Approaches for Selective Oxidation of Methane to Methanol



Abstract
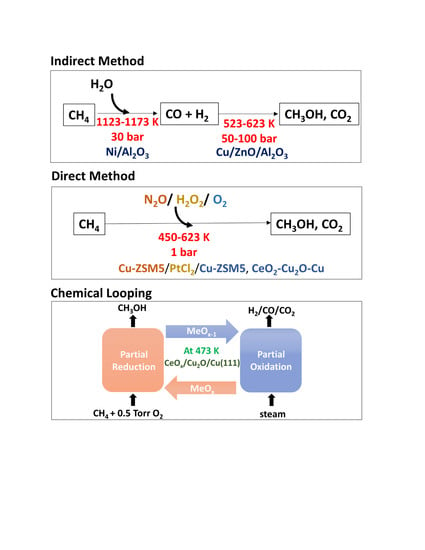
:
1. Introduction
2. Direct Methane to Methanol (DMTM) Synthesis
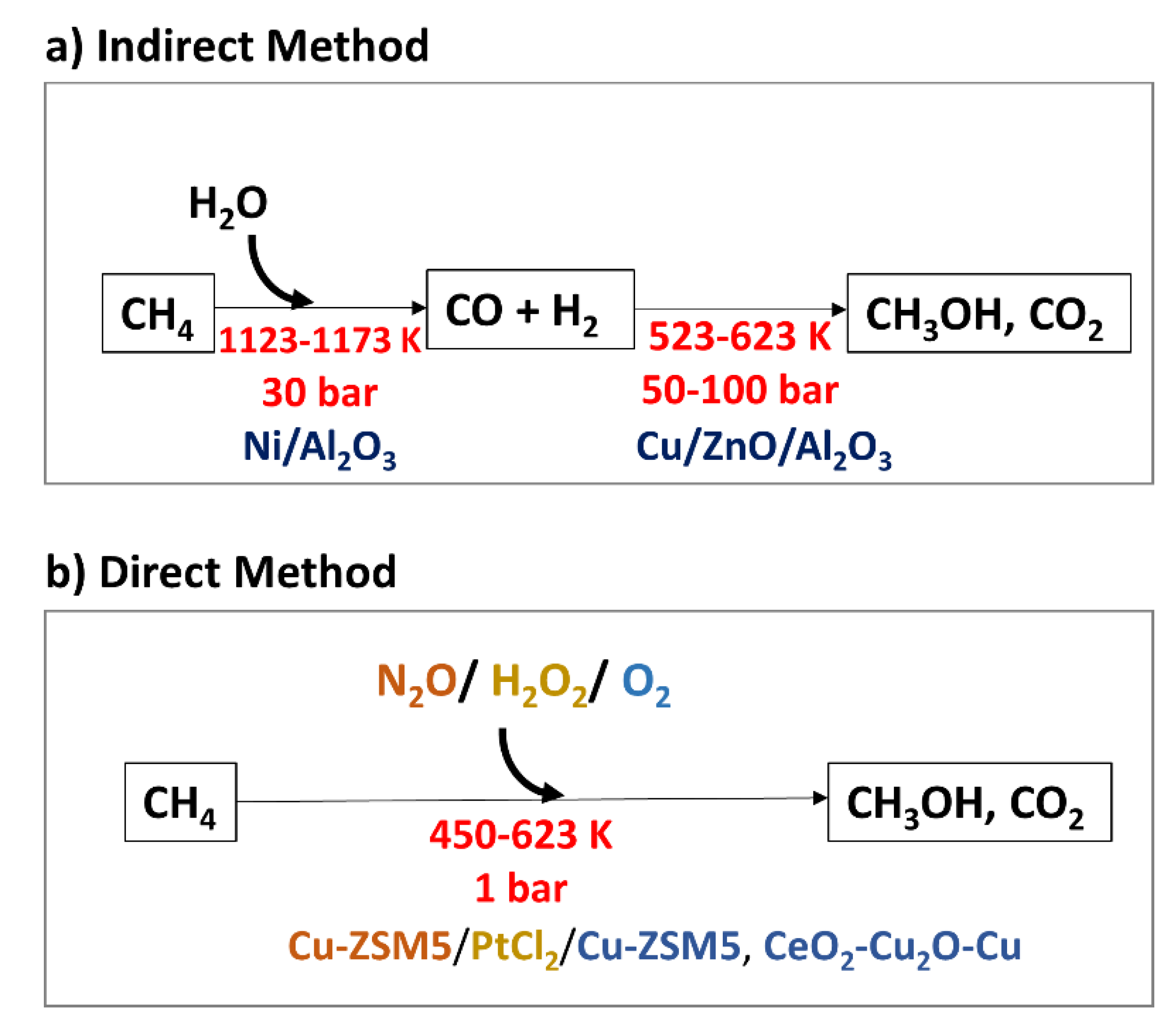
2.1. DMTM in Nature
2.2. Bio-Mimetic Zeolites: H2SO4, H2O2, and N2O
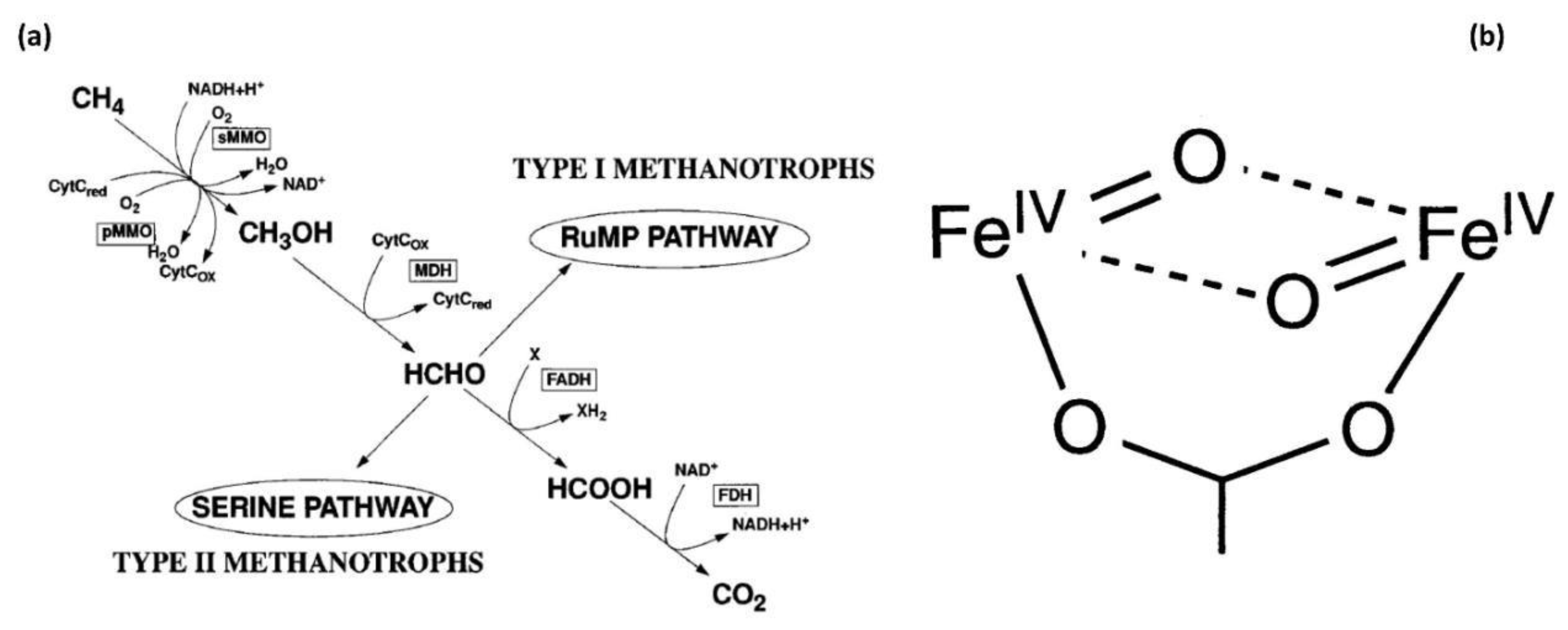
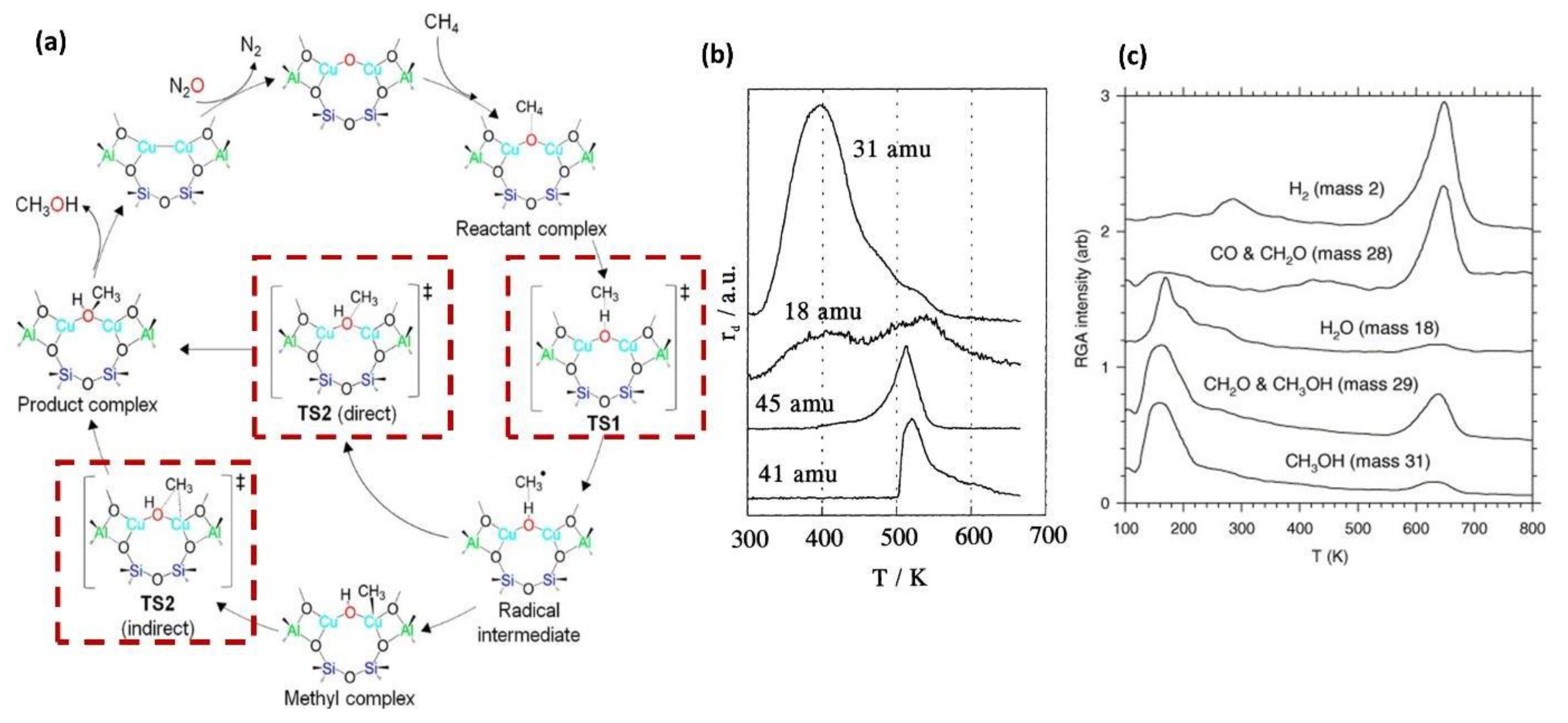
2.3. Bio-Mimetic Zeolites: O2 as Oxidant
2.4. Metal Oxides: O2 as Oxidant
2.4.1. Ceria Surfaces
2.4.2. Doping
3. From Methane Activation to Methanol Selectivity
4. Beyond Catalysis: Chemical Looping
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zakaria, Z.; Kamarudin, S.K. Direct conversion technologies of methane to methanol: An overview. Renew. Sustain. Energy Rev. 2016, 65, 250–261. [Google Scholar] [CrossRef]
- Park, D.; Lee, J. Biological conversion of methane to methanol. Korean J. Chem. Eng. 2013, 30, 977–987. [Google Scholar] [CrossRef]
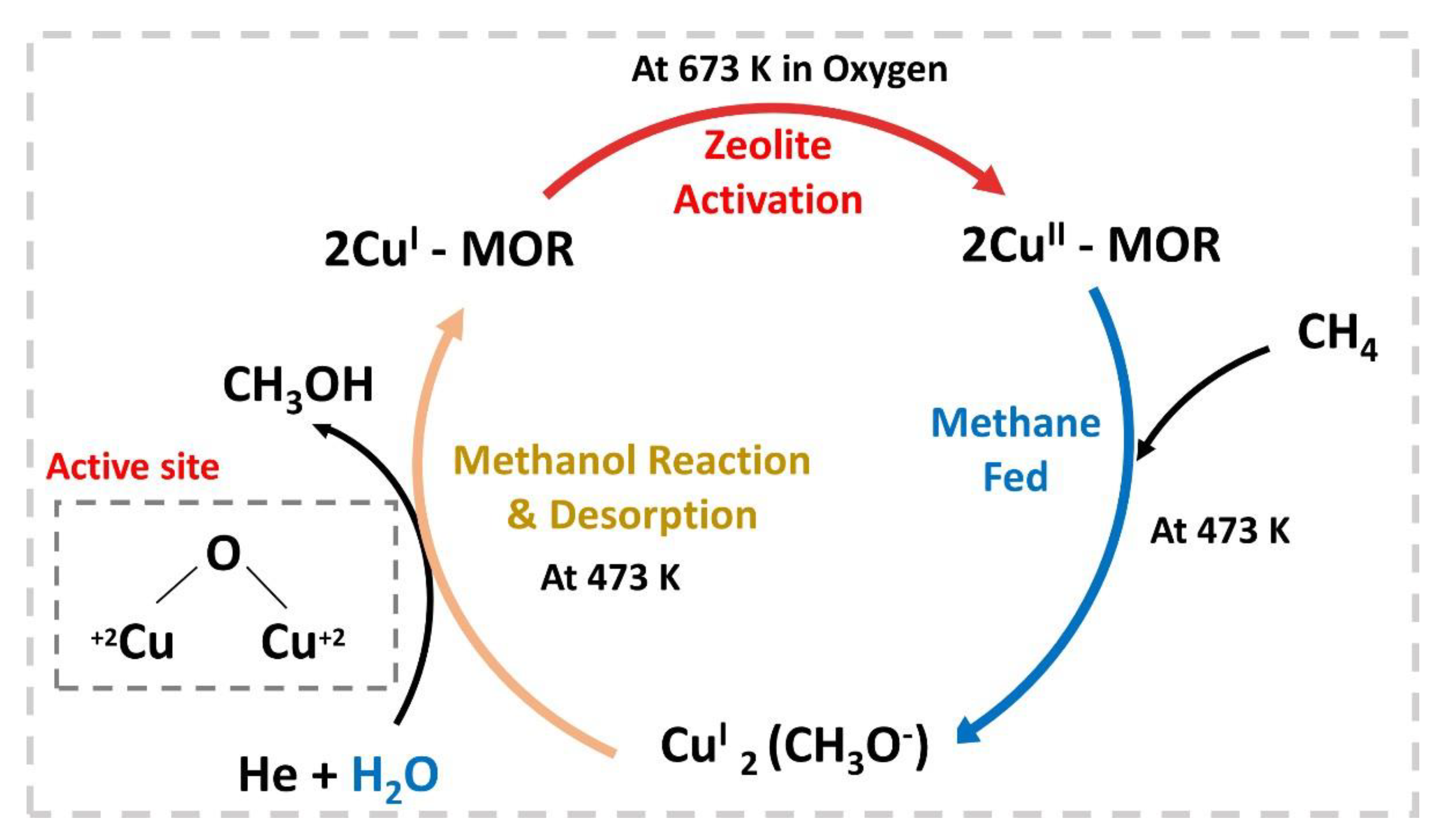
- Tomkins, P.; Mansouri, A.; Bozbag, S.E.; Krumeich, F.; Park, M.B.; Alayon, E.M.C.; Ranocchiari, M.; van Bokhoven, J.A. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew. Chem. Int. Ed. 2016, 55, 5467–5471. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. The Changing Landscape of Hydrocarbon Feedstocks for Chemical Production: Implications for Catalysis: Proceedings of a Workshop; The National Academies Press: Washington, DC, USA, 2016; p. 136. [Google Scholar]
- Kennedy, T.F.; Shanks, D. Methanol: Manufacture and Uses. In Monohydric Alcohols; American Chemical Society: Washington, DC, USA, 1981; Volume 159, pp. 19–27. [Google Scholar]
- Wickson, E.J. Monohydric Alcohols; American Chemical Society: Washington, DC, USA, 1981; Volume 159, p. 244. [Google Scholar]
- Dolan, G. Overview of Global Methanol Fuel Blending, Touching on Benefits of Methanol Blending Methanol Fuel Forum in Trinidad and Tobago in Association with Proman and MI Member MHTL [Online]. 2019. Available online: https://www.methanol.org/wp-content/uploads/2019/02/4.-Greg-Dolan-Overview-of-Global-Methanol-Fuel-Blending.pdf. (accessed on 1 December 2018).
- Sheldon, D. Methanol Production—A Technical History. Johns. Matthey Technol. Rev. 2017, 61, 172–182. [Google Scholar] [CrossRef]
- Mittasch, A.P.M.; Winkler, K. Ausfuhrung Organischer Katalysen. Germany Patent DE415686, 24 July 1923. [Google Scholar]
