CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study



Abstract
:1. Introduction
2. Results
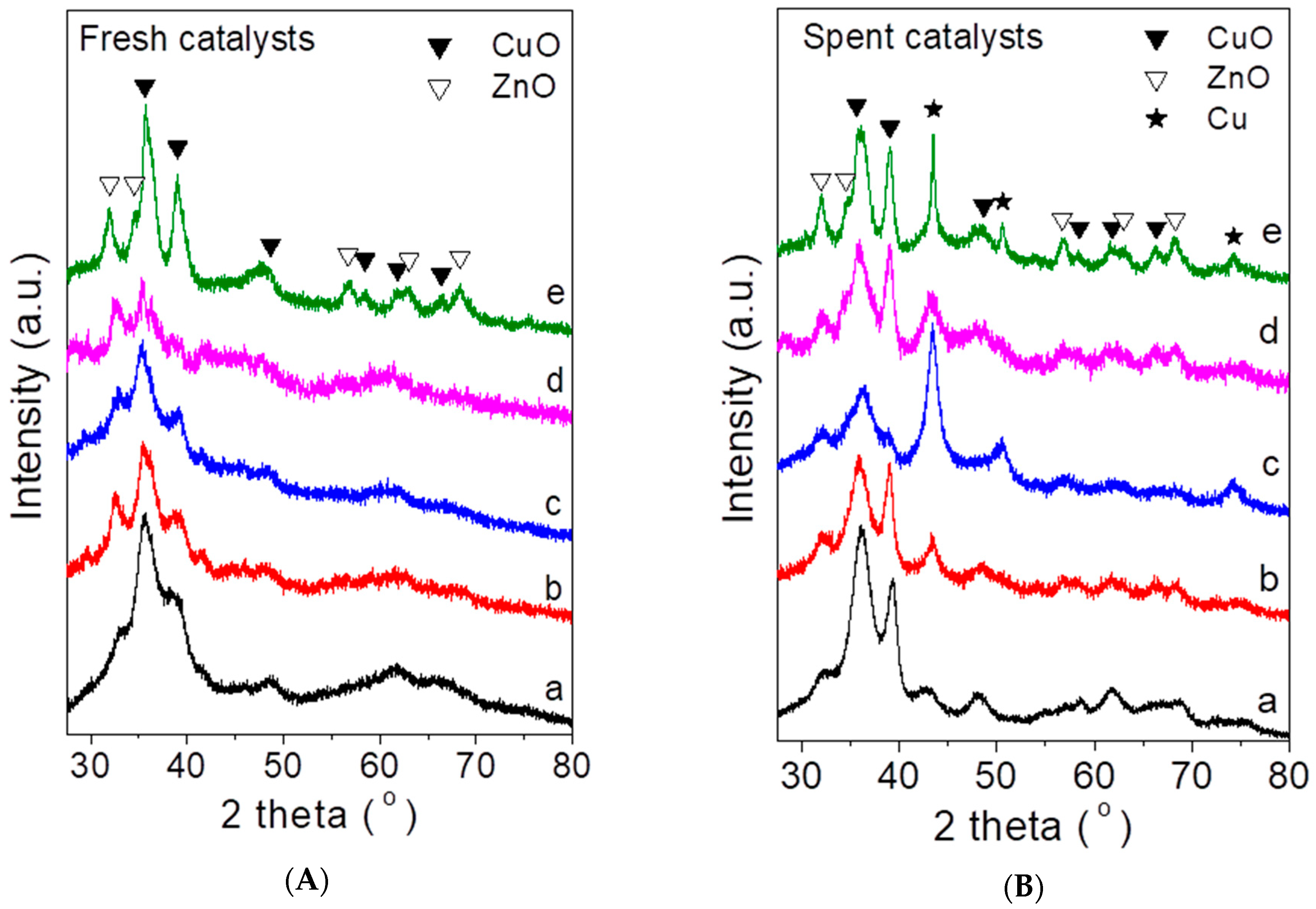
2.1. Characteristics of the Synthesized Catalysts
2.2. Catalytic Performance Tests
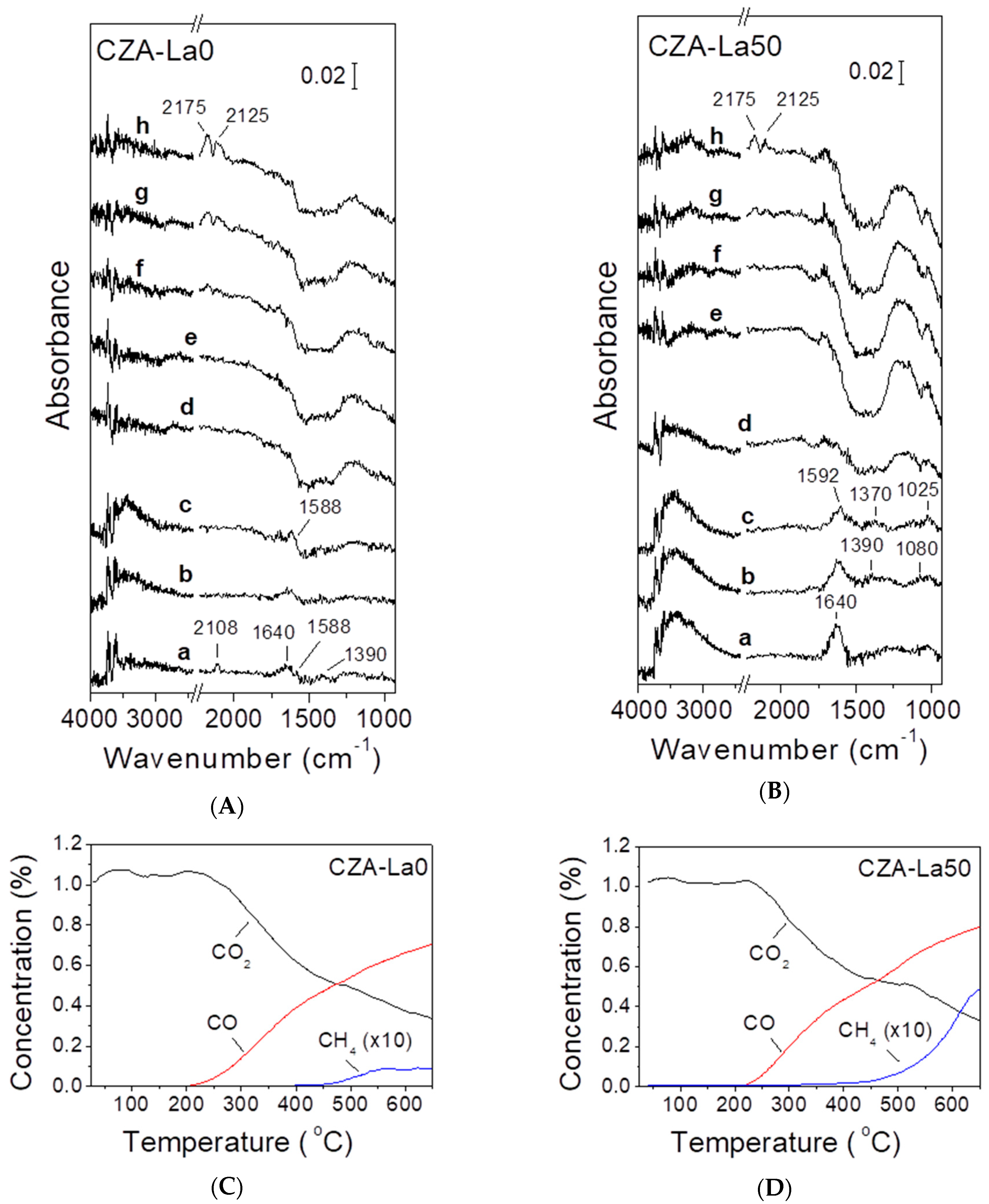
2.3. In Situ DRIFTS Studies
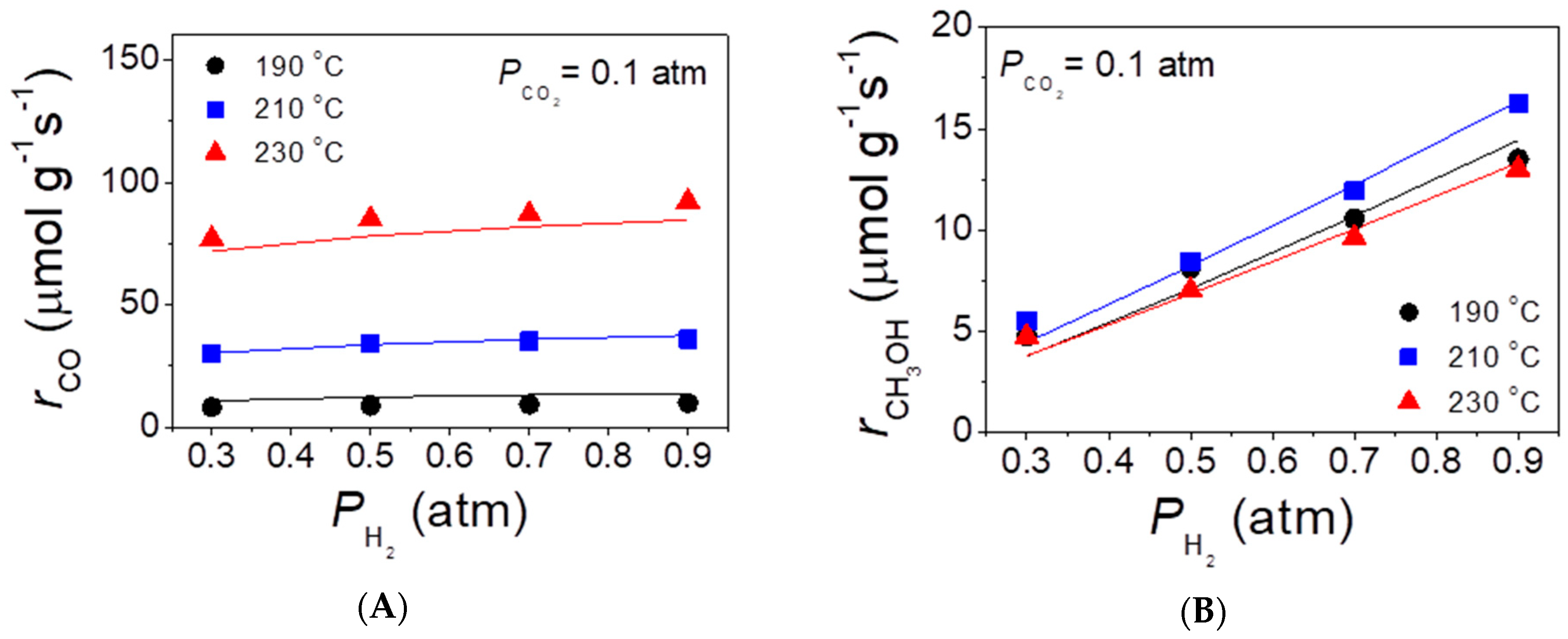
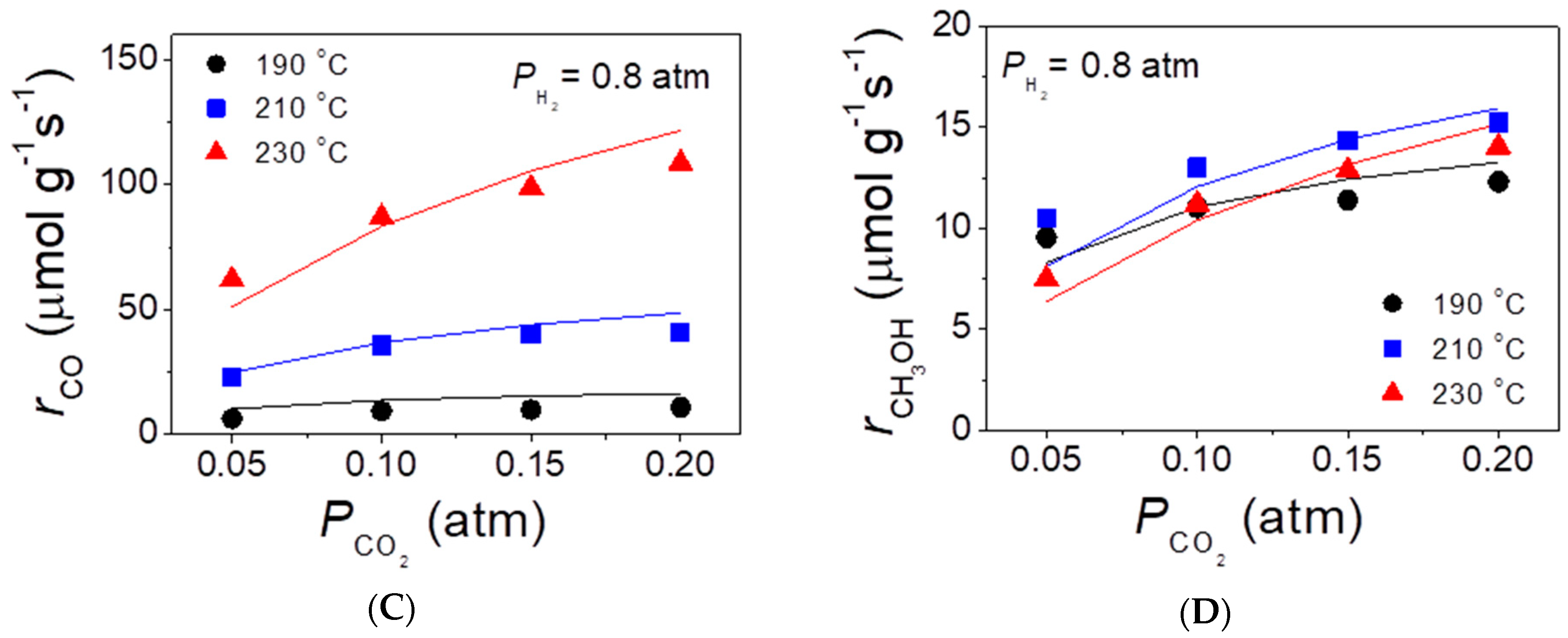
2.4. Reaction Kinetics
3. Discussion
3.1. Effects of La2O3 Addition on the Characteristics and Performance of Catalysts
3.2. Mechanistic Implications and Development of the Kinetic Model
4. Materials and Methods
4.1. Catalyst Preparation
4.2. Catalyst Characterization
4.3. In Situ DRIFTS Experiments
4.4. Transient-MS and CO2-TPD Experiments
4.5. Catalytic Performance Tests and Kinetic Measurements
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Kobayashi, H.; Taylor, J.M.; Mitsuka, Y.; Ogiwara, N.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Kitagawa, H. Charge transfer dependence on CO2 hydrogenation activity to methanol in Cu nanoparticles covered with metal–organic framework systems. Chem. Sci. 2019, 10, 3289–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowker, M. Methanol synthesis from CO2 hydrogenation. ChemCatChem 2019, 11, 4238–4246. [Google Scholar] [CrossRef] [Green Version]
- Tabatabaei, J.; Sakakini, B.H.; Waugh, K.C. On the mechanism of methanol synthesis and the water-gas shift reaction on ZnO. Catal. Lett. 2006, 110, 77–84. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, J.; Uchijima, T.; Kanai, Y.; Fujitani, T. The role of ZnO in Cu/ZnO methanol synthesis catalysts. Catal. Today 1996, 28, 223–230. [Google Scholar] [CrossRef]
