels. Nat. Cell Biol. 2008, 454, 841–845. [Google Scholar] [CrossRef]
- Farag, S. Production of Chemicals by Microwave Thermal Treatment of Lignin. Ph.D. Thesis, University of Montreal, Montreal, QC, Canada, December 2013. [Google Scholar]
- De Wild, P.J.; Huijgen, W.J.J.; Heeres, H. Pyrolysis of wheat straw-derived organosolv lignin. J. Anal. Appl. Pyrolysis 2012, 93, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.; Ben, H.; Ragauskas, A.; Deng, Y. Lignin Pyrolysis Components and Upgrading—Technology Review. BioEnergy Res. 2013, 6, 1183–1204. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Farag, S.; Chaouki, J. Economics evaluation for on-site pyrolysis of kraft lignin to value-added chemicals. Bioresour. Technol. 2015, 175, 254–261. [Google Scholar] [CrossRef]
- Farag, S.; Chaouki, J. Technical and Economical Feasibility of Pyrolysis of Kraft Lignin. In Proceedings of the TechConnect Briefs, Washington, DC, USA, 14–17 June 2015. [Google Scholar]
- Farag, S.; Fu, D.; Jessop, P.G.; Chaouki, J. Detailed compositional analysis and structural investigation of a bio-oil from microwave pyrolysis of kraft lignin. J. Anal. Appl. Pyrolysis 2014, 109, 249–257. [Google Scholar] [CrossRef]
- Farag, S.; Kouisni, L.; Chaouki, J. Lumped Approach in Kinetic Modeling of Microwave Pyrolysis of Kraft Lignin. Energy Fuels 2014, 28, 1406–1417. [Google Scholar] [CrossRef]
- Naik, S.; Goud, V.V.; Rout, P.K.; Dalai, A.K. Production of first and second generation biofuels: A comprehensive review. Renew. Sustain. Energy Rev. 2010, 14, 578–597. [Google Scholar] [CrossRef]
- Sims, R.E.; Mabee, W.; Saddler, J.N.; Taylor, M. An overview of second generation biofuel technologies. Bioresour. Technol. 2010, 101, 1570–1580. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [Green Version]
- Huber, G.W.; Dumesic, J.A. An overview of aqueous-phase catalytic processes for production of hydrogen and alkanes in a biorefinery. Catal. Today 2006, 111, 119–132. [Google Scholar] [CrossRef]
- Demirbas, A.; Arin, G. An Overview of Biomass Pyrolysis. Energy Sources 2002, 24, 471–482. [Google Scholar] [CrossRef]
- Demirbas, A. Biofuels securing the planet’s future energy needs. Energy Convers. Manag. 2009, 50, 2239–2249. [Google Scholar] [CrossRef]
- Attia, M.; Farag, S.; Habibzadeh, S.; Chaouki, J. Fast Pyrolysis of Lignocellulosic Biomass for the Production of Energy and Chemicals: A Critical Review. Curr. Org. Chem. 2016, 20, 1. [Google Scholar] [CrossRef] [Green Version]
- Bridgwater, T. Fast pyrolysis of biomass for the production of liquids. Biomass Combust. Sci. Technol. Eng. 2013, 130–171. [Google Scholar] [CrossRef]
- Attia, M.; Farag, S.; Jaffer, S.A.; Chaouki, J. Metal and sulfur removal from petroleum oil using a novel demetallization-desulfurization agent and process. J. Clean. Prod. 2020, 275, 124177. [Google Scholar] [CrossRef]
- Shang, H.; Liu, Y.; Shi, J.-C.; Shi, Q.; Zhang, W.-H. Microwave-assisted nickel and vanadium removal from crude oil. Fuel Process. Technol. 2016, 142, 250–257. [Google Scholar] [CrossRef]
- Dedeles, G.R.; Abe, A.; Saito, K.; Asano, K.; Saito, K.; Yokota, A.; Tomita, F. Microbial demetallization of crude oil: Nickel protoporphyrin disodium as a model organo-metallic substrate. J. Biosci. Bioeng. 2000, 90, 515–521. [Google Scholar] [CrossRef]
- Si, Z.; Zhang, X.; Wang, C.; Ma, L.; Dong, R. An Overview on Catalytic Hydrodeoxygenation of Pyrolysis Oil and Its Model Compounds. Catalysts 2017, 7, 169. [Google Scholar] [CrossRef] [Green Version]
- Saber, M.; Nakhshiniev, B.; Yoshikawa, K. A review of production and upgrading of algal bio-oil. Renew. Sustain. Energy Rev. 2016, 58, 918–930. [Google Scholar] [CrossRef]
- Jin, W.; Pérez, L.P.; Yu, J.; Odriozola, J.; Gu, S.; Reina, T. Cost-effective routes for catalytic biomass upgrading. Curr. Opin. Green Sustain. Chem. 2020, 23, 1–9. [Google Scholar] [CrossRef]
