Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition

 ,
,  , , , , ,
, , , , , 
Abstract
1. Introduction
2. Results and Discussion
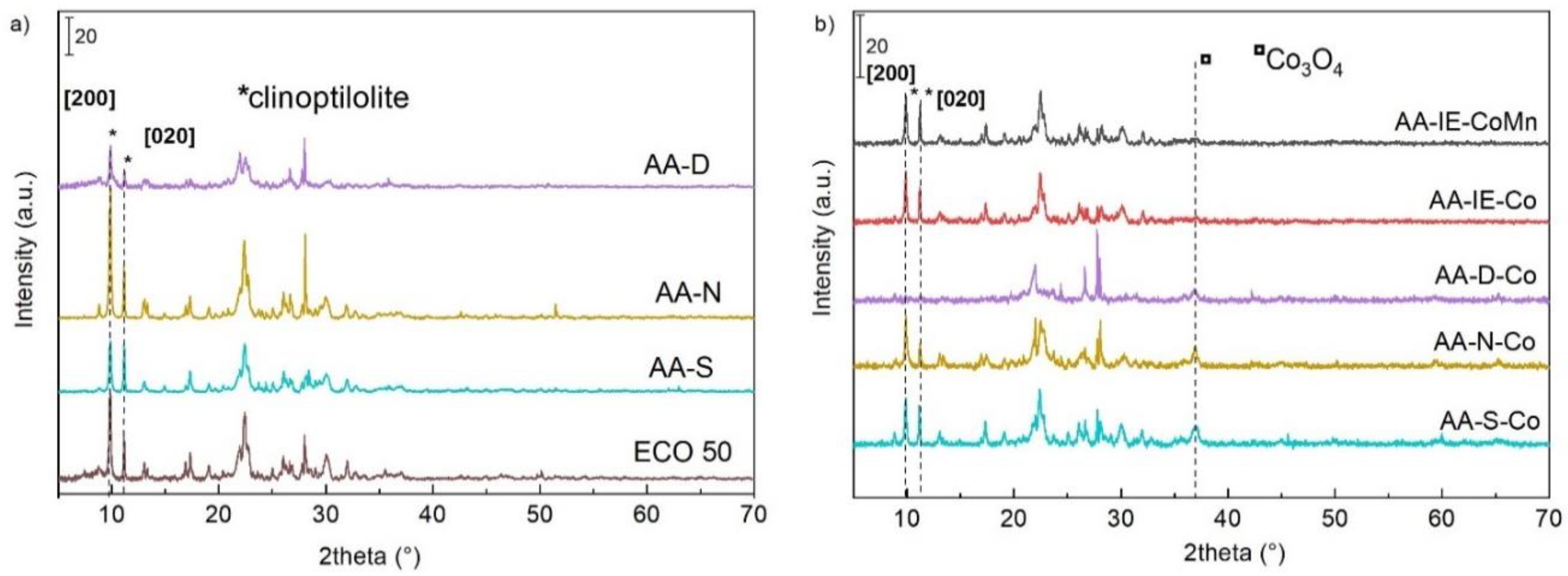
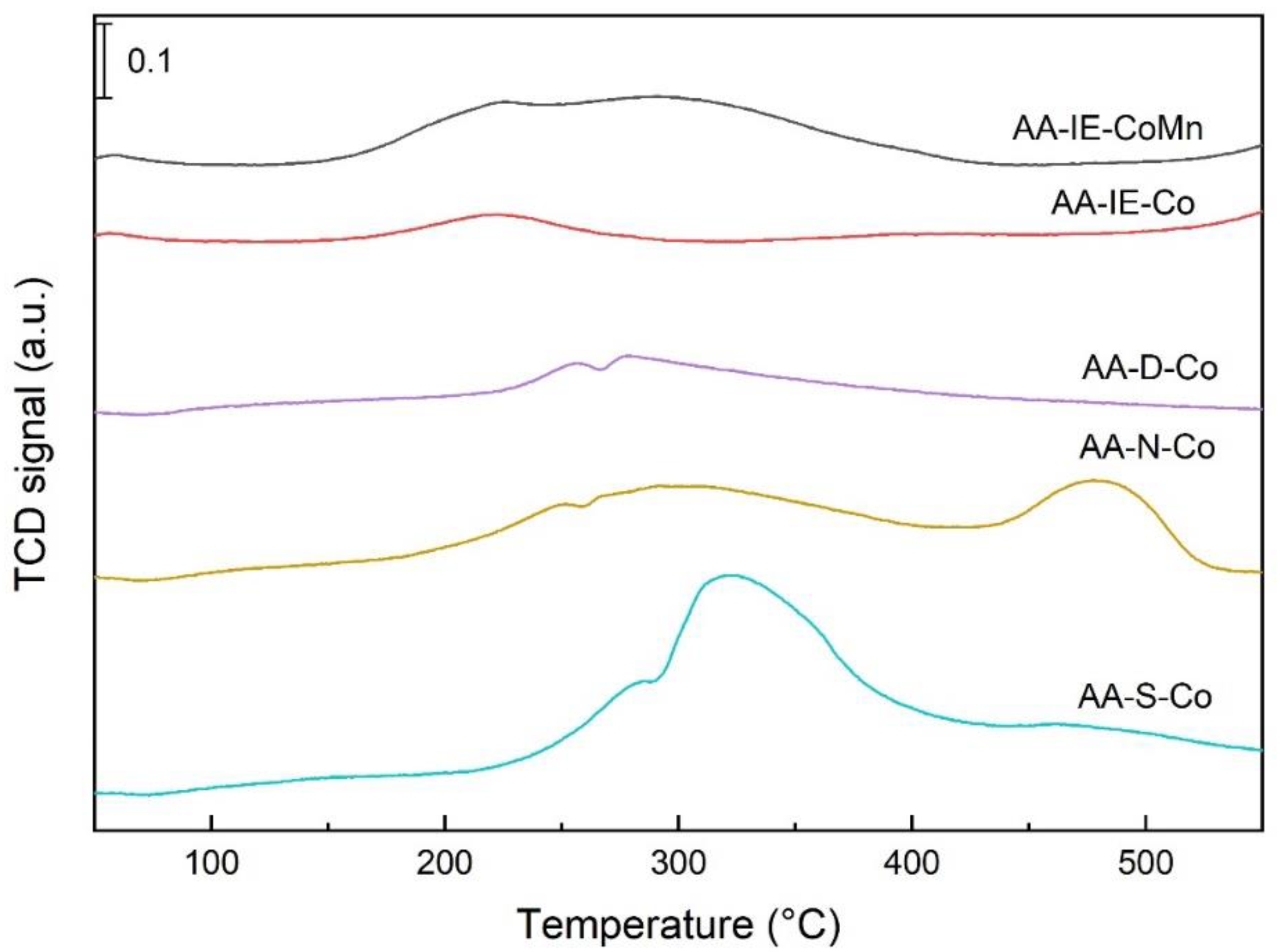
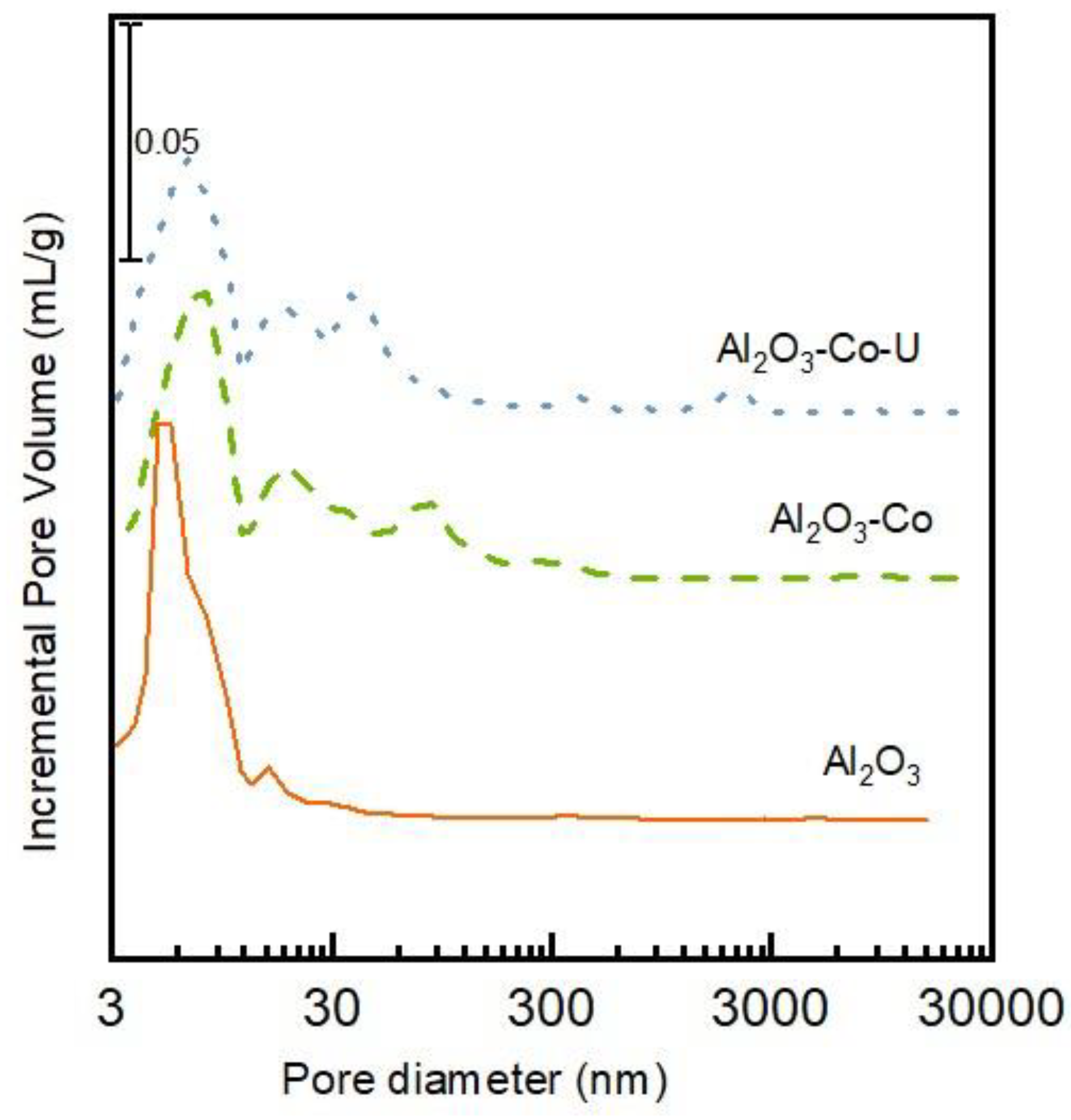
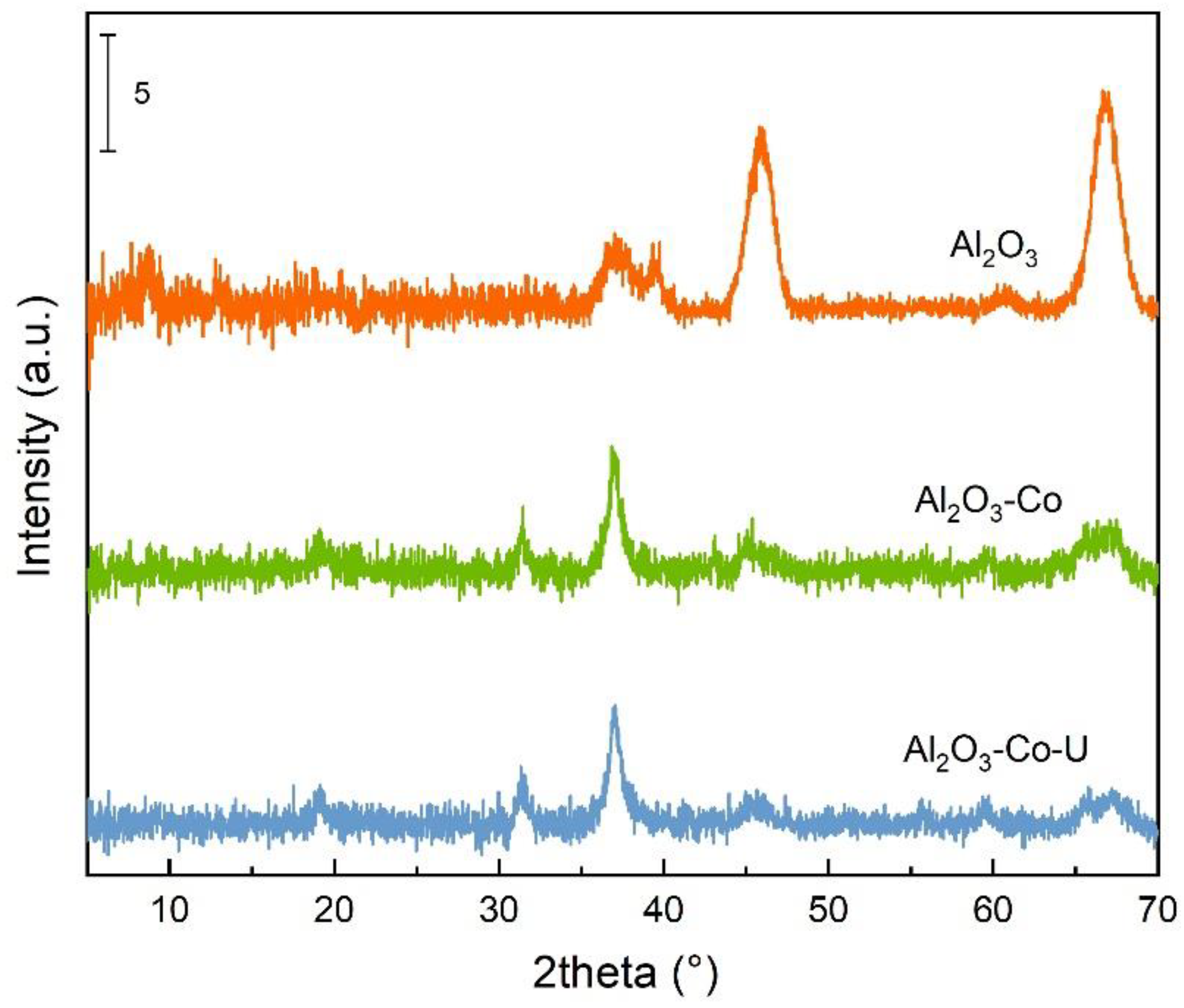
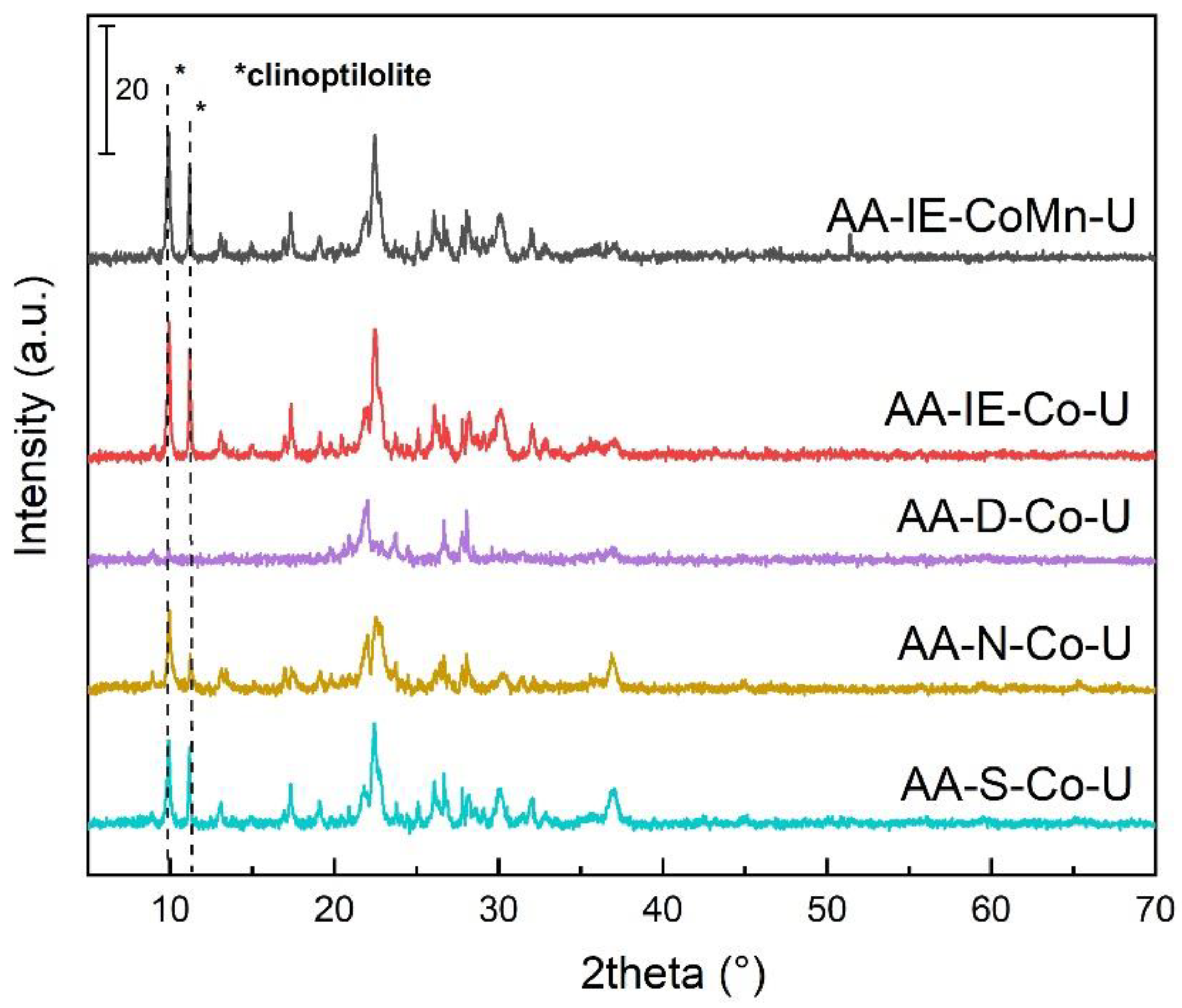
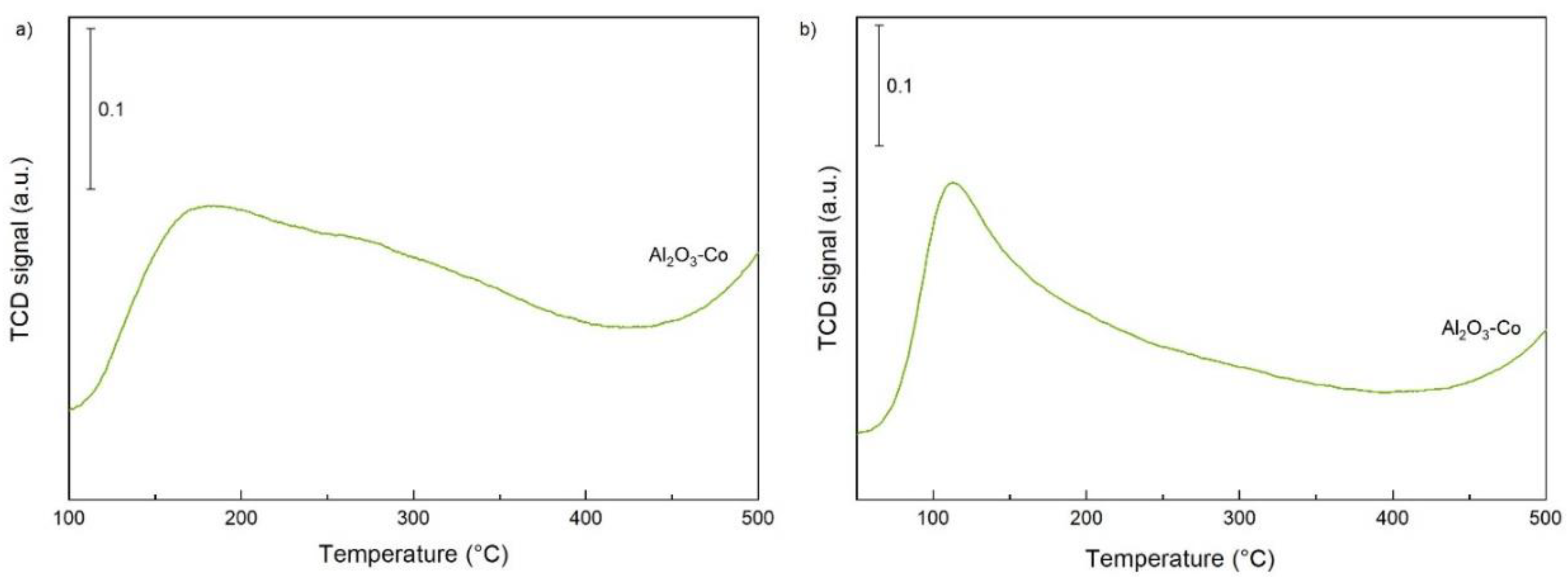
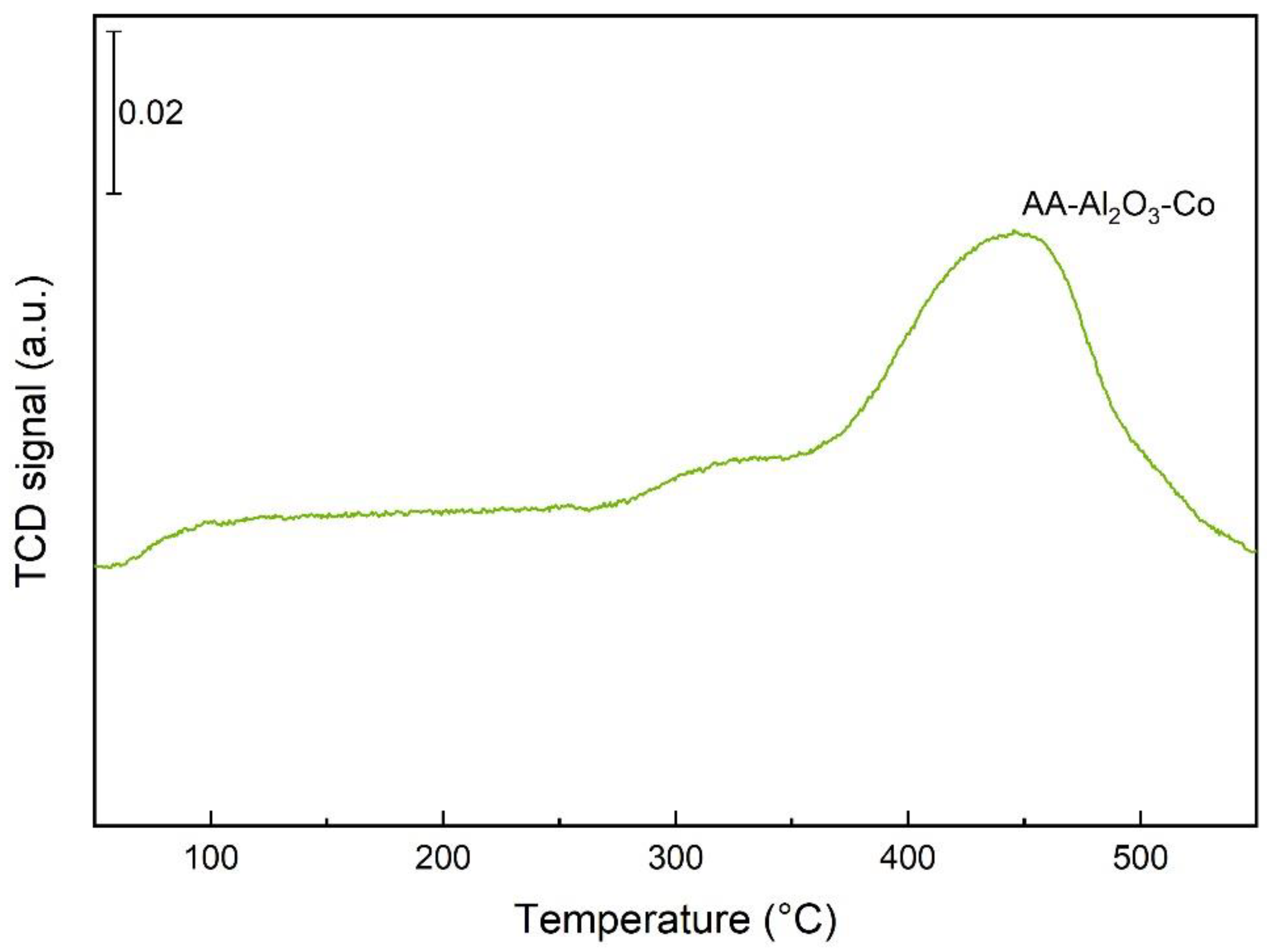
2.1. Characterisation of the Catalysts and Supports
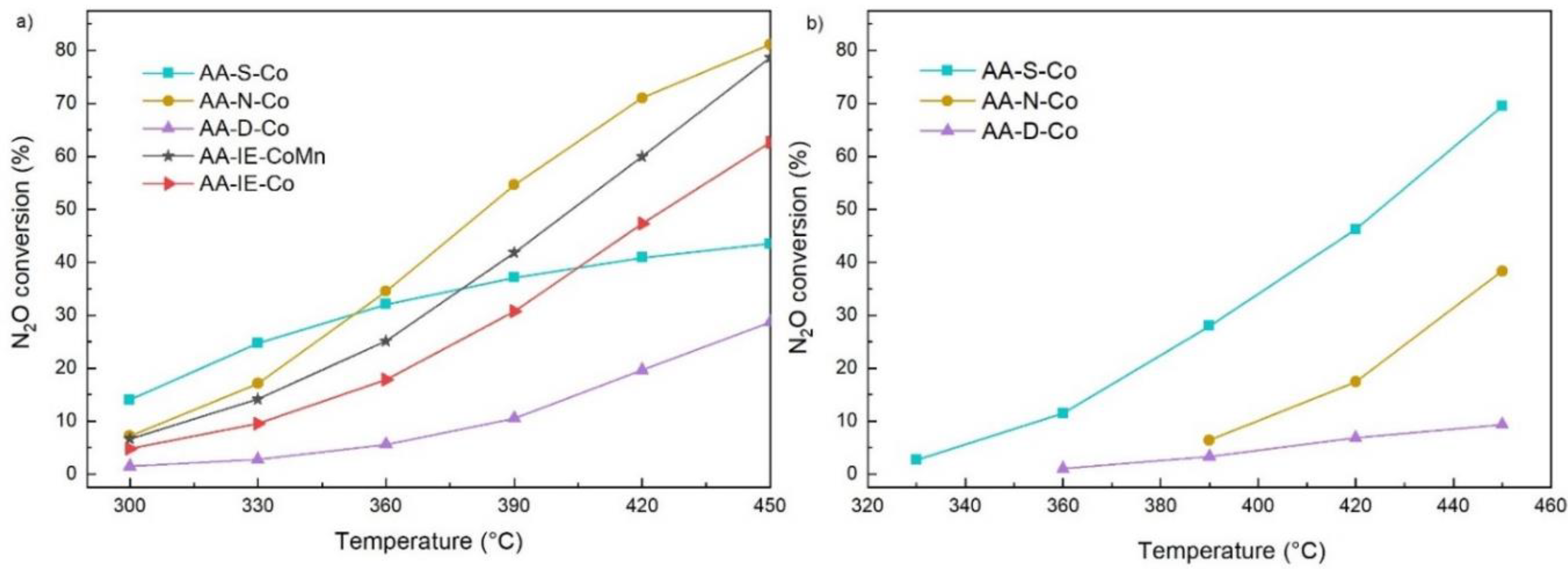
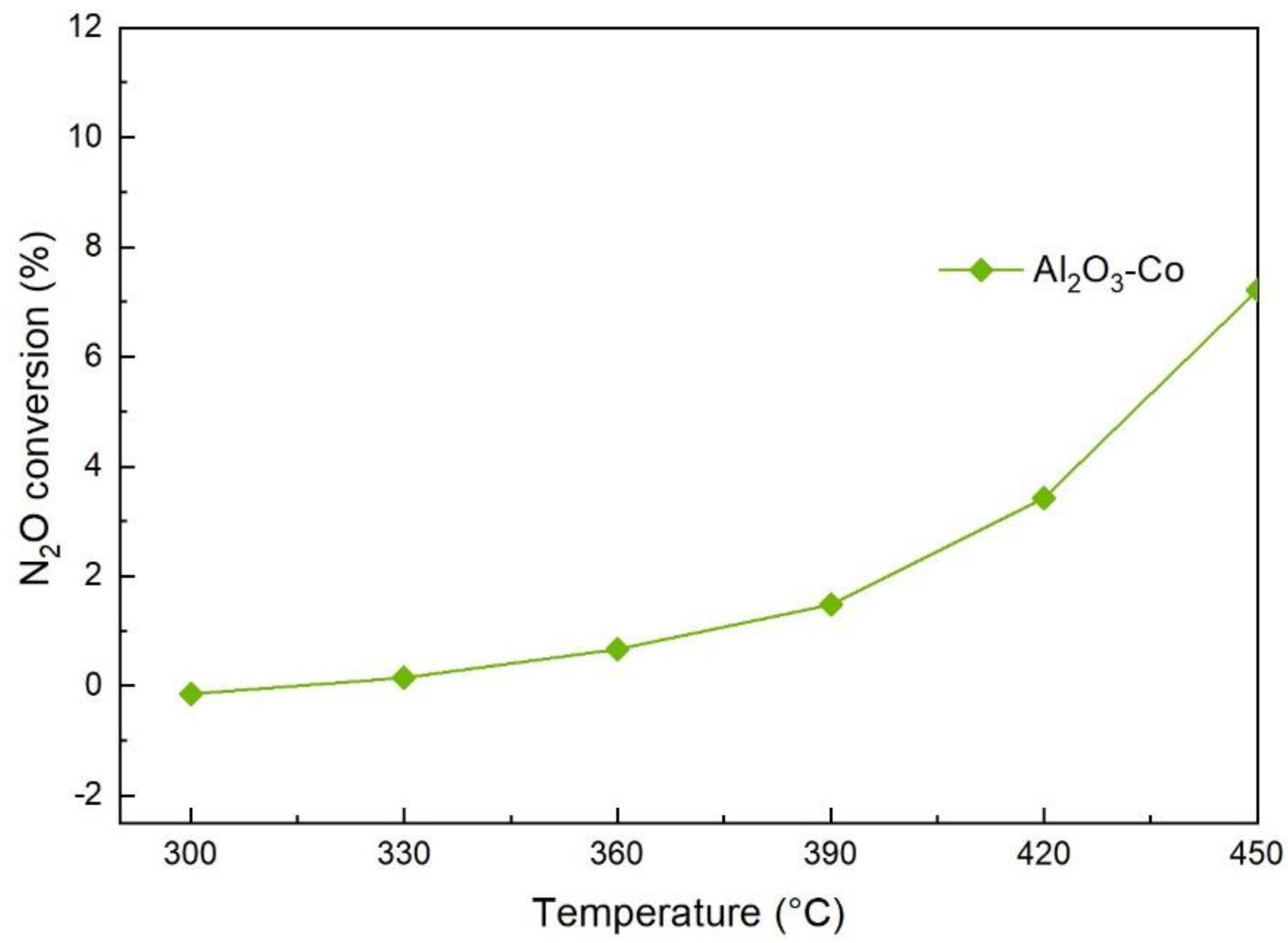
2.2. N2O Catalytic Decomposition
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Zeolite Foams
3.3. Post-Synthesis Modifications and Catalyst Synthesis
3.4. Characterisation of Supports and Prepared Catalysts
3.5. Catalytic Tests
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Chemical Composition (wt.%) | Si/Al Ratio (mol/mol) | Co/AM * Ratio | SSA ** (m2/g) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Si | Al | K | Na | Ca | Fe | Co | Mn | ||||
| Al2O3-Co | 0.1 | 49.0 | - | 0.3 | 0.1 | - | 5.2 | - | - | - | 212.9 |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3-Co | 296 | 173 | 178 | 60 | 283 | 118 | 40 | - | - | - |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3-Co | 145 | 109 | 31 | 21 | 144 | 89 | 61 | 263 | 25 | 18 |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Al2O3-Co | 109 | - | - | - | 435 | 85 | 80 | 479 | 24 | 20 |






References
- Choya, A.; De Rivas, B.; Gutiérrez-Ortiz, J.I.; Velasco, J.R.G.; López-Fonseca, R. Synthesis, Characterization and Kinetic Behavior of Supported Cobalt Catalysts for Oxidative after-Treatment of Methane Lean Mixtures. Materials 2019, 12, 3174. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-K.; Wang, C.-B.; Chiu, H.-C.; Chien, S.-H. In situ FTIR Study of Cobalt Oxides for the Oxidation of Carbon Monoxide. Catal. Lett. 2003, 86, 63–68. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Wang, L.; Zhang, C.; He, H. Catalytic oxidation of formaldehyde over manganese oxides with different crystal structures. Catal. Sci. Technol. 2015, 5, 2305–2313. [Google Scholar] [CrossRef]
- Hrachovcová, K.; Tišler, Z.; Svobodová, E.; Šafář, J. Modified Alkali Activated Zeolite Foams with Improved Textural and Mechanical Properties. Minerals 2020, 10, 483. [Google Scholar] [CrossRef]