- Yoshihara, J.; Parker, S.C.; Schafer, A.; Campbell, C.T. Methanol synthesis and reverse water-gas shift kinetics over clean polycrystalline copper. Catal. Lett. 1995, 31, 313–324. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of methanol synthesis on Cu through CO2 and CO hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Huš, M.; Kopač, D.; Štefančič, N.S.; Jurković, D.L.; Dasireddy, V.D.B.C.; Likozar, B. Unravelling the mechanisms of CO2 hydrogenation to methanol on Cu-based catalysts using first-principles multiscale modelling and experiments. Catal. Sci. Technol. 2017, 7, 5900–5913. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yao, H.; Jiang, Z.; Fang, T. Theoretical study of methanol synthesis from CO2 hydrogenation on PdCu3(111) surface. Appl. Surf. Sci. 2018, 451, 333–345. [Google Scholar] [CrossRef]
- Kakumoto, T.; Watanabe, T. A theoretical study for methanol synthesis by CO2 hydrogenation. Catal. Today 1997, 36, 39–44. [Google Scholar] [CrossRef]
- Liu, L.; Fan, F.; Bai, M.; Xue, F.; Ma, X.; Jiang, Z.; Fang, T. Mechanistic study of methanol synthesis from CO2 hydrogenation on Rh-doped Cu(111) surfaces. Mol. Catal. 2019, 466, 26–36. [Google Scholar] [CrossRef]
- Bahruji, H.; Bowker, M.; Hutchings, G.J.; Dimitratos, N.; Wells, P.; Gibson, E.; Jones, W.; Brookes, C.; Morgan, D.; Lalev, G.M. Pd/ZnO catalysts for direct CO2 hydrogenation to methanol. J. Catal. 2016, 343, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Chen, J.G. CO2 hydrogenation to methanol over ZrO2-containing catalysts: Insights into ZrO2 induced synergy. ACS Catal. 2019, 9, 7840–7861. [Google Scholar] [CrossRef]
- Phongamwong, T.; Chantaprasertporn, U.; Witoon, T.; Numpilai, T.; Poo-arporn, Y.; Limphirat, W.; Donphai, W.; Dittanet, P.; Chareonpanich, M.; Limtrakul, J. CO2 hydrogenation to methanol over CuO–ZnO–ZrO2–SiO2 catalysts: Effects of SiO2 contents. Chem. Eng. J. 2017, 316, 692–703. [Google Scholar] [CrossRef]