- Hu, X.; Li, C.; Xu, Y.; Wang, Q.; Zhu, X. On the thermal oxidation stability of pyrolysis biomass oil. Int. J. Renew. Energy Technol. 2011, 2, 155. [Google Scholar] [CrossRef]
- Valle, B.; Remiro, A.; García-Gómez, N.; Gayubo, A.G.; Bilbao, J. Recent research progress on bio-oil conversion into bio-fuels and raw chemicals: A review. J. Chem. Technol. Biotechnol. 2019, 94, 670–689. [Google Scholar] [CrossRef]
- Fermoso, J.; Pizarro, P.; Coronado, J.M.; Serrano, D. Advanced biofuels production by upgrading of pyrolysis bio-oil. Wiley Interdiscip. Rev. Energy Environ. 2017, 6, e245. [Google Scholar] [CrossRef]
- Pawar, A.; Panwar, N.L.; Salvi, B.L. Comprehensive review on pyrolytic oil production, upgrading and its utilization. J. Mater. Cycles Waste Manag. 2020, 22, 1712–1722. [Google Scholar] [CrossRef]
- Mostafazadeh, A.K.; Solomatnikova, O.; Drogui, P.; Tyagi, R.D. A review of recent research and developments in fast pyrolysis and bio-oil upgrading. Biomass Convers. Biorefin. 2018, 8, 739–773. [Google Scholar] [CrossRef]
- Liu, R.; Fei, W.; Shen, C. Influence of acetone addition on the physicochemical properties of bio-oils. J. Energy Inst. 2014, 87, 127–133. [Google Scholar] [CrossRef]
- Jiang, X.; Ellis, N. Upgrading Bio-oil through Emulsification with Biodiesel: Mixture Production. Energy Fuels 2010, 24, 1358–1364. [Google Scholar] [CrossRef]
- Kanhounnon, W.G.; Kuevi, U.A.; Kpotin, G.A.; Koudjina, S.; Houngue, A.K.; Atohoun, G.Y.S.; Mensah, J.-B.; Badawi, M. Quantum mechanistic study of furan and 2-methylfuran hydrodeoxygenation on molybdenum and tungsten sulfide clusters. J. Mol. Model. 2019, 25, 237. [Google Scholar] [CrossRef]
- Sundqvist, T.; Oasmaa, A.; Koskinen, A. Upgrading Fast Pyrolysis Bio-Oil Quality by Esterification and Azeotropic Water Removal. Energy Fuels 2015, 29, 2527–2534. [Google Scholar] [CrossRef] [Green Version]
- Weerachanchai, P.; Tangsathitkulchai, C.; Tangsathitkulchai, M. Effect of reaction conditions on the catalytic esterification of bio-oil. Korean J. Chem. Eng. 2012, 29, 182–189. [Google Scholar] [CrossRef]
- Jo, H.; Verma, D.; Kim, J. Excellent aging stability of upgraded fast pyrolysis bio-oil in supercritical ethanol. Fuel 2018, 232, 610–619. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, I.-G.; Park, J.-Y.; Lee, K.-Y. Efficient upgrading of pyrolysis bio-oil over Ni-based catalysts in supercritical ethanol. Fuel 2019, 241, 207–217. [Google Scholar] [CrossRef]
- Quan, C.; Xu, S.; Zhou, C. Steam reforming of bio-oil from coconut shell pyrolysis over Fe/olivine catalyst. Energy Convers. Manag. 2017, 141, 40–47. [Google Scholar] [CrossRef]
- Lan, P.; Xu, Q.; Zhou, M.; Lan, L.; Zhang, S.; Yan, Y. Catalytic Steam Reforming of Fast Pyrolysis Bio-Oil in Fixed Bed and Fluidized Bed Reactors. Chem. Eng. Technol. 2010, 33, 2021–2028. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.-D.; Jensen, P.A.; Knudsen, K.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Tanneru, S.K.; Steele, P.H. Direct hydrocracking of oxidized bio-oil to hydrocarbons. Fuel 2015, 154, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Morales-Delarosa, S.; Campos-Martin, J. Catalytic processes and catalyst development in biorefining. Adv. Biorefin. 2014, 152–198. [Google Scholar] [CrossRef]
- Dabros, T.M.; Stummann, M.Z.; Høj, M.; Jensen, P.A.; Grunwaldt, J.-D.; Gabrielsen, J.; Mortensen, P.M.; Jensen, A.D. Transportation fuels from biomass fast pyrolysis, catalytic hydrodeoxygenation, and catalytic fast hydropyrolysis. Prog. Energy Combust. Sci. 2018, 68, 268–309. [Google Scholar] [CrossRef]