- Atkins, P.W.; Jones, L.L. Chemistry: Molecules, Matter, and Change, 3rd ed.; W. H. Freeman and Company: New York, NY, USA, 1997. [Google Scholar]
- Hidalgo-Herrador, J.M.; Tišler, Z.; Vráblík, A.; Velvarská, R.; Lederer, J. Acid-modified phonolite and foamed zeolite as supports for NiW catalysts for deoxygenation of waste rendering fat. React. Kinet. Mech. Catal. 2019, 126, 773–793. [Google Scholar] [CrossRef]
- Tišler, Z.; Velvarská, R.; Skuhrovcová, L.; Pelíšková, L.; Akhmetzyanova, U. Key Role of Precursor Nature in Phase Composition of Supported Molybdenum Carbides and Nitrides. Materials 2019, 12, 415. [Google Scholar] [CrossRef] [PubMed]
- Tišler, Z.; Horacek, J.; Safar, J.; Velvarska, R.; Peliskova, L.; Kocik, J.; Gherib, Y.; Marklova, K.; Bulánek, R.; Kubička, D. Clinoptilolite foams prepared by alkali activation of natural zeolite and their post-synthesis modifications. Microporous Mesoporous Mater. 2019, 282, 169–178. [Google Scholar] [CrossRef]
- Maniak, G.; Stelmachowski, P.; Kotarba, A.; Sojka, Z.; Pérez, V.R.; López, A.B. Rationales for the selection of the best precursor for potassium doping of cobalt spinel based deN2O catalyst. Appl. Catal. B Environ. 2013, 136, 302–307. [Google Scholar] [CrossRef]
- Sun, M.; Wang, L.; Feng, B.; Zhang, Z.; Lu, G.; Guo, Y. The role of potassium in K/Co3O4 for soot combustion under loose contact. Catal. Today 2011, 175, 100–105. [Google Scholar] [CrossRef]
- Obalová, L.; Pacultová, K.; Balabánová, J.; Jirátová, K.; Bastl, Z.; Valášková, M.; Lacný, Z.; Kovanda, F. Effect of Mn/Al ratio in Co–Mn–Al mixed oxide catalysts prepared from hydrotalcite-like precursors on catalytic decomposition of N2O. Catal. Today 2007, 119, 233–238. [Google Scholar] [CrossRef]
- Ramírez, J.P.; Kapteijn, F.; Schöffel, K.; Moulijn, J.A. Formation and control of N2O in nitric acid production: Where do we stand today? Appl. Catal. B Environ. 2003, 44, 117–151. [Google Scholar] [CrossRef]
- Kapteijn, F.; Marbán, G.; Mirasol, J.R.; Moulijn, J.A. Kinetic Analysis of the Decomposition of Nitrous Oxide over ZSM 5 Catalysts. J. Catal. 1997, 167, 256–265. [Google Scholar] [CrossRef]
- Asano, K.; Ohnishi, C.; Iwamoto, S.; Shioya, Y.; Inoue, M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl. Catal. B Environ. 2008, 78, 242–249. [Google Scholar] [CrossRef]
- Maniak, G.; Stelmachowski, P.; Zasada, F.; Piskorz, W.; Kotarba, A.; Sojka, Z. Guidelines for optimization of catalytic activity of 3d transition metal oxide catalysts in N2O decomposition by potassium promotion. Catal. Today 2011, 176, 369–372. [Google Scholar] [CrossRef]