- Słoczyński, J.; Grabowski, R.; Olszewski, P.; Kozłowska, A.; Stoch, J.; Lachowska, M.; Skrzypek, J. Effect of metal oxide additives on the activity and stability of Cu/ZnO/ZrO2 catalysts in the synthesis of methanol from CO2 and H2. Appl. Catal. A 2006, 310, 127–137. [Google Scholar] [CrossRef]
- Hu, X.; Zhao, C.; Guan, Q.; Hu, X.; Li, W.; Chen, J. Selective hydrogenation of CO2 over a Ce promoted Cu-based catalyst confined by SBA-15. Inorg. Chem. Front. 2019, 6, 1799–1812. [Google Scholar] [CrossRef]
- Allam, D.; Bennici, S.; Limousy, L.; Hocine, S. Improved Cu- and Zn-based catalysts for CO2 hydrogenation to methanol. C. R. Chim. 2019, 22, 227–237. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Influence of modifier (Mn, La, Ce, Zr and Y) on the performance of Cu/Zn/Al catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. Appl. Catal. A 2013, 468, 442–452. [Google Scholar] [CrossRef]
- Lam, E.; Corral-Pérez, J.J.; Larmier, K.; Noh, G.; Wolf, P.; Comas-Vives, A.; Urakawa, A.; Copéret, C. CO2 hydrogenation on Cu/Al2O3: Role of the metal/support interface in driving activity and selectivity of a bifunctional catalyst. Angew. Chem. Int. Ed. 2019, 58, 13989–13996. [Google Scholar] [CrossRef]
- Natesakhawat, S.; Lekse, J.W.; Baltrus, J.P.; Ohodnicki, P.R.; Howard, B.H.; Deng, X.; Matranga, C. Active sites and structure–activity relationships of copper-based catalysts for carbon dioxide hydrogenation to methanol. ACS Catal. 2012, 2, 1667–1676. [Google Scholar] [CrossRef]
- Zhan, H.; Li, F.; Gao, P.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Methanol synthesis from CO2 hydrogenation over La–M–Cu–Zn–O (M = Y, Ce, Mg, Zr) catalysts derived from perovskite-type precursors. J. Power Sources 2014, 251, 113–121. [Google Scholar] [CrossRef]