- Li, F.; Srivatsa, S.C.; Bhattacharya, S. A review on catalytic pyrolysis of microalgae to high-quality bio-oil with low oxygeneous and nitrogenous compounds. Renew. Sustain. Energy Rev. 2019, 108, 481–497. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, R.; Yin, R.; Mei, Y. Upgrading of bio-oil from biomass fast pyrolysis in China: A review. Renew. Sustain. Energy Rev. 2013, 24, 66–72. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Li, N.; Tompsett, G.A.; Huber, G. Renewable High-Octane Gasoline by Aqueous-Phase Hydrodeoxygenation of C5 and C6 Carbohydrates over Pt/Zirconium Phosphate Catalysts. ChemSusChem 2010, 3, 1154–1157. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Pourzolfaghar, H.; Abnisa, F.; Daud, W.M.A.W.; Aroua, M.K. Atmospheric hydrodeoxygenation of bio-oil oxygenated model compounds: A review. J. Anal. Appl. Pyrolysis 2018, 133, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Marker, T.L.; Felix, L.G.; Linck, M.B.; Roberts, M.J. Integrated hydropyrolysis and hydroconversion (IH2) for the direct production of gasoline and diesel fuels or blending components from biomass, Part 1: Proof of principle testing. Environ. Prog. Sustain. Energy 2011, 31, 191–199. [Google Scholar] [CrossRef]
- Regmi, Y.N.; Mann, J.K.; McBride, J.R.; Tao, J.; Barnes, C.E.; Labbé, N.; Chmely, S.C. Catalytic transfer hydrogenolysis of organosolv lignin using B-containing FeNi alloyed catalysts. Catal. Today 2018, 302, 190–195. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, E.E.; Kim, Y.T.; Jung, S.; Kim, H.J.; Huber, G.W.; Lee, J. Recent advances in hydrodeoxygenation of biomass-derived oxygenates over heterogeneous catalysts. Green Chem. 2019, 21, 3715–3743. [Google Scholar] [CrossRef]
- Elkasabi, Y.; Mullen, C.A.; Pighinelli, A.L.; Boateng, A.A. Hydrodeoxygenation of fast-pyrolysis bio-oils from various feedstocks using carbon-supported catalysts. Fuel Process. Technol. 2014, 123, 11–18. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.; Zacher, A.H.; Wang, L.; Ren, S.; Liang, J.; Wei, Y.; Liu, Y.; Tang, J.; Zhang, Q.; et al. A review of catalytic hydrodeoxygenation of lignin-derived phenols from biomass pyrolysis. Bioresour. Technol. 2012, 124, 470–477. [Google Scholar] [CrossRef]
- Elliott, D.; Neuenschwander, G. Liquid Fuels by Low-Severity Hydrotreating of Biocrude. In Developments in Thermochemical Biomass Conversion; Springer: Berlin/Heidelberg, Germany, 1997; pp. 611–621. [Google Scholar]
- Elliott, D.C.; Oasmaa, A. Catalytic hydrotreating of black liquor oils. Energy Fuels 1991, 5, 102–109. [Google Scholar] [CrossRef]
- Ly, H.V.; Kim, J.; Hwang, H.T.; Choi, J.H.; Woo, H.C.; Kim, S.-S. Catalytic Hydrodeoxygenation of Fast Pyrolysis Bio-Oil from Saccharina japonica Alga for Bio-Oil Upgrading. Catalysts 2019, 9, 1043. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, C.C.; Reolon, M.B.G.; Zimmermann, M.; Raffelt, K.; Grunwaldt, J.-D.; Dahmen, N. Synthesis and Regeneration of Nickel-Based Catalysts for Hydrodeoxygenation of Beech Wood Fast Pyrolysis Bio-Oil. Catalysts 2018, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Centeno, A.; Laurent, E.; Delmon, B. Influence of the Support of CoMo Sulfide Catalysts and of the Addition of Potassium and Platinum on the Catalytic Performances for the Hydrodeoxygenation of Carbonyl, Carboxyl, and Guaiacol-Type Molecules. J. Catal. 1995, 154, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Laurent, E.; Delmon, B. Study of the hydrodeoxygenation of carbonyl, car☐ ylic and guaiacyl groups over sulfided CoMo/γ-Al2O3 and NiMo/γ-Al2O3 catalysts: I. Catalytic reaction schemes. Appl. Catal. A Gen. 1994, 109, 77–96. [Google Scholar] [CrossRef]
- Laurent, E.; Delmon, B. Study of the hydrodeoxygenation of carbonyl, car☐ ylic and guaiacyl groups over sulfided CoMo/γ-Al2O3 and NiMo/γ-Al2O3 catalyst: II. Influence of water, ammonia and hydrogen sulfide. Appl. Catal. A Gen. 1994, 109, 97–115. [Google Scholar] [CrossRef]
- Laurent, E.; Delmon, B. Influence of water in the deactivation of a sulfided NiMo/γ-Al2O3 catalyst during hydrodeoxygenation. J. Catal. 1994, 146, 281–291. [Google Scholar] [CrossRef]