- Maniak, G.; Stelmachowski, P.; Stanek, J.J.; Kotarba, A.; Sojka, Z. Catalytic properties in N2O decomposition of mixed cobalt–iron spinels. Catal. Commun. 2011, 15, 127–131. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Maniak, G.; Kotarba, A.; Sojka, Z. Strong electronic promotion of Co3O4 towards N2O decomposition by surface alkali dopants. Catal. Commun. 2009, 10, 1062–1065. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, Y.; Wang, A.; Li, L.; Wang, X.; Zhang, T. N2O decomposition over K-promoted Co-Al catalysts prepared from hydrotalcite-like precursors. Appl. Catal. B Environ. 2009, 89, 391–397. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, Q.; He, C.; Wang, Y.; Cheng, J.; Hao, Z. CoMOR zeolite catalyst prepared by buffered ion exchange for effective decomposition of nitrous oxide. J. Hazard. Mater. 2011, 192, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Pasha, N.; Lingaiah, N.; Babu, N.S.; Reddy, P.S.S.; Prasad, P.S. Studies on cesium doped cobalt oxide catalysts for direct N2O decomposition in the presence of oxygen and steam. Catal. Commun. 2008, 10, 132–136. [Google Scholar] [CrossRef]
- Da Cruz, R.; Mascarenhas, A.; Andrade, H.M.C. Co-ZSM-5 catalysts for N2O decomposition. Appl. Catal. B Environ. 1998, 18, 223–231. [Google Scholar] [CrossRef]
- Shen, Q.; Li, L.; Li, J.; Tian, H.; Hao, Z. A study on N2O catalytic decomposition over Co/MgO catalysts. J. Hazard. Mater. 2009, 163, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Karásková, K.; Obalová, L.; Jirátová, K.; Kovanda, F. Effect of promoters in Co–Mn–Al mixed oxide catalyst on N2O decomposition. Chem. Eng. J. 2010, 160, 480–487. [Google Scholar] [CrossRef]
- Klyushina, A.; Pacultová, K.; Karásková, K.; Jirátová, K.; Ritz, M.; Fridrichová, D.; Volodarskaja, A.; Obalová, L. Effect of preparation method on catalytic properties of Co-Mn-Al mixed oxides for N2O decomposition. J. Mol. Catal. A Chem. 2016, 425, 237–247. [Google Scholar] [CrossRef]
- Campa, M.C.; Indovina, V.; Pietrogiacomi, D. The dependence of catalytic activity for N2O decomposition on the exchange extent of cobalt or copper in Na-MOR, H-MOR and Na-MFI. Appl. Catal. B Environ. 2009, 91, 347–354. [Google Scholar] [CrossRef]