- Hansen, J.; Kharecha, P.; Sato, M.; Masson-Delmotte, V.; Ackerman, F.; Beerling, D.J.; Hearty, P.J.; Hoegh-Guldberg, O.; Hsu, S.-L.; Parmesan, C.; et al. Assessing “Dangerous Climate Change”: Required Reduction of Carbon Emissions to Protect Young People, Future Generations and Nature. PLoS ONE 2013, 8, e81648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, J.; Sato, M.; Ruedy, R.; Lacis, A.; Oinas, V. Global warming in the twenty-first century: An alternative scenario. Proc. Natl. Acad. Sci. USA 2000, 97, 9875–9880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunsford, J.H. Catalytic conversion of methane to more useful chemicals and fuels: A challenge for the 21st century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Da Silva, M.J. Synthesis of methanol from methane: Challenges and advances on the multi-step (syngas) and one-step routes (DMTM). Fuel Process. Technol. 2016, 145, 42–61. [Google Scholar] [CrossRef]
- Methanol Market: Global Industry Trends, Share, Size, Growth, Opportunity and Forecast 2019–2024; IMARC Services Pvt. Ltd.: Uttar Pradesh, India, April 2019; Market Research.com.
- Usman, M.; Daud, W.M.A.W. Recent advances in the methanol synthesis via methane reforming processes. RSC Adv. 2015, 5, 21945–21972. [Google Scholar] [CrossRef]
- Iglesias, I.; Forti, M.; Baronetti, G.; Mariño, F. Zr-enhanced stability of ceria based supports for methane steam reforming at severe reaction conditions. Int. J. Hydrog. Energy 2019, 44, 8121–8132. [Google Scholar] [CrossRef]
- Agrafiotis, C.; von Storch, H.; Roeb, M.; Sattler, C. Solar thermal reforming of methane feedstocks for hydrogen and syngas production—A review. Renew. Sustain. Energy Rev. 2014, 29, 656–682. [Google Scholar] [CrossRef]
- Pen˜a, M.A.; Gómez, J.P.; Fierro, J.L.G. New catalytic routes for syngas and hydrogen production. Appl. Catal. A 1996, 144, 7–57. [Google Scholar] [CrossRef]
- Miguel, C.V.; Soria, M.A.; Mendes, A.; Madeira, L.M. Direct CO2 hydrogenation to methane or methanol from post-combustion exhaust streams—A thermodynamic study. J. Nat. Gas Sci. Eng. 2015, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bozzano, G.; Manenti, F. Efficient methanol synthesis: Perspectives, technologies and optimization strategies. Prog. Energy Combust. Sci. 2016, 56, 71–105. [Google Scholar] [CrossRef]
- Sehested, J. Industrial and scientific directions of methanol catalyst development. J. Catal. 2019, 371, 368–375. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; van Bokhoven, J.A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523. [Google Scholar] [CrossRef]
- Schüth, F. Making more from methane. Science 2019, 363, 1282. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Theoretical Overview of Methane Hydroxylation by Copper–Oxygen Species in Enzymatic and Zeolitic Catalysts. Acc. Chem. Res. 2018, 51, 2382–2390. [Google Scholar] [CrossRef]