- Ferrari, M.; Maggi, R.; Delmon, B.; Grange, P. Influences of the Hydrogen Sulfide Partial Pressure and of a Nitrogen Compound on the Hydrodeoxygenation Activity of a CoMo/Carbon Catalyst. J. Catal. 2001, 198, 47–55. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F.A. Hydrodeoxygenation of Aqueous-Phase Catalytic Pyrolysis Oil to Liquid Hydrocarbons Using Multifunctional Nickel Catalyst. Ind. Eng. Chem. Res. 2018, 57, 13257–13268. [Google Scholar] [CrossRef] [Green Version]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Zhang, Y.; Monnier, J.; Ikura, M. Bio-oil upgrading using dispersed unsupported MoS2 catalyst. Fuel Process. Technol. 2020, 206, 106403. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, K.; Qiao, Z.; Li, L.; Liu, P.; Yang, Y. Influence of Surfactants on the Synthesis of MoS2 Catalysts and Their Activities in the Hydrodeoxygenation of 4-Methylphenol. Ind. Eng. Chem. Res. 2014, 53, 10301–10309. [Google Scholar] [CrossRef]
- Yoosuk, B.; Tumnantong, D.; Prasassarakich, P. Amorphous unsupported Ni–Mo sulfide prepared by one step hydrothermal method for phenol hydrodeoxygenation. Fuel 2012, 91, 246–252. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of guaiacol with CoMo catalysts. Part I: Promoting effect of cobalt on HDO selectivity and activity. Appl. Catal. B Environ. 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Yoosuk, B.; Tumnantong, D.; Prasassarakich, P. Unsupported MoS2 and CoMoS2 catalysts for hydrodeoxygenation of phenol. Chem. Eng. Sci. 2012, 79, 1–7. [Google Scholar] [CrossRef]
- Bunch, A.Y.; Ozkan, U.S. Investigation of the Reaction Network of Benzofuran Hydrodeoxygenation over Sulfided and Reduced Ni–Mo/Al2O3 Catalysts. J. Catal. 2002, 206, 177–187. [Google Scholar] [CrossRef]
- Toba, M.; Abe, Y.; Kuramochi, H.; Osako, M.; Mochizuki, T.; Yoshimura, Y. Hydrodeoxygenation of waste vegetable oil over sulfide catalysts. Catal. Today 2011, 164, 533–537. [Google Scholar] [CrossRef]
- Loricera, C.; Pawelec, B.; Infantes-Molina, A.; Álvarez-Galván, M.; Huirache-Acuña, R.; Nava, R.; Fierro, J.L.G. Hydrogenolysis of anisole over mesoporous sulfided CoMoW/SBA-15(16) catalysts. Catal. Today 2011, 172, 103–110. [Google Scholar] [CrossRef]
- Yang, Y.; Gilbert, A.; Xu, C. Hydrodeoxygenation of bio-crude in supercritical hexane with sulfided CoMo and CoMoP catalysts supported on MgO: A model compound study using phenol. Appl. Catal. A Gen. 2009, 360, 242–249. [Google Scholar] [CrossRef]
- Sepúlveda, C.; García, R.; Reyes, P.; Ghampson, I.T.; Fierro, J.L.G.; Laurenti, D.; Vrinat, M.; Escalona, N. Hydrodeoxygenation of guaiacol over ReS2/activated carbon catalysts. Support and Re loading effect. Appl. Catal. A Gen. 2014, 475, 427–437. [Google Scholar]
- Prasomsri, T.; Nimmanwudipong, T.; Román-Leshkov, Y. Effective hydrodeoxygenation of biomass-derived oxygenates into unsaturated hydrocarbons by MoO3 using low H2 pressures. Energy Environ. Sci. 2013, 6, 1732–1738. [Google Scholar] [CrossRef]
- Gutierrez, A.; Kaila, R.K.; Honkela, M.; Slioor, R.; Krause, A. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- Han, Y.; Gholizadeh, M.; Tran, C.-C.; Kaliaguine, S.; Li, C.-Z.; Olarte, M.; Garcia-Perez, M. Hydrotreatment of pyrolysis bio-oil: A review. Fuel Process. Technol. 2019, 195, 106140. [Google Scholar] [CrossRef]
- Mendes, F.L.; Da Silva, V.T.; Pacheco, M.E.; Toniolo, F.S.; Henriques, C.A. Bio-oil hydrotreating using nickel phosphides supported on carbon-covered alumina. Fuel 2019, 241, 686–694. [Google Scholar] [CrossRef]
- Gutiérrez-Rubio, S.; Moreno, I.; Serrano, D.P.; Coronado, J.M. Hydrotreating of Guaiacol and Acetic Acid Blends over Ni2P/ZSM-5 Catalysts: Elucidating Molecular Interactions during Bio-Oil Upgrading. ACS Omega 2019, 4, 21516–21528. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Hernández, D.; LaVerde, J.; Pérez, S.; Lopez, D. Catalytic Upgrading of Residual Biomass Derived Bio-oil over Molybdenum Carbide. Waste Biomass Valoris. 2019, 11, 2849–2856. [Google Scholar] [CrossRef]