- Fellah, M.F.; Onal, I. N2O decomposition on Fe- and Co-ZSM-5: A density functional study. Catal. Today 2008, 137, 410–417. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Jirátová, K.; Kovanda, F. Effect of potassium in calcined Co–Mn–Al layered double hydroxide on the catalytic decomposition of N2O. Appl. Catal. B Environ. 2009, 90, 132–140. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Ciura, K.; Grzybek, G. Morphology-dependent reactivity of cobalt oxide nanoparticles in N2O decomposition. Catal. Sci. Technol. 2016, 6, 5554–5560. [Google Scholar] [CrossRef]
- Gudyka, S.; Grzybek, G.; Gryboś, J.; Indyka, P.; Leszczyński, B.; Kotarba, A.; Sojka, Z. Enhancing the deN2O activity of the supported Co3O4|α-Al2O3 catalyst by glycerol-assisted shape engineering of the active phase at the nanoscale. Appl. Catal. B Environ. 2017, 201, 339–347. [Google Scholar] [CrossRef]
- Grzybek, G.; Wójcik, S.; Legutko, P.; Gryboś, J.; Indyka, P.; Leszczyński, B.; Kotarba, A.; Sojka, Z. Thermal stability and repartition of potassium promoter between the support and active phase in the K Co2.6Zn0.4O4|α Al2O3 catalyst for N2O decomposition: Crucial role of activation temperature on catalytic performance. Appl. Catal. B Environ. 2017, 205, 597–604. [Google Scholar] [CrossRef]
- Boroń, P.; Chmielarz, L.; Casale, S.; Calers, C.; Krafft, J.-M.; Dzwigaj, S. Effect of Co content on the catalytic activity of CoSiBEA zeolites in N2O decomposition and SCR of NO with ammonia. Catal. Today 2015, 258, 507–517. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, R.; Chen, B.; Li, Y.; Li, Y. Comparative study on the direct decomposition of nitrous oxide over M (Fe, Co, Cu)–BEA zeolites. J. Catal. 2012, 294, 99–112. [Google Scholar] [CrossRef]
- Smeets, P.J.; Meng, Q.; Corthals, S.; Leeman, H.; Schoonheydt, R.A. Co–ZSM-5 catalysts in the decomposition of N2O and the SCR of NO with CH4: Influence of preparation method and cobalt loading. Appl. Catal. B Environ. 2008, 84, 505–513. [Google Scholar] [CrossRef]
- Ghahri, A.; Golbabaei, F.; Vafajoo, L.; Mireskandari, S.M.; Yaseri, M.; Shahtaheri, S.J. Removal of Greenhouse Gas (N2O) by Catalytic Decomposition on Natural Clinoptilolite Zeolites Impregnated with Cobalt. Int. J. Environ. Res. 2017, 11, 327–337. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Wach, A.; Kustrowski, P.; Kutláková, K.M.; Michalik, S.; Jirátová, K. Alkali metals as promoters in Co–Mn–Al mixed oxide for N2O decomposition. Appl. Catal. A Gen. 2013, 462–463, 227–235. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Soliman, S.A.; Abdellah, S.E. Pure and Ni-substituted Co3O4 spinel catalysts for direct N2O decomposition. Chin. J. Catal. 2014, 35, 1105–1112. [Google Scholar] [CrossRef]