- Hanson, R.S.; Hanson, T.E. Methanotrophic bacteria. Microbiol. Rev. 1996, 60, 439–471. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.Y.; Lee, S.H.; Choi, Y.S.; Park, S.J.; Na, J.G.; Chang, I.S.; Kim, C.; Kim, H.C.; Kim, Y.H.; Lee, J.W.; et al. Biocatalytic conversion of methane to methanol as a key step for development of methane-based biorefineries. J. Microbiol. Biotechnol. 2014, 24, 1597–1605. [Google Scholar] [CrossRef] [Green Version]
- Friedle, S.; Reisner, E.; Lippard, S.J. Current challenges of modeling diiron enzyme active sites for dioxygen activation by biomimetic synthetic complexes. Chem. Soc. Rev. 2010, 39, 2768–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knops-Gerrits, P.-P.H.; Goddard, W.A., III. The structure–activity relationships of methane mono-oxygenase mimics in alkane activation. Catal. Today 2003, 81, 263–286. [Google Scholar] [CrossRef]
- Shu, L.; Nesheim, J.C.; Kauffmann, K.; Münck, E.; Lipscomb, J.D.; Que, L. An Fe2IVO2 Diamond Core Structure for the Key Intermediate Q of Methane Monooxygenase. Science 1997, 275, 515–518. [Google Scholar] [CrossRef]
- Ross, M.O.; Rosenzweig, A.C. A tale of two methane monooxygenases. J. Biol. Inorg. Chem. JBIC 2017, 22, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Tinberg, C.E.; Lippard, S.J. Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res. 2011, 44, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-K.; Lipscomb, J.D. Oxygen Activation Catalyzed by Methane Monooxygenase Hydroxylase Component: Proton Delivery during the O−O Bond Cleavage Steps. Biochemistry 1999, 38, 4423–4432. [Google Scholar] [CrossRef] [PubMed]
- Skulan, A.J.; Brunold, T.C.; Baldwin, J.; Saleh, L.; Bollinger, J.M.; Solomon, E.I. Nature of the Peroxo Intermediate of the W48F/D84E Ribonucleotide Reductase Variant: Implications for O2 Activation by Binuclear Non-Heme Iron Enzymes. J. Am. Chem. Soc. 2004, 126, 8842–8855. [Google Scholar] [CrossRef] [PubMed]
- Han, W.-G.; Noodleman, L. Structural Model Studies for the Peroxo Intermediate P and the Reaction Pathway from P → Q of Methane Monooxygenase Using Broken-Symmetry Density Functional Calculations. Inorg. Chem. 2008, 47, 2975–2986. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Rosenzweig, A.C. Structural and mechanistic insights into methane oxidation by particulate methane monooxygenase. Acc. Chem. Res. 2007, 40, 573–580. [Google Scholar] [CrossRef]
- Ross, M.O.; MacMillan, F.; Wang, J.; Nisthal, A.; Lawton, T.J.; Olafson, B.D.; Mayo, S.L.; Rosenzweig, A.C.; Hoffman, B.M. Particulate methane monooxygenase contains only mononuclear copper centers. Science 2019, 364, 566. [Google Scholar] [CrossRef] [Green Version]
- Kamachi, T.; Okura, I. Methanol Biosynthesis Using Methanotrophs. In Methane Biocatalysis: Paving the Way to Sustainability; Kalyuzhnaya, M.G., Xing, X.-H., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 169–182. [Google Scholar]