- Shu, R.; Li, R.; Lin, B.; Wang, C.; Cheng, Z.; Chen, Y. A review on the catalytic hydrodeoxygenation of lignin-derived phenolic compounds and the conversion of raw lignin to hydrocarbon liquid fuels. Biomass Bioenergy 2020, 132, 105432. [Google Scholar] [CrossRef]
- Patil, M.L.; Lali, A.M.; Dalai, A. Catalytic hydrodeoxygenation of bio-oil model compound for production of fuel grade oil. Asia Pac. J. Chem. Eng. 2019, 14. [Google Scholar] [CrossRef]
- Sergeev, A.G.; Hartwig, J.F. Selective, Nickel-Catalyzed Hydrogenolysis of Aryl Ethers. Science 2011, 332, 439–443. [Google Scholar] [CrossRef] [Green Version]
- Basudeb, S.; Saha, B.; Luque, R. Hydrodeoxygenation processes: Advances on catalytic transformations of biomass-derived platform chemicals into hydrocarbon fuels. Bioresour. Technol. 2015, 178, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef] [Green Version]
- Grilc, M.; Likozar, B. Levulinic acid hydrodeoxygenation, decarboxylation and oligmerization over NiMo/Al2O3 catalyst to bio-based value-added chemicals: Modelling of mass transfer, thermodynamics and micro-kinetics. Chem. Eng. J. 2017, 330, 383–397. [Google Scholar] [CrossRef]
- Rocha, A.S.; Souza, L.A.; Oliveira, R.R.; Rocha, A.B.; Da Silva, V.T. Hydrodeoxygenation of acrylic acid using Mo2C/Al2O3. Appl. Catal. A Gen. 2017, 531, 69–78. [Google Scholar] [CrossRef]
- Zheng, H.; Zhu, Y.-L.; Teng, B.-T.; Bai, Z.-Q.; Zhang, C.-H.; Xiang, H.-W.; Li, Y.-W. Towards understanding the reaction pathway in vapour phase hydrogenation of furfural to 2-methylfuran. J. Mol. Catal. A Chem. 2006, 246, 18–23. [Google Scholar] [CrossRef]
- Chen, G.; Liu, J.; Li, X.; Zhang, J.; Yin, H.; Su, Z. Investigation on catalytic hydrodeoxygenation of eugenol blend with light fraction in bio-oil over Ni-based catalysts. Renew. Energy 2020, 157, 456–465. [Google Scholar] [CrossRef]
- Lin, B.; Li, R.; Shu, R.; Wang, C.; Yuan, Z.; Liu, Y.; Chen, Y. Synergistic effect of highly dispersed Ru and moderate acid site on the hydrodeoxygenation of phenolic compounds and raw bio-oil. J. Energy Inst. 2019, 93. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, A.; Biswas, B.; Kumar, J.; Yenumala, S.R.; Bhaskar, T. Hydrodeoxygenation of m-Cresol over Ru based catalysts: Influence of catalyst support on m-Cresol conversion and methylcyclohexane selectivity. Renew. Energy 2020, 151, 687–697. [Google Scholar] [CrossRef]
- Mukundan, S.; Sriganesh, G.; Kumar, P. Upgrading Prosopis juliflora to biofuels via a two-step pyrolysis—Catalytic hydrodeoxygenation approach. Fuel 2020, 267, 117320. [Google Scholar] [CrossRef]
- Bakhtyari, A.; Rahimpour, M.; Raeissi, S. Cobalt-molybdenum catalysts for the hydrodeoxygenation of cyclohexanone. Renew. Energy 2020, 150, 443–455. [Google Scholar] [CrossRef]
- Bakhtyari, A.; Sakhayi, A.; Rahimpour, M.; Raeissi, S. Upgrading of cyclohexanone to hydrocarbons by hydrodeoxygenation over nickel–molybdenum catalysts. Int. J. Hydrogen Energy 2020, 45, 11062–11076. [Google Scholar] [CrossRef]
- Eschenbacher, A.; Saraeian, A.; Shanks, B.H.; Jensen, P.A.; Li, C.; Duus, J.Ø.; Hansen, A.B.; Mentzel, U.V.; Henriksen, U.B.; Ahrenfeldt, J.; et al. Enhancing bio-oil quality and energy recovery by atmospheric hydrodeoxygenation of wheat straw pyrolysis vapors using Pt and Mo-based catalysts. Sustain. Energy Fuels 2020, 4, 1991–2008. [Google Scholar] [CrossRef]
- Resende, K.; Noronha, F.; Hori, C. Hydrodeoxygenation of phenol over metal supported niobia catalysts. Renew. Energy 2020, 149, 198–207. [Google Scholar] [CrossRef]
- Jacobson, K.; Maheria, K.C.; Dalai, A.K. Bio-oil valorization: A review. Renew. Sustain. Energy Rev. 2013, 23, 91–106. [Google Scholar] [CrossRef]
- Cheng, S.; Wei, L.; Zhao, X.; Julson, J. Application, Deactivation, and Regeneration of Heterogeneous Catalysts in Bio-Oil Upgrading. Catalysts 2016, 6, 195. [Google Scholar] [CrossRef]