- Del Río, L.; Marbán, G. Stainless steel wire mesh supported potassium doped cobalt oxide catalysts for the catalytic decomposition of nitrous oxide. Appl. Catal. B Environ. 2012, 126, 39–46. [Google Scholar] [CrossRef]
- Karásková, K.; Obalová, L.; Kovanda, F. N2O catalytic decomposition and temperature programmed desorption tests on alkali metals promoted Co–Mn–Al mixed oxide. Catal. Today 2011, 176, 208–211. [Google Scholar] [CrossRef]
- Žaneta, C.; Obalová, L.; Kovanda, F.; Legut, D.; Titov, A.; Ritz, M.; Fridrichová, D.; Michalik, S.; Kuśtrowski, P.; Jirátová, K. Effect of precursor synthesis on catalytic activity of Co3O4 in N2O decomposition. Catal. Today 2015, 257, 18–25. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst. Appl. Catal. B Environ. 2007, 75, 167–174. [Google Scholar] [CrossRef]
- Piskorz, W.; Zasada, F.; Stelmachowski, P.; Kotarba, A.; Sojka, Z. Decomposition of N2O over the surface of cobalt spinel: A DFT account of reactivity experiments. Catal. Today 2008, 137, 418–422. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Promotion effect of residual K on the decomposition of N2O over cobalt–cerium mixed oxide catalyst. Catal. Today 2007, 126, 449–455. [Google Scholar] [CrossRef]
- Klegova, A.; Inayat, A.; Indyka, P.; Gryboś, J.; Sojka, Z.; Pacultová, K.; Schwieger, W.; Volodarskaja, A.; Kuśtrowski, P.; Rokicińska, A.; et al. Cobalt mixed oxides deposited on the SiC open-cell foams for nitrous oxide decomposition. Appl. Catal. B Environ. 2019, 255, 255. [Google Scholar] [CrossRef]
- Iwanek, E.; Krawczyk, K.; Petryk, J.; Sobczak, J.W.; Kaszkur, Z. Direct nitrous oxide decomposition with CoOx CeO2 catalysts. Appl. Catal. B Environ. 2011, 106, 416–422. [Google Scholar] [CrossRef]
- Thangavelu, K.; Parameswari, K.; Kuppusamy, K.; Haldorai, Y. A simple and facile method to synthesize Co3O4 nanoparticles from metal benzoate dihydrazinate complex as a precursor. Mater. Lett. 2011, 65, 1482–1484. [Google Scholar] [CrossRef]
- Russo, N.; Fino, D.; Saracco, G.; Specchia, V. N2O catalytic decomposition over various spinel-type oxides. Catal. Today 2007, 119, 228–232. [Google Scholar] [CrossRef]