- Blanchette, C.D.; Knipe, J.M.; Stolaroff, J.K.; DeOtte, J.R.; Oakdale, J.S.; Maiti, A.; Lenhardt, J.M.; Sirajuddin, S.; Rosenzweig, A.C.; Baker, S.E. Printable enzyme- embedded materials for methane to methanol conversion. Nat. Commun. 2016, 7, 11900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirajuddin, S.; Barupala, D.; Helling, S.; Marcus, K.; Stemmler, T.L.; Rosenzweig, A.C. Effects of zinc on particulate methane monooxygenase activity and structure. J. Biol. Chem. 2014, 289, 21782–21794. [Google Scholar] [CrossRef] [Green Version]
- Lange, J.-P.; Sushkevich, V.L.; Knorpp, A.J.; van Bokhoven, J.A. Methane-to- Methanol via Chemical Looping: Economic Potential and Guidance for Future Research. Ind. Eng. Chem. Res. 2019, 58, 8674–8680. [Google Scholar] [CrossRef]
- Szécsényi, Á.; Khramenkova, E.; Chernyshov, I.Y.; Li, G.; Gascon, J.; Pidko, E.A. Breaking Linear Scaling Relationships with Secondary Interactions in Confined Space: A Case Study of Methane Oxidation by Fe/ZSM-5 Zeolite. ACS Catal. 2019, 9, 9276–9284. [Google Scholar] [CrossRef] [Green Version]
- Prikhod’ko, R.V.; Astrelin, I.M.; Sychev, M.V.; Hensen, E.J.M. Influence of preparation procedure on the surface chemistry and catalytic characteristics of FeZSM-5 zeolite in selective oxidation of benzene to phenol. Russ. J. Appl. Chem. 2006, 79, 1115–1121. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Evitt, E.R.; Loffler, D.G.; Wentrcek, P.R.; Voss, G.; Masuda, T. A mercury-catalyzed, high-yield system for the oxidation of methane to methanol. Science 1993, 259, 340–343. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum catalysts for the high-yield oxidation of methane to a methanol derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Lopez-Sanchez, J.A.; Jenkins, R.L.; Whiting, G.; Kondrat, S.A.; ab Rahim, M.H.; Forde, M.M.; Thetford, A.; Hagen, H.; et al. Aqueous-Phase Methane Oxidation over Fe-MFI Zeolites; Promotion through Isomorphous Framework Substitution. ACS Catal. 2013, 3, 1835–1844. [Google Scholar] [CrossRef]
- Forde, M.M.; Armstrong, R.D.; McVicker, R.; Wells, P.P.; Dimitratos, N.; He, Q.; Lu, L.; Jenkins, R.L.; Hammond, C.; Lopez-Sanchez, J.A.; et al. Light alkane oxidation using catalysts prepared by chemical vapour impregnation: Tuning alcohol selectivity through catalyst pre-treatment. Chem. Sci. 2014, 5, 3603–3616. [Google Scholar] [CrossRef] [Green Version]
- Hammond, C.; Dimitratos, N.; Jenkins, R.L.; Lopez-Sanchez, J.A.; Kondrat, S.A.; Hasbi ab Rahim, M.; Forde, M.M.; Thetford, A.; Taylor, S.H.; Hagen, H.; et al. Elucidation and Evolution of the Active Component within Cu/Fe/ZSM-5 for Catalytic Methane Oxidation: From Synthesis to Catalysis. ACS Catal. 2013, 785, 689–699. [Google Scholar] [CrossRef]