- Hayward, J.S.; Smith, P.J.; Kondrat, S.A.; Bowker, M.; Hutchings, G.J. The effects of secondary oxides on copper-based catalysts for green methanol synthesis. ChemCatChem 2017, 9, 1655–1662. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. The influence of La doping on the catalytic behavior of Cu/ZrO2 for methanol synthesis from CO2 hydrogenation. J. Mol. Catal. A 2011, 345, 60–68. [Google Scholar] [CrossRef]
- Yang, C.; Ren, J.; Sun, Y. Role of La2O3 in Pd-supported catalysts for methanol decomposition. Catal. Lett. 2002, 84, 123–129. [Google Scholar] [CrossRef]
- Borodko, Y.; Somorjai, G.A. Catalytic hydrogenation of carbon oxides—A 10-year perspective. Appl. Catal. A 1999, 186, 355–362. [Google Scholar] [CrossRef]
- Toyir, J.; Miloua, R.; Elkadri, N.E.; Nawdali, M.; Toufik, H.; Miloua, F.; Saito, M. Sustainable process for the production of methanol from CO2 and H2 using Cu/ZnO-based multicomponent catalyst. Phys. Procedia 2009, 2, 1075–1079. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Arena, F.; Frusteri, F. The changing nature of the active site of Cu-Zn-Zr catalysts for the CO2 hydrogenation reaction to methanol. Appl. Catal. B 2014, 152–153, 152–161. [Google Scholar] [CrossRef]
- An, X.; Li, J.; Zuo, Y.; Zhang, Q.; Wang, D.; Wang, J. A Cu/Zn/Al/Zr fibrous catalyst that is an improved CO2 hydrogenation to methanol catalyst. Catal. Lett. 2007, 118, 264–269. [Google Scholar] [CrossRef]
- Yoshihara, J.; Campbell, C.T. Methanol synthesis and reverse water–gas shift kinetics over Cu(110) model catalysts: Structural sensitivity. J. Catal. 1996, 161, 776–782. [Google Scholar] [CrossRef]