- Grzybek, G.; Stelmachowski, P.; Indyka, P.; Inger, M.; Wilk, M.; Kotarba, A.; Sojka, Z. Cobalt–zinc spinel dispersed over cordierite monoliths for catalytic N2O abatement from nitric acid plants. Catal. Today 2015, 257, 93–97. [Google Scholar] [CrossRef]
- Zhang, R.; Hedjazi, K.; Chen, B.; Li, Y.; Lei, Z.; Liu, N. M(Fe, Co)-BEA washcoated honeycomb cordierite for N 2 O direct decomposition. Catal. Today 2016, 273, 273–285. [Google Scholar] [CrossRef]
- Jirátová, K.; Balabánová, J.; Kovanda, F.; Klegová, A.; Obalová, L.; Fajgar, R. Cobalt Oxides Supported Over Ceria–Zirconia Coated Cordierite Monoliths as Catalysts for Deep Oxidation of Ethanol and N2O Decomposition. Catal. Lett. 2017, 147, 1379–1391. [Google Scholar] [CrossRef]
- Klyushina, A.; Pacultová, K.; Krejčová, S.; Słowik, G.; Jirátová, K.; Kovanda, F.; Ryczkowski, J.; Obalová, L. Advantages of stainless steel sieves as support for catalytic N2O decomposition over K doped Co3O4. Catal. Today 2015, 257 Pt 1, 2–10. [Google Scholar] [CrossRef]
- Klegová, A.; Pacultová, K.; Fridrichová, D.; Volodarskaja, A.; Kovanda, F.; Jirátová, K. Cobalt Oxide Catalysts on Commercial Supports for N2O Decomposition. Chem. Eng. Technol. 2017, 40, 981–990. [Google Scholar] [CrossRef]
- Pacultová, K.; Klegova, A.; Kiška, T.; Fridrichová, D.; Martaus, A.; Rokicińska, A.; Kuśtrowski, P.; Obalová, L. Effect of support on the catalytic activity of Co3O4-Cs deposited on open-cell ceramic foams for N2O decomposition. Mater. Res. Bull. 2020, 129, 110892. [Google Scholar] [CrossRef]
- Rutkowska, M.; Piwowarska, Z.; Micek, E.; Chmielarz, L. Hierarchical Fe-, Cu- and Co-Beta zeolites obtained by mesotemplate-free method. Part I: Synthesis and catalytic activity in N2O decomposition. Microporous Mesoporous Mater. 2015, 209, 54–65. [Google Scholar] [CrossRef]
- Sadek, R.; Chalupka, K.A.; Mierczynski, P.; Rynkowski, J.; Gurgul, J.; Dzwigaj, S. Cobalt Based Catalysts Supported on Two Kinds of Beta Zeolite for Application in Fischer-Tropsch Synthesis. Catalysts 2019, 9, 497. [Google Scholar] [CrossRef]
- Kang, S.-H.; Ryu, J.-H.; Kim, J.-H.; Jang, I.H.; Kim, A.R.; Han, G.Y.; Bae, J.W.; Ha, K.-S. Role of ZSM5 Distribution on Co/SiO2 Fischer–Tropsch Catalyst for the Production of C5–C22 Hydrocarbons. Energy Fuels 2012, 26, 6061–6069. [Google Scholar] [CrossRef]