- Agarwal, N.; Freakley, S.J.; McVicker, R.U.; Althahban, S.M.; Dimitratos, N.; He, Q.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 2017, 358, 223–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ab Rahim, M.H.; Forde, M.M.; Jenkins, R.L.; Hammond, C.; He, Q.; Dimitratos, N.; Lopez- Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold–Palladium Alloy Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Ab Rahim, M.H.; Forde, M.M.; Hammond, C.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; Hutchings, G.J. Systematic Study of the Oxidation of Methane Using Supported Gold Palladium Nanoparticles Under Mild Aqueous Conditions. Top. Catal. 2013, 56, 1843–1857. [Google Scholar] [CrossRef]
- Ab Rahim, M.H.; Armstrong, R.D.; Hammond, C.; Dimitratos, N.; Freakley, S.J.; Forde, M.M.; Morgan, D.J.; Lalev, G.; Jenkins, R.L.; Lopez-Sanchez, J.A.; et al. Low temperature selective oxidation of methane to methanol using titania supported gold palladium copper catalysts. Catal. Sci. Technol. 2016, 6, 3410–3418. [Google Scholar] [CrossRef] [Green Version]
- Pannov, G.I.; Sobolev, V.I.; Kharitonov, A.S. The role of iron in N2O decomposition on ZSM-5 zeolite and reactivity of the surface oxygen formed. J. Mol. Catal. 1990, 61, 85–97. [Google Scholar] [CrossRef]
- Panov, G.I.; Sobolev, V.I.; Dubkov, K.A.; Parmon, V.N.; Ovanesyan, N.S.; Shilov, A.E.; Shteinman, A.A. Iron complexes in zeolites as a new model of methane monooxygenase. React. Kinet. Catal. Lett. 1997, 61, 251–258. [Google Scholar] [CrossRef]
- Starokon, E.V.; Parfenov, M.V.; Arzumanov, S.S.; Pirutko, L.V.; Stepanov, A.G.; Panov, G.I. Oxidation of methane to methanol on the surface of FeZSM-5 zeolite. J. Catal. 2013, 300, 47–54. [Google Scholar] [CrossRef]
- Starokon, E.V.; Parfenov, M.V.; Pirutko, L.V.; Abornev, S.I.; Panov, G.I. Room-Temperature Oxidation of Methane by α-Oxygen and Extraction of Products from the FeZSM-5 Surface. J. Phys. Chem. C 2011, 115, 2155–2161. [Google Scholar] [CrossRef]
- Malykhin, S. Hydrogen abstraction reactions of the [FeO]2+ moiety: The role of the electronic state. Chem. Phys. Lett. 2015, 622, 69–74. [Google Scholar] [CrossRef]
- Rosa, A.; Ricciardi, G.; Jan Baerends, E. Is [FeO](2+) the active center also in iron containing zeolites? A density functional theory study of methane hydroxylation catalysis by Fe-ZSM-5 zeolite. Inorg. Chem. 2010, 49, 3866–3880. [Google Scholar] [CrossRef] [PubMed]