- Yang, Y.; Mims, C.A.; Disselkamp, R.S.; Kwak, J.-H.; Peden, C.H.F.; Campbell, C.T. (Non)formation of methanol by direct hydrogenation of formate on copper catalysts. J. Phys. Chem. C 2010, 114, 17205–17211. [Google Scholar] [CrossRef]
- Ramli, M.Z.; Syed-Hassan, S.S.A.; Hadi, A. Performance of Cu-Zn-Al-Zr catalyst prepared by ultrasonic spray precipitation technique in the synthesis of methanol via CO2 hydrogenation. Fuel Process. Technol. 2018, 169, 191–198. [Google Scholar] [CrossRef]
- Cai, W.; Chen, Q.; Wang, F.; Li, Z.; Yu, H.; Zhang, S.; Cui, L.; Li, C. Comparison of the promoted CuZnMxOy (M: Ga, Fe) catalysts for CO2 hydrogenation to methanol. Catal. Lett. 2019, 149, 2508–2518. [Google Scholar] [CrossRef]
- Ban, H.; Li, C.; Asami, K.; Fujimoto, K. Influence of rare-earth elements (La, Ce, Nd and Pr) on the performance of Cu/Zn/Zr catalyst for CH3OH synthesis from CO2. Catal. Commun. 2014, 54, 50–54. [Google Scholar] [CrossRef]
- Arena, F.; Mezzatesta, G.; Zafarana, G.; Trunfio, G.; Frusteri, F.; Spadaro, L. How oxide carriers control the catalytic functionality of the Cu–ZnO system in the hydrogenation of CO2 to methanol. Catal. Today 2013, 210, 39–46. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, C.-W.; Pan, W.; Zhu, Q.-M.; Deng, J.-F. In situ IR studies on the mechanism of methanol synthesis over an ultrafine Cu/ZnO/Al2O3 catalyst. Appl. Catal. A 1998, 171, 301–308. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Mechanistic study of the selective methanation of CO over Ru/TiO2 catalyst. Identification of active surface species and reaction pathways. J. Phys. Chem. C 2011, 115, 1220–1230. [Google Scholar] [CrossRef]
- Le Peltier, F.; Chaumette, P.; Saussey, J.; Bettahar, M.M.; Lavalley, J.C. In situ FT-IR and kinetic study of methanol synthesis from CO/H2 over ZnAl2O4 and Cu–ZnAl2O4 catalysts. J. Mol. Catal. A 1998, 132, 91–100. [Google Scholar] [CrossRef]