- Kurian, M.; Thankachan, S.; Nair, D.S.; Aswathy, E.K.; Babu, A.; Thomas, A.; KT, B.K. Structural, magnetic, and acidic properties of cobalt ferrite nanoparticles synthesised by wet chemical methods. J. Adv. Ceram. 2015, 4, 199–205. [Google Scholar] [CrossRef]







| Sample | Chemical Composition (wt.%) | Si/Al Ratio (mol/mol) | Co/AM * Ratio | SSA ** (m2/g) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Si | Al | K | Na | Ca | Fe | Co | Mn | ||||
| AA-S | 33.4 | 5.5 | 7.2 | 2.9 | 2.3 | 0.9 | - | - | 5.8 | - | 15.0 |
| AA-N | 37.8 | 6.3 | 1.6 | 0.3 | 1.7 | 1.1 | - | - | 5.8 | - | 23.4 |
| AA-D | 42.0 | 3.4 | 1.1 | 0.2 | 0.5 | 0.7 | - | - | 11.8 | - | 111.7 |
| AA-S-Co | 30.5 | 5.1 | 6.6 | 2.6 | 2.1 | 0.9 | 6.4 | - | 5.7 | 0.4 | 13.5 |
| AA-N-Co | 35.0 | 6.0 | 1.4 | 0.2 | 1.6 | 1.0 | 5.7 | - | 5.6 | 2.1 | 27.7 |
| AA-D-Co | 38.7 | 3.5 | 1.1 | 0.2 | 0.4 | 0.7 | 5.4 | - | 10.6 | 2.5 | 49.9 |
| AA-IE-Co | 32.0 | 5.2 | 4.1 | 0.5 | 1.7 | 0.9 | 6.3 | - | 5.9 | 0.9 | 115.8 |
| AA-IE-CoMn | 31.9 | 5.2 | 4.2 | 0.5 | 1.7 | 0.9 | 5.4 | 0.8 | 5.9 | 0.7 | 118.6 |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| AA-S-Co | 501 | 175 | 501 | 100 | - | - | - | - | - | - |
| AA-N-Co | 1542 | 173 | 1313 | 85 | 390 | 229 | 15 | - | - | - |
| AA-D-Co | 593 | 158 | 446 | 75 | 255 | 70 | 12 | 382 | 77 | 13 |
| AA-IE-Co | 1446 | 178 | 1379 | 95 | 247 | 27 | 2 | 362 | 40 | 3 |
| AA-IE-MnCo | 1211 | 187 | 1211 | 100 | - | - | - | - | - | - |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| AA-S-Co | 213 | 105 | 42 | 20 | 186 | 161 | 76 | 321 | 10 | 14 |
| AA-N-Co | 125 | 110 | 82 | 66 | 363 | 14 | 11 | 437 | 30 | 23 |
| AA-D-Co | 156 | 112 | 36 | 23 | 204 | 44 | 28 | 289 | 76 | 49 |
| AA-IE-Co | 306 | 111 | 215 | 70 | 338 | 91 | 30 | - | - | - |
| AA-IE-MnCo | 408 | 113 | 227 | 56 | 273 | 181 | 44 | - | - | - |
| Sample | cSUM (μmol/g) | Tmax1 (°C) | cmax1 (μmol/g) | Pmax1 * (%) | Tmax2 (°C) | cmax2 (μmol/g) | Pmax2 * (%) | Tmax3 (°C) | cmax3 (μmol/g) | Pmax3 * (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| AA-S-Co | 360 | 289 | 77 | 21 | 330 | 218 | 61 | 478 | 65 | 19 |
| AA-N-Co | 875 | 304 | 704 | 80 | 483 | 171 | 20 | - | - | - |
| AA-D-Co | 260 | 284 | 76 | 29 | 306 | 184 | 71 | - | - | |
| AA-IE-Co | 176 | 222 | 60 | 34 | 418 | 116 | 66 | - | - | - |
| AA-IE-MnCo | 500 | 239 | 337 | 67 | 303 | 163 | 33 | - | - | - |
| Sample | k390 (m3 s−1 kg−1) | k390 * (m3 s−1 m−2) | EA (J mol−1) | EA */EA |
|---|---|---|---|---|