- Zhidomirov, G.; Shubin, A.; Larin, A.; Malykhin, S.; Rybakov, A. Molecular Models of the Stabilization of Bivalent Metal Cations in Zeolite Catalysts; Springer: Dordrecht, 2011; pp. 579–643. [Google Scholar]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc. Natl. Acad. Sci. USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef] [PubMed]
- Smeets, P.J.; Groothaert, M.H.; Schoonheydt, R.A. Cu based zeolites: A UV–vis study of the active site in the selective methane oxidation at low temperatures. Catal. Today 2005, 110, 303–309. [Google Scholar] [CrossRef]
- Groothaert, M.H.; Lievens, K.; van Bokhoven, J.A.; Battiston, A.A.; Weckhuysen, B.M.; Pierloot, K.; Schoonheydt, R.A. Bis(μ-oxo)dicopper as Key Intermediate in the Catalytic Decomposition of Nitric Oxide. Chemphyschem 2003, 4, 626–630. [Google Scholar] [CrossRef]
- Groothaert, M.H.; van Bokhoven, J.A.; Battiston, A.A.; Weckhuysen, B.M.; Schoonheydt, R.A. Bis(μ-oxo)dicopper in Cu-ZSM-5 and Its Role in the Decomposition of NO: A Combined in Situ XAFS, UV−Vis−Near-IR, and Kinetic Study. J. Am. Chem. Soc. 2003, 125, 7629–7640. [Google Scholar] [CrossRef]
- Li, G.; Vassilev, P.; Sanchez-Sanchez, M.; Lercher, J.A.; Hensen, E.J.M.; Pidko, E.A. Stability and reactivity of copper oxo-clusters in ZSM-5 zeolite for selective methane oxidation to methanol. J. Catal. 2016, 338, 305–312. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Miyanishi, M.; Yoshizawa, K. Roles of Zeolite Confinement and Cu–O–Cu Angle on the Direct Conversion of Methane to Methanol by [Cu2(μ-O)]2+-Exchanged AEI, CHA, AFX, and MFI Zeolites. ACS Catal. 2017, 7, 3741–3751. [Google Scholar] [CrossRef]
- Chan, S.I.; Wang, V.C.C.; Lai, J.C.H.; Yu, S.S.F.; Chen, P.P.Y.; Chen, K.H.C.; Chen, C.-L.; Chan, M.K. Redox Potentiometry Studies of Particulate Methane Monooxygenase: Support for a Trinuclear Copper Cluster Active Site. Angew. Chem. Int. Ed. 2007, 46, 1992–1994. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Yoshizawa, K. Direct Conversion of Methane to Methanol by Metal-Exchanged ZSM-5 Zeolite (Metal = Fe, Co, Ni, Cu). ACS Catal. 2016, 6, 8321–8331. [Google Scholar] [CrossRef]
- Hunger, B.; Matysik, S.; Heuchel, M.; Einicke, W.-D. Adsorption of Methanol on ZSM-5 Zeolites. Langmuir 1997, 13, 6249–6254. [Google Scholar] [CrossRef]
- Mullins, D.R.; Robbins, M.D.; Zhou, J. Adsorption and reaction of methanol on thin-film cerium oxide. Surf. Sci. 2006, 600, 1547–1558. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Román-Leshkov, Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Dandu, N.K.; Adeyiga, O.; Panthi, D.; Bird, S.A.; Odoh, S.O. Performance of density functional theory for describing hetero-metallic active-site motifs for methane-to- methanol conversion in metal-exchanged zeolites. J. Comput. Chem. 2018, 39, 2667–2678. [Google Scholar] [CrossRef] [PubMed]