- Furimsky, E. Deactivation of hydroprocessing catalysts. Catal. Today 1999, 52, 381–495. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Palos, R.; Hita, I.; Arandes, J.M.; Rodríguez-Mirasol, J.; Cordero, T.; Bilbao, J.; Castaño, P. Revealing the pathways of catalyst deactivation by coke during the hydrodeoxygenation of raw bio-oil. Appl. Catal. B Environ. 2018, 239, 513–524. [Google Scholar] [CrossRef]
- Bagnato, G.; Sanna, A. Process and Techno-Economic Analysis for Fuel and Chemical Production by Hydrodeoxygenation of Bio-Oil. Catalysts 2019, 9, 1021. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, J.L.; Gunukula, S.; Boateng, A.A.; Mullen, C.A.; DeSisto, W.J.; Wheeler, M.C. Pyrolysis of forest residues: An approach to techno-economics for bio-fuel production. Fuel 2017, 193, 477–484. [Google Scholar] [CrossRef]
- Farag, S.; Mudraboyina, B.P.; Jessop, P.G.; Chaouki, J. Impact of the heating mechanism on the yield and composition of bio-oil from pyrolysis of kraft lignin. Biomass Bioenergy 2016, 95, 344–353. [Google Scholar] [CrossRef]
- Samih, S.; Latifi, M.; Farag, S.; Leclerc, P.; Chaouki, J. From complex feedstocks to new processes: The role of the newly developed micro-reactors. Chem. Eng. Process. Process Intensif. 2018, 131, 92–105. [Google Scholar] [CrossRef]
- Shekara, B.C.; Prakash, B.J.; Bhat, Y.S. Microwave-induced deactivation-free catalytic activity of BEA zeolite in acylation reactions. J. Catal. 2012, 290, 101–107. [Google Scholar] [CrossRef]
- Paixão, V.; Monteiro, R.; Andrade, M.; Fernandes, A.; Rocha, J.; Carvalho, A.; Leitão, R.A.D.S.E. Desilication of MOR zeolite: Conventional versus microwave assisted heating. Appl. Catal. A Gen. 2011, 402, 59–68. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Z.K. Microwave-assisted conversion of lignocellulosic biomass into furans in ionic liquid. Bioresour. Technol. 2010, 101, 1111–1114. [Google Scholar] [CrossRef]
- Pan, R.; Wu, Y.; Wang, Q.; Hong, Y. Preparation and catalytic properties of platinum dioxide nanoparticles: A comparison between conventional heating and microwave-assisted method. Chem. Eng. J. 2009, 153, 206–210. [Google Scholar] [CrossRef]
- Budarin, V.L.; Clark, J.H.; Lanigan, B.A.; Shuttleworth, P.; Breeden, S.W.; Wilson, A.J.; MacQuarrie, D.J.; Milkowski, K.; Jones, J.; Bridgeman, T.; et al. The preparation of high-grade bio-oils through the controlled, low temperature microwave activation of wheat straw. Bioresour. Technol. 2009, 100, 6064–6068. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Balasubramanian, R.; See, S.W. Optimization and validation of a low temperature microwave-assisted extraction method for analysis of polycyclic aromatic hydrocarbons in airborne particulate matter. Talanta 2006, 69, 79–86. [Google Scholar] [CrossRef]
- Lucchesi, M.E.; Chemat, F.; Smadja, J. Solvent-free microwave extraction of essential oil from aromatic herbs: Comparison with conventional hydro-distillation. J. Chromatogr. A 2004, 1043, 323–327. [Google Scholar] [CrossRef]
- Patil, P.; Gude, V.G.; Pinappu, S.; Deng, S. Transesterification kinetics of Camelina sativa oil on metal oxide catalysts under conventional and microwave heating conditions. Chem. Eng. J. 2011, 168, 1296–1300. [Google Scholar] [CrossRef]
- Guiotoku, M.; Rambo, C.; Hansel, F.; Magalhães, W.; Hotza, D. Microwave-assisted hydrothermal carbonization of lignocellulosic materials. Mater. Lett. 2009, 63, 2707–2709. [Google Scholar] [CrossRef]
- Kržan, A.; Žagar, E. Microwave driven wood liquefaction with glycols. Bioresour. Technol. 2009, 100, 3143–3146. [Google Scholar] [CrossRef]
- Dogan, H.; Hilmioglu, N.D. Dissolution of cellulose with NMMO by microwave heating. Carbohydr. Polym. 2009, 75, 90–94. [Google Scholar] [CrossRef]
- Sithambaram, S.; Nyutu, E.K.; Suib, S.L. OMS-2 catalyzed oxidation of tetralin: A comparative study of microwave and conventional heating under open vessel conditions. Appl. Catal. A Gen. 2008, 348, 214–220. [Google Scholar] [CrossRef]
- Orozco, A.; Ahmad, M.; Rooney, D.; Walker, G. Dilute Acid Hydrolysis of Cellulose and Cellulosic Bio-Waste Using a Microwave Reactor System. Process. Saf. Environ. Prot. 2007, 85, 446–449. [Google Scholar] [CrossRef]