- Sanchez-Escribano, V.; Larrubia Vargas, M.A.; Finocchio, E.; Busca, G. On the mechanisms and the selectivity determining steps in syngas conversion over supported metal catalysts: An IR study. Appl. Catal. A 2007, 316, 68–74. [Google Scholar] [CrossRef]
- Schumann, J.; Eichelbaum, M.; Lunkenbein, T.; Thomas, N.; Alvarez Galvan, M.C.; Schlogl, R.; Behrens, M. Promoting strong metal support interaction: Doping ZnO for enhanced activity of Cu/ZnO:M (M=Al, Ga, Mg) catalysts. ACS Catal. 2015, 5, 3260–3270. [Google Scholar] [CrossRef]
- Fang, X.; Men, Y.; Wu, F.; Zhao, Q.; Singh, R.; Xiao, P.; Du, T.; Webley, P.A. Moderate-pressure conversion of H2 and CO2 to methanol via adsorption enhanced hydrogenation. Int. J. Hydrogen Energy 2019, 44, 21913–21925. [Google Scholar] [CrossRef]
- Xiao, J.; Mao, D.; Guo, X.; Yu, J. Effect of TiO2, ZrO2, and TiO2–ZrO2 on the performance of CuO–ZnO catalyst for CO2 hydrogenation to methanol. Appl. Surf. Sci. 2015, 338, 146–153. [Google Scholar] [CrossRef]
- Li, C.; Yuan, X.; Fujimoto, K. Development of highly stable catalyst for methanol synthesis from carbon dioxide. Appl. Catal. A 2014, 469, 306–311. [Google Scholar] [CrossRef]
- Din, I.U.; Shaharun, M.S.; Alotaibi, M.A.; Alharthi, A.I.; Naeem, A. Recent developments on heterogeneous catalytic CO2 reduction to methanol. J. CO₂ Util. 2019, 34, 20–33. [Google Scholar] [CrossRef]
- Saito, M.; Fujitani, T.; Takeuchi, M.; Watanabe, T. Development of copper/zinc oxide-based multicomponent catalysts for methanol synthesis from carbon dioxide and hydrogen. Appl. Catal. A 1996, 138, 311–318. [Google Scholar] [CrossRef]
- Liu, Y.-M.; Liu, J.-T.; Liu, S.-Z.; Li, J.; Gao, Z.-H.; Zuo, Z.-J.; Huang, W. Reaction mechanisms of methanol synthesis from CO/CO2 hydrogenation on Cu2O(111): Comparison with Cu(111). J. CO₂ Util. 2017, 20, 59–65. [Google Scholar] [CrossRef]