- Ison, A.; Gorte, R.J. The adsorption of methanol and water on H-ZSM-5. J. Catal. 1984, 89, 150–158. [Google Scholar] [CrossRef]
- Omojola, T.; Cherkasov, N.; McNab, A.I.; Lukyanov, D.B.; Anderson, J.A.; Rebrov, E.V.; van Veen, A.C. Mechanistic Insights into the Desorption of Methanol and Dimethyl Ether Over ZSM-5 Catalysts. Catal. Lett. 2018, 148, 474–488. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; He, D.; Zhang, Q.; Xu, B.; Zhu, Q. Comparative studies on direct conversion of methane to methanol/formaldehyde over La–Co–O and ZrO2 supported molybdenum oxide catalysts. Top. Catal. 2005, 32, 215–223. [Google Scholar] [CrossRef]
- Fu, G.; Xu, X.; Lu, X.; Wan, H. Mechanisms of Methane Activation and Transformation on Molybdenum Oxide Based Catalysts. J. Am. Chem. Soc. 2005, 127, 3989–3996. [Google Scholar] [CrossRef]
- Rocha, R.S.; Reis, R.M.; Lanza, M.R.V.; Bertazzoli, R. Electrosynthesis of methanol from methane: The role of V2O5 in the reaction selectivity for methanol of a TiO2/RuO2/V2O5 gas diffusion electrode. Electrochim. Acta 2013, 87, 606–610. [Google Scholar] [CrossRef]
- Wachs, I.E.; Routray, K. Catalysis Science of Bulk Mixed Oxides. ACS Catal. 2012, 2, 1235–1246. [Google Scholar] [CrossRef]
- Wang, X.M.N.M.; Nilsson, J.; Carlson, S.; Gustafson, J.; Skoglundh, M.; Carlsson, P.-A. Copper-Modified Zeolites and Silica for Conversion of Methane to Methanol. Catalysts 2018, 8, 545. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Ohmae, M.; Nanba, T.; Takeishi, K.; Azuma, N.; Ueno, A.; Ohfune, H.; Hayashi, H.; Udagawa, Y. Direct conversion of methane into methanol over MoO3/SiO2 catalyst in an excess amount of water vapor. Catal. Today 1998, 45, 29–33. [Google Scholar] [CrossRef]
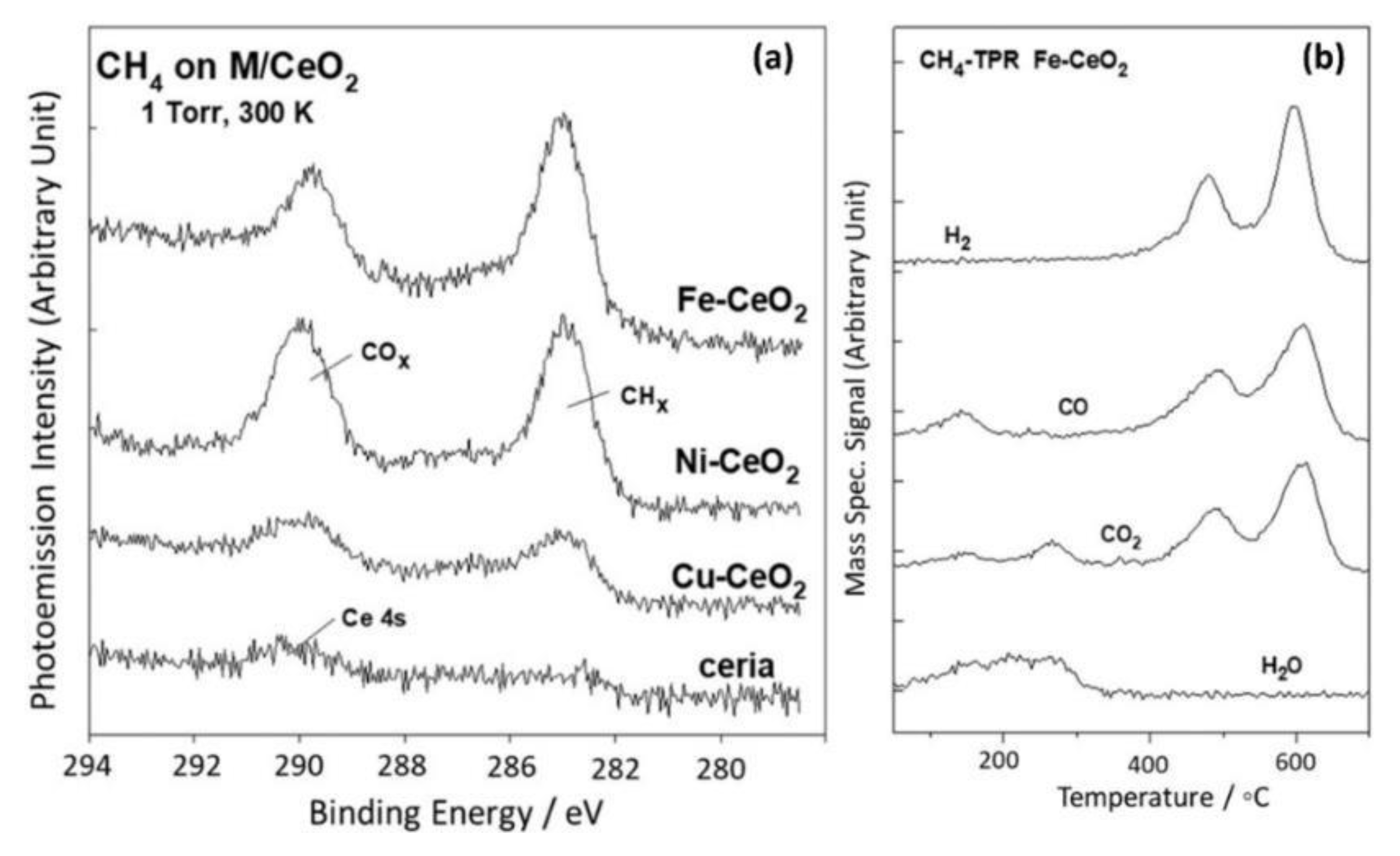
- Zhang, F.; Yao, S.; Liu, Z.; Gutiérrez, R.A.; Vovchok, D.; Cen, J.; Xu, W.; Ramírez, P.J.; Kim, T.; Senanayake, S.D.; et al. Reaction of Methane with MOx/CeO2 (M = Fe, Ni, and Cu) Catalysts: In Situ Studies with Time-Resolved X-ray Diffraction. J. Phys. Chem. C 2018, 122, 28739–28747. [Google Scholar] [CrossRef]
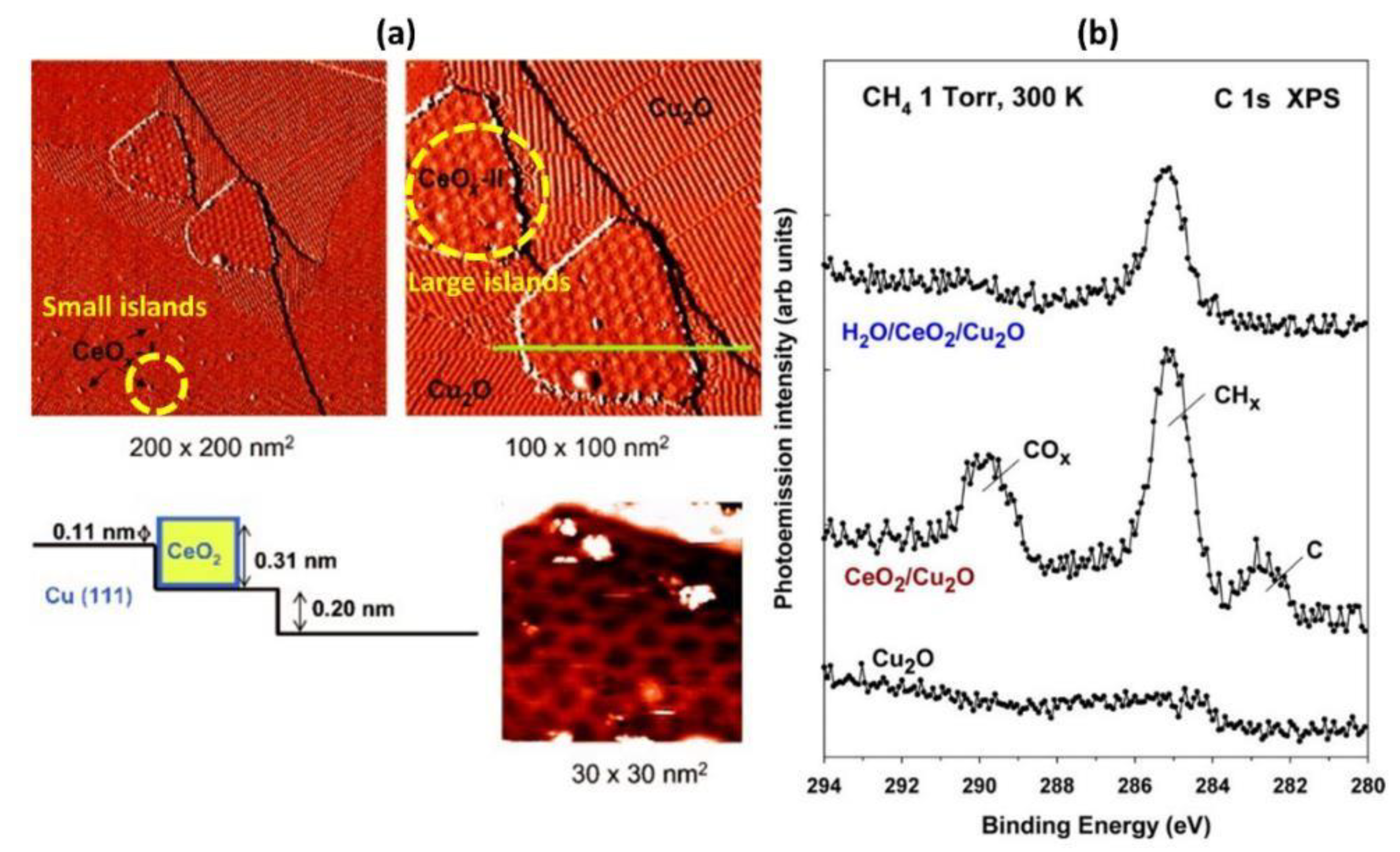
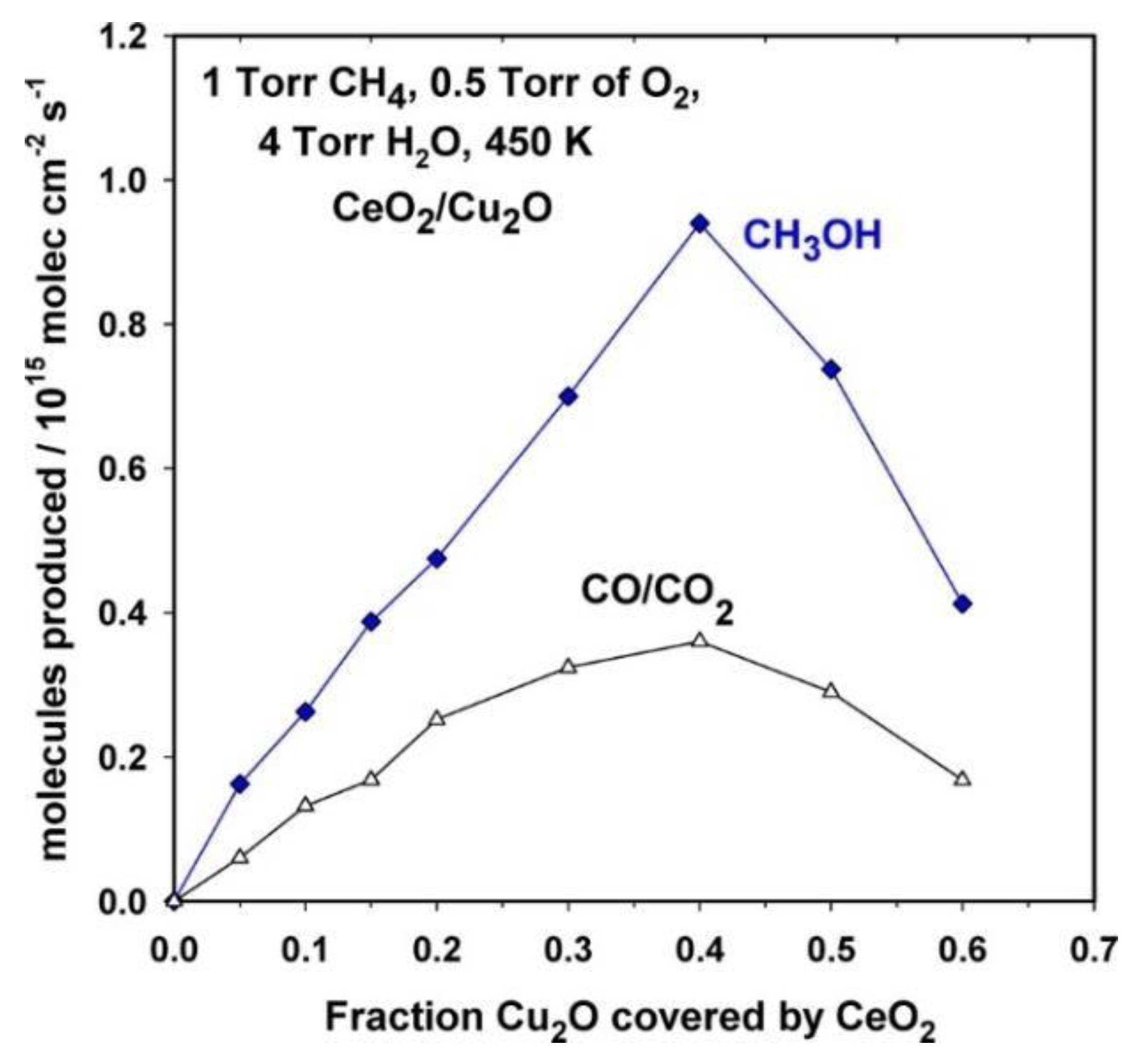
- Zuo, Z.; Ramírez, P.J.; Senanayake, S.D.; Liu, P.; Rodriguez, J.A. Low-Temperature Conversion of Methane to Methanol on CeOx/Cu2O Catalysts: Water Controlled Activation of the C–H Bond. J. Am. Chem. Soc. 2016, 138, 13810–13813. [Google Scholar] [CrossRef]
- Nibbelke, R.H.; Scheerova, J.; Decroon, M.H.J.M.; Marin, G.B. The Oxidative Coupling of Methane over MgO-Based Catalysts: A Steady-State Isotope Transient Kinetic Analysis. J. Catal. 1995, 156, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Haber, J.; Grzybowska, B. Mechanism of the oxidation of olefins on mixed oxide catalysts. J. Catal. 1973, 28, 489–492. [Google Scholar] [CrossRef]