- Menéndez, J.; Domínguez, A.; Inguanzo, M.; Pis, J. Microwave pyrolysis of sewage sludge: Analysis of the gas fraction. J. Anal. Appl. Pyrolysis 2004, 71, 657–667. [Google Scholar] [CrossRef]
- Zhu, S.; Wu, Y.; Yu, Z.; Liao, J.; Zhang, Y. Pretreatment by microwave/alkali of rice straw and its enzymic hydrolysis. Process. Biochem. 2005, 40, 3082–3086. [Google Scholar] [CrossRef]
- Motasemi, F.; Afzal, M.T. A review on the microwave-assisted pyrolysis technique. Renew. Sustain. Energy Rev. 2013, 28, 317–330. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conventional | Fast | Flash | |
|---|---|---|---|
| Pyrolysis Temperature (K) | 550–950 | 850–1250 | 1050–1300 |
| Heating Rate (K/s) | 0.1–1 | 10–200 | >1000 |
| Particle Size (mm) | 5–10 | <1 | <0.2 |
| Solid Residence Time (s) | 450–550 | 0.5–10 | <0.5 |
| Material | C | H | O | N | S |
|---|---|---|---|---|---|
| Ash | 49.7 | 6.9 | 3.0 | - | - |
| Beech | 51.6 | 6.3 | 41.4 | - | - |
| Sawdust | 48.3 | 6.22 | 45.2 | 0.22 | 0 |
| Barley Straw | 45.7 | 6.1 | 38.3 | 0.4 | 0.1 |
| Bituminous Coal | 73.1 | 5.5 | 8.7 | 1.4 | 1.7 |
| Cypress | 55.0 | 6.5 | 38.1 | - | - |
| Kraft lignin | 63.3 | 5.8 | 29.2 | 0.1 | 1.6 |
| Miscanthus | 48.1 | 5.4 | 42.2 | 0.5 | <0.1 |
| Rice Straw | 41.4 | 5 | 39.9 | 0.7 | 0.1 |
| Wood | 51.6 | 6.3 | 41.5 | - | 0.1 |
| Wheat Straw | 48.5 | 5.5 | 3.9 | 0.3 | 0.1 |
| Material | C | H | O | N | S |
| Component | Structure | Area wt.% |
|---|---|---|
| Formaldehyde |  | 3.14 |
| Hydroxyacetaldehyde |  | 3.14 |
| Hydroxypropanone |  | 2.70 |
| Butyric acid |  | 0.96 |
| Acetic acid |  | 29.76 |
| Glyceraldehyde |  | 3.54 |
| 2,2-dimethoxy-ethanol |  | 6.83 |
| Furfural |  | 6.56 |
| 2,5-dimethoxy-tetrahydrofuran |  | 3.47 |
| 4-hydroxy-butyric acid |  | 0.43 |
| 5H-furan-2-one |  | 0.74 |
| 2-Furoic acid |  | - |
| 2,3-dimethyl-cyclohexanol |  | 1.31 |
| 3-methyl-5H-furan-2-one |  | 0.38 |
| Phenol |  | 1.57 |
| o-cresol |  | 1.12 |
| m-cresol |  | 1.46 |
| 2-methoxy-6-methyl-phenol |  | 1.78 |
| 3,4-dimethyl-phenol |  | 1.14 |
| 4-ethyl-phenol |  | 1.31 |
| Catechol |  | 3.53 |
| 3-methyl-catechol |  | 1.36 |
| Vanillin |  | 0.24 |
| 4-ethyl-catechol |  | 0.71 |
| Levoglucosan |  | 9.95 |
| 2,3,4-trimethoxy-benzaldehyde |  | 0.20 |
| 3-(4-hydroxy-2-methoxy-phenyl)-propenal |  | 0.15 |
| Properties | Bio-Oil from Fast Pyrolysis | Heavy Fuel |
|---|---|---|
| Water (wt.%) | 15–30 | 0.1 |
| pH | 2–4 | - |
| Density (kg/m3) | 1200 | 940 |
| Elemental composition (wt.%) | ||
| C | 48–65 | 83–86 |
| H | 5.5–7 | 11–14 |
| O | 30–45 | <1 |
| N | 0–0.3 | <0.3 |
| S | <0.05 | <3.0 |
| HHV (MJ/kg) | 16–19 | 40 |
| Viscosity (cP, 50 ℃) | 40–100 | 180 |
| Ash (wt.%) | 0–0.2 | 0.1 |
| Solids (wt.%) | 0.2–1 | 1 |
| Distillation residue (wt.%) | <50 | 1 |
| NOx emission | <0.7 | 1.4 |
| SOx emission | 0 | 0.28 |
| Upgrading Approach | Advantages | Disadvantages |
|---|---|---|
| Emulsion | Easy-operation | High-cost of surfactant; high energy consumption; cannot remove unfavourable substances |
| Solvent addition | Easy-operation | Cannot remove unfavourable substances |
| Hydrocracking | The formation of light components | The requirement for a high-pressure resistant reactor, coke formation; catalyst deactivation; high-pressure H2 is needed |
| Hydrotreatment | Well-established treatment in oil refineries; effectively removes heteroatoms | The requirement for a high-pressure resistant reactor; coke formation; catalyst deactivation; high-pressure H2 is needed |
| Steam reforming | Highly energy-dense H2 as the main product | The requirement for high-temperature-resistant reactor |
| Supercritical fluids | Significantly lowers the viscosity and increases HHV of bio-oil | High-cost of the solvent; requirement for high-pressure resistant reactor |