- Dasireddy, V.B.C.D.; Likozar, B. The role of copper oxidation state in Cu/ZnO/Al2O3 catalysts in CO2 hydrogenation and methanol productivity. Renew. Energy 2019, 140, 452–460. [Google Scholar] [CrossRef]
- Fisher, I.A.; Woo, H.C.; Bell, A.T. Effects of zirconia promotion on the activity of Cu/SiO2 for methanol synthesis from CO/H2 and CO2/H2. Catal. Lett. 1997, 44, 11–17. [Google Scholar] [CrossRef]
- Hong, Q.J.; Liu, Z.P. Mechanism of CO2 hydrogenation over Cu/ZrO2(212) interface from first-principles kinetics Monte Carlo simulations. Surf. Sci. 2010, 604, 1869–1876. [Google Scholar]
- Graaf, G.H.; Stamhuis, E.J.; Beenackers, A.A.C.M. Kinetics of low-pressure methanol synthesis. Chem. Eng. Sci. 1988, 43, 3185–3195. [Google Scholar] [CrossRef]
- Graaf, G.H.; Scholtens, H.; Stamhuis, E.J.; Beenackers, A.A.C.M. Intra-particle diffusion limitations in low-pressure methanol synthesis. Chem. Eng. Sci. 1988, 45, 773–783. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Belin, T.; Ahouari, H.; Soualah, A.; Pouilloux, Y.; Le Valant, A. The Cu–ZnO synergy in methanol synthesis from CO2, Part 2: Origin of the methanol and CO selectivities explained by experimental studies and a sphere contact quantification model in randomly packed binary mixtures on Cu–ZnO coprecipitate catalysts. J. Catal. 2015, 330, 533–544. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Štefančič, N.S.; Huš, M.; Likozar, B. Effect of alkaline earth metal oxide (MO) Cu/MO/Al2O3 catalysts on methanol synthesis activity and selectivity via CO2 reduction. Fuel 2018, 233, 103–112. [Google Scholar] [CrossRef]