- Haber, J.; Janas, J.; Schiavello, M.; Tilley, R.J.D. Tungsten oxides as catalysts in selective oxidation. J. Catal. 1983, 82, 395–403. [Google Scholar] [CrossRef]
- Védrine, J.C.; Fechete, I. Heterogeneous partial oxidation catalysis on metal oxides. Comptes Rendus Chim. 2016, 19, 1203–1225. [Google Scholar] [CrossRef]
- Pacchioni, G. Oxygen Vacancy: The Invisible Agent on Oxide Surfaces. Chemphyschem 2003, 4, 1041–1047. [Google Scholar] [CrossRef]
- Grootendorst, E.J.; Verbeek, Y.; Ponec, V. The Role of the Mars and Van Krevelen Mechanism in the Selective Oxidation of Nitrosobenzene and the Deoxygenation of Nitrobenzene on Oxidic Catalysts. J. Catal. 1995, 157, 706–712. [Google Scholar] [CrossRef]
- Liang, Z.; Li, T.; Kim, M.; Asthagiri, A.; Weaver, J.F. Low-temperature activation of methane on the IrO2(110) surface. Science 2017, 356, 299. [Google Scholar] [PubMed]
- Senftle, T.P.; van Duin, A.C.T.; Janik, M.J. Role of Site Stability in Methane Activation on PdxCe1–xOδ Surfaces. ACS Catal. 2015, 5, 6187–6199. [Google Scholar] [CrossRef]
- Mayernick, A.D.; Janik, M.J. Methane Activation and Oxygen Vacancy Formation over CeO2 and Zr, Pd Substituted CeO2 Surfaces. J. Phys. Chem. C 2008, 112, 14955–14964. [Google Scholar] [CrossRef]
- Arnold, E.W.; Sundaresan, S. The role of lattice oxygen in the dynamic behavior of oxide catalysts. Chem. Eng. Commun. 1987, 58, 213–230. [Google Scholar] [CrossRef]
- Wang, L.; Yu, Y.; He, H.; Zhang, Y.; Qin, X.; Wang, B. Oxygen vacancy clusters essential for the catalytic activity of CeO2 nanocubes for o-xylene oxidation. Sci. Rep. 2017, 7, 12845. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Nolan, M.; Parker, S.C.; Watson, G.W. The electronic structure of oxygen vacancy defects at the low index surfaces of ceria. Surf. Sci. 2005, 595, 223–232. [Google Scholar] [CrossRef]
- Sayle, T.X.T.; Parker, S.C.; Catlow, C.R.A. Surface oxygen vacancy formation on CeO2 and its role in the oxidation of carbon monoxide. J. Chem. Soc. Chem. Commun. 1992, 14, 977–978. [Google Scholar] [CrossRef]
- Mullins, D.R. The surface chemistry of cerium oxide. Surf. Sci. Rep. 2015, 70, 42–85. [Google Scholar] [CrossRef] [Green Version]
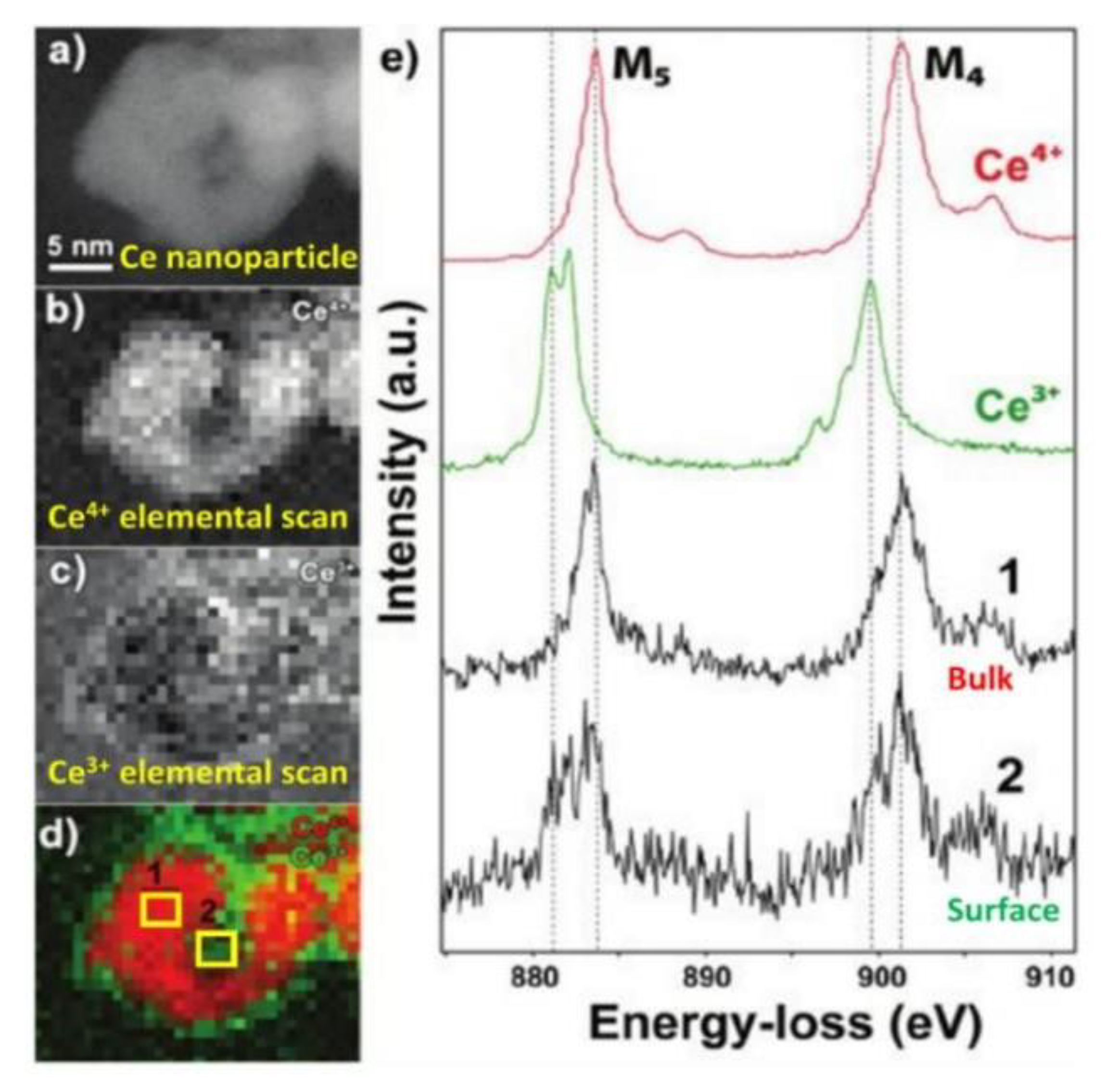
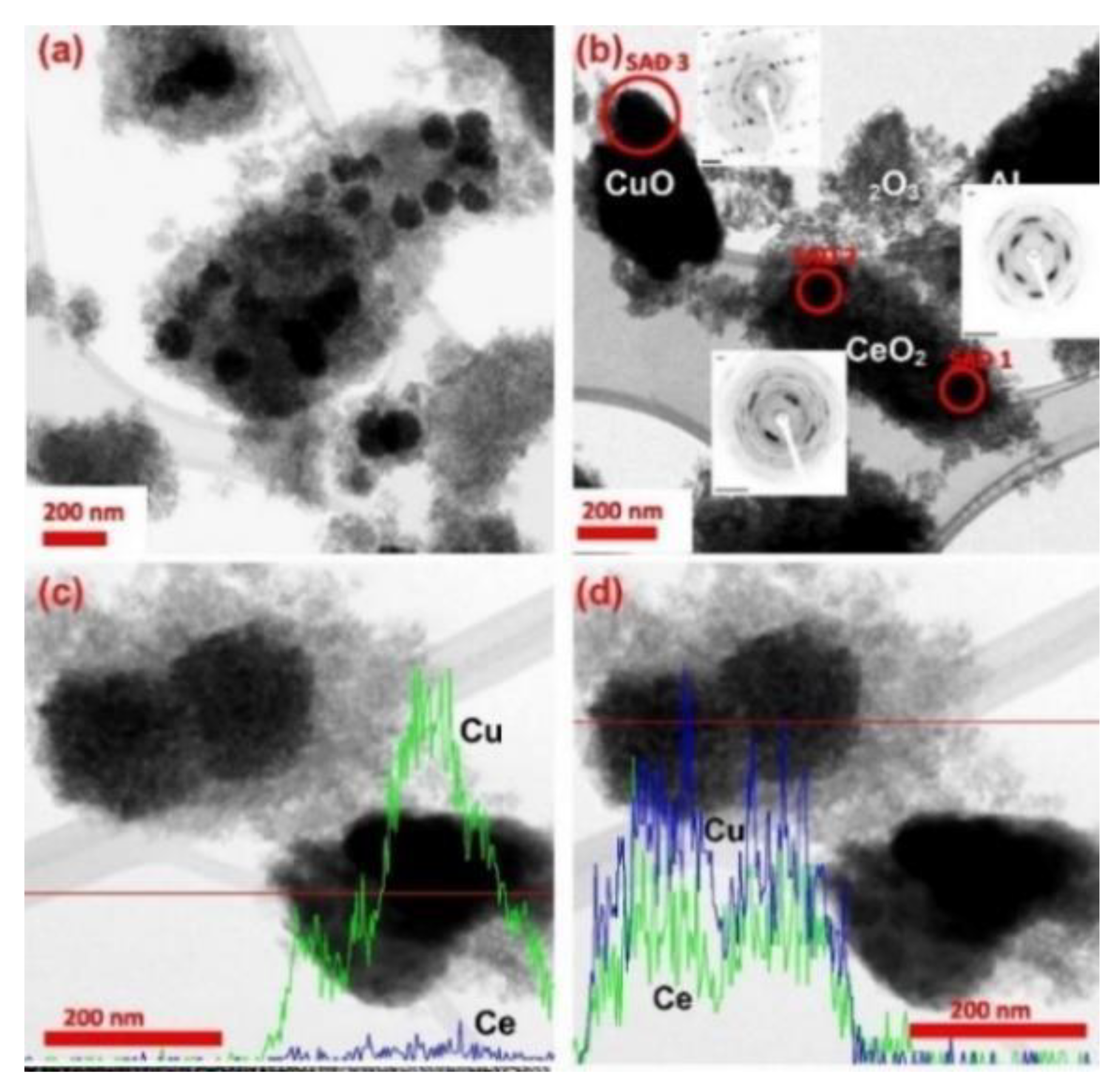
- Meledina, M.; Turner, S.; Galvita, V.V.; Poelman, H.; Marin, G.B.; Van Tendeloo, G. Local environment of Fe dopants in nanoscale Fe : CeO2−x oxygen storage material. Nanoscale 2015, 7, 3196–3204. [Google Scholar] [CrossRef] [PubMed]