| Esterification | Easy-operation; moderate reaction conditions | Cannot remove nitrogenates and thus cannot be used for algae-derived bio-oil |
| Catalyst | Oxygenate Compound | Deoxygenated Compound | Reference |
|---|---|---|---|
| NiMoS | Phenol | Cyclohexane, Benzene, Cyclohexene | [68] |
| MoS2 | Guaiacol | Phenol, cyclohexane, benzene, methyl cyclopentane | [69] |
| MoS | Phenol | Benzene, cyclohexene, cyclohexane | [70] |
| NiMoS | 2,3-dihudro benzofuran | 2-Ethylphenol, ethyl cyclohexane | [71] |
| NiMo, CoMo | Waste cooking oils | n-paraffins, isoparaffins, a small amount of olefins | [72] |
| CoMoWS/SBA-15 | Anisole | Phenol, cresol, xylenol | [73] |
| CoMoS/MgO | Phenol | 2-Cyclohexylphenol, cyclohexylbenzene, cyclohexanol | [74] |
| CoMoS | Phenol | Benzene, cyclohexane, Cyclohexene | [70] |
| Ni-Mo | 2,3-dihydro benzofuran | Ethyl benzene, ethyl-cyclohexane, ethylcyclohexene, methylcyclohexane, and cyclohexane | [71] |
| ReS2/AC | Guaiacol | Phenol, catechol | [75] |
| Feedstock | Catalyst | Conclusions | Ref. |
|---|---|---|---|
| Eugenol + Light fraction of bio-oil | Ni-based | Highest product selectivity of propyl-cyclohexane was 67.9% and all eugenol can be converted Al-SBA-15 was mixed with ZSM-5 as the support to enhance the catalytic activity The presence of acetic acid and furfural affected the HDO of eugenol | [90] |
| Fast pyrolysis bio-oil | MoS2 | 90% of O from the bio-oil was removed The formation of coke was not obvious | [66] |
| Phenolics and bio-oil | Ru-based | Guaiacol was converted into cycloalkanes 29.1% of hydrocarbons and 41.3% of alkylphenols were produced from HDO of bio-oil Dispersed Ru and moderate acid site improved the HDO reaction | [91] |
| Fast pyrolysis bio-oil | β-Mo2C | The HHV of upgraded bio-oil was 41.1 MJ/kg; Catalyst was highly stable upon recycling and it was easy to recycle | [81] |
| m-Cresol | Ru-based | Ru/ZSM-5 was the best catalyst in terms of the 100% product yield, 240 h of lifetime, and was used for 4 cycles | [92] |
| Fast pyrolysis bio-oil | NbMo/C | HDO at 300 °C for 60 min lower the O and moisture content to 19% and 0.1%, respectively; Coke formation was observed. The viscosity, density, and stability of upgraded bio-oil were comparable to those of petroleum | [93] |
| Cyclohexanone | Co-based | Catalyst had high catalytic activity and selectivity for cyclohexanone to produce cyclohexane, cyclohexene, benzene, and cyclohexylbenzene 100% of hydrocarbon selectivity and 89% of cyclohexanone conversion were obtained at 400 °C and 15 bar of H2 | [94] |
| Cyclohexanone | NiMo-based | C6, C7, C12 cyclic, aromatic, and bicyclics were the main products; 87% of cyclohexanone conversion was obtained at 400 °C and 8 bar of H2 | [95] |
| Fast pyrolysis bio-oil | Pt and MoO3-based and Mo-based industrial catalyst | All catalysts reduced O content of bio-oil to 7–12 wt.%; Pt catalyst showed better performance to lower acidic value; more coke was formed using Mo-based industrial catalyst | [96] |
| Phenol | Rh, Pd, Ni-based | The order for the catalytic activity was: Pd > Rh > Ni when performing reduction at 300 °C while no difference in the catalytic activity at reduction of 500 °C | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attia, M.; Farag, S.; Chaouki, J. Upgrading of Oils from Biomass and Waste: Catalytic Hydrodeoxygenation. Catalysts 2020, 10, 1381. https://doi.org/10.3390/catal10121381
Attia M, Farag S, Chaouki J. Upgrading of Oils from Biomass and Waste: Catalytic Hydrodeoxygenation. Catalysts. 2020; 10(12):1381. https://doi.org/10.3390/catal10121381
Chicago/Turabian StyleAttia, Mai, Sherif Farag, and Jamal Chaouki. 2020. "Upgrading of Oils from Biomass and Waste: Catalytic Hydrodeoxygenation" Catalysts 10, no. 12: 1381. https://doi.org/10.3390/catal10121381
APA StyleAttia, M., Farag, S., & Chaouki, J. (2020). Upgrading of Oils from Biomass and Waste: Catalytic Hydrodeoxygenation. Catalysts, 10(12), 1381. https://doi.org/10.3390/catal10121381