| AA-S-Co | 6.59 × 10−4 | 4.88 × 10−2 | 60,750 | 2.29 |
| AA-N-Co | 1.27 × 10−3 | 4.58 × 10−2 | 87,322 | 1.52 |
| AA-D-Co | 1.89 × 10−4 | 3.80 × 10−3 | 74,589 | 1.77 |
| AA-S-Co * | 1.82 × 10−3 | 1.35 × 10−1 | 139,135 | - |
| AA-N-Co * | 3.65 × 10−4 | 1.32 × 10−2 | 132,484 | - |
| AA-D-Co * | 1.84 × 10−4 | 3.69 × 10−3 | 131,677 | - |
| AA-IE-Co | 5.97 × 10−4 | 5.15 × 10−3 | 69,927 | - |
| AA-IE-MnCo | 8.75 × 10−4 | 7.38 × 10−3 | 71,050 | - |
| Sample | Chemical Composition (wt.%) | Si/Al Ratio (mol/mol) | SSA * (m2/g) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Si | Al | K | Fe | Ca | Na | Mg | Ti | |||
| ECO 50 | 35.2 | 6.7 | 3.5 | 1.3 | 3.1 | 0.2 | 0.5 | 0.1 | 5.2 | 26.4 |
| γ-Al2O3 | 0.1 | 51.8 | - | - | - | 0.4 | 0.3 | - | - | 309.0 |
| Parameter | Value |
|---|---|
| Silicate modulus of the alkaline activator | Ms = 1.51 |
| Water coefficient: | w = 0.7 |
| Alkali content | Me2O * = 8.2 wt.% |
| Alkali molar ratio | Na2O:K2O = 0.56 |
| Catalyst | Support | Preparation Method |
|---|---|---|
| Al2O3-Co | Commercial (γ-Al2O3) | IWI |
| AA-S-Co | AZF (AA-S) | IWI |
| AA-N-Co | Ion exchanged AZF (AA-N) | IWI |
| AA-D-Co | Acid leached AZF (AA-D) | IWI |
| AA-IE-Co | AZF (AA-S) | DIE |
| AA-IE-CoMn | AZF (AA-S) | DIE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tišler, Z.; Klegová, A.; Svobodová, E.; Šafář, J.; Strejcová, K.; Kohout, J.; Šlang, S.; Pacultová, K.; Rodríguez-Padrón, D.; Bulánek, R. Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition. Catalysts 2020, 10, 1398. https://doi.org/10.3390/catal10121398
Tišler Z, Klegová A, Svobodová E, Šafář J, Strejcová K, Kohout J, Šlang S, Pacultová K, Rodríguez-Padrón D, Bulánek R. Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition. Catalysts. 2020; 10(12):1398. https://doi.org/10.3390/catal10121398
Chicago/Turabian StyleTišler, Zdeněk, Anna Klegová, Eliška Svobodová, Jan Šafář, Kateřina Strejcová, Jan Kohout, Stanislav Šlang, Kateřina Pacultová, Daily Rodríguez-Padrón, and Roman Bulánek. 2020. "Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition" Catalysts 10, no. 12: 1398. https://doi.org/10.3390/catal10121398
APA StyleTišler, Z., Klegová, A., Svobodová, E., Šafář, J., Strejcová, K., Kohout, J., Šlang, S., Pacultová, K., Rodríguez-Padrón, D., & Bulánek, R. (2020). Cobalt Based Catalysts on Alkali-Activated Zeolite Foams for N2O Decomposition. Catalysts, 10(12), 1398. https://doi.org/10.3390/catal10121398