- Liu, C.; Guo, X.; Guo, Q.; Mao, D.; Yu, J.; Lu, G. Methanol synthesis from CO2 hydrogenation over copper catalysts supported on MgO-modified TiO2. J. Mol. Catal. A 2016, 425, 86–93. [Google Scholar] [CrossRef]
- Graaf, G.H.; Sijtsema, P.J.J.M.; Stamhuis, E.J.; Joosten, G.E.H. Chemical equilibria in methanol synthesis. Chem. Eng. Sci. 1986, 41, 2883–2890. [Google Scholar] [CrossRef]
- Boudart, M.; Djega-Mariadassou, G. Kinetics of Heterogeneous Catalytic Reactions; Princeton University Press: Princeton, NJ, USA, 1984. [Google Scholar]
- Karelovic, A.; Ruiz, P. The role of copper particle size in low pressure methanol synthesis via CO2 hydrogenation over Cu/ZnO catalysts. Catal. Sci. Technol. 2015, 5, 869–881. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Specific Surface Area (m2 g−1) | Pore Volume (cm3 g−1) | Pore Size (nm) |
|---|---|---|---|
| CZA-La0 | 111 | 0.70 | 20 |
| CZA-La25 | 88 | 0.78 | 30 |
| CZA-La50 | 96 | 0.79 | 30 |
| CZA-La75 | 61 | 0.45 | 27 |
| CZA-La100 | 47 | 0.32 | 25 |
| k5 | k9 | K1 | K2 | K = K7.K8 |
|---|---|---|---|---|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kourtelesis, M.; Kousi, K.; Kondarides, D.I. CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study. Catalysts 2020, 10, 183. https://doi.org/10.3390/catal10020183
Kourtelesis M, Kousi K, Kondarides DI. CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study. Catalysts. 2020; 10(2):183. https://doi.org/10.3390/catal10020183
Chicago/Turabian StyleKourtelesis, Marios, Kalliopi Kousi, and Dimitris I. Kondarides. 2020. "CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study" Catalysts 10, no. 2: 183. https://doi.org/10.3390/catal10020183
APA StyleKourtelesis, M., Kousi, K., & Kondarides, D. I. (2020). CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study. Catalysts, 10(2), 183. https://doi.org/10.3390/catal10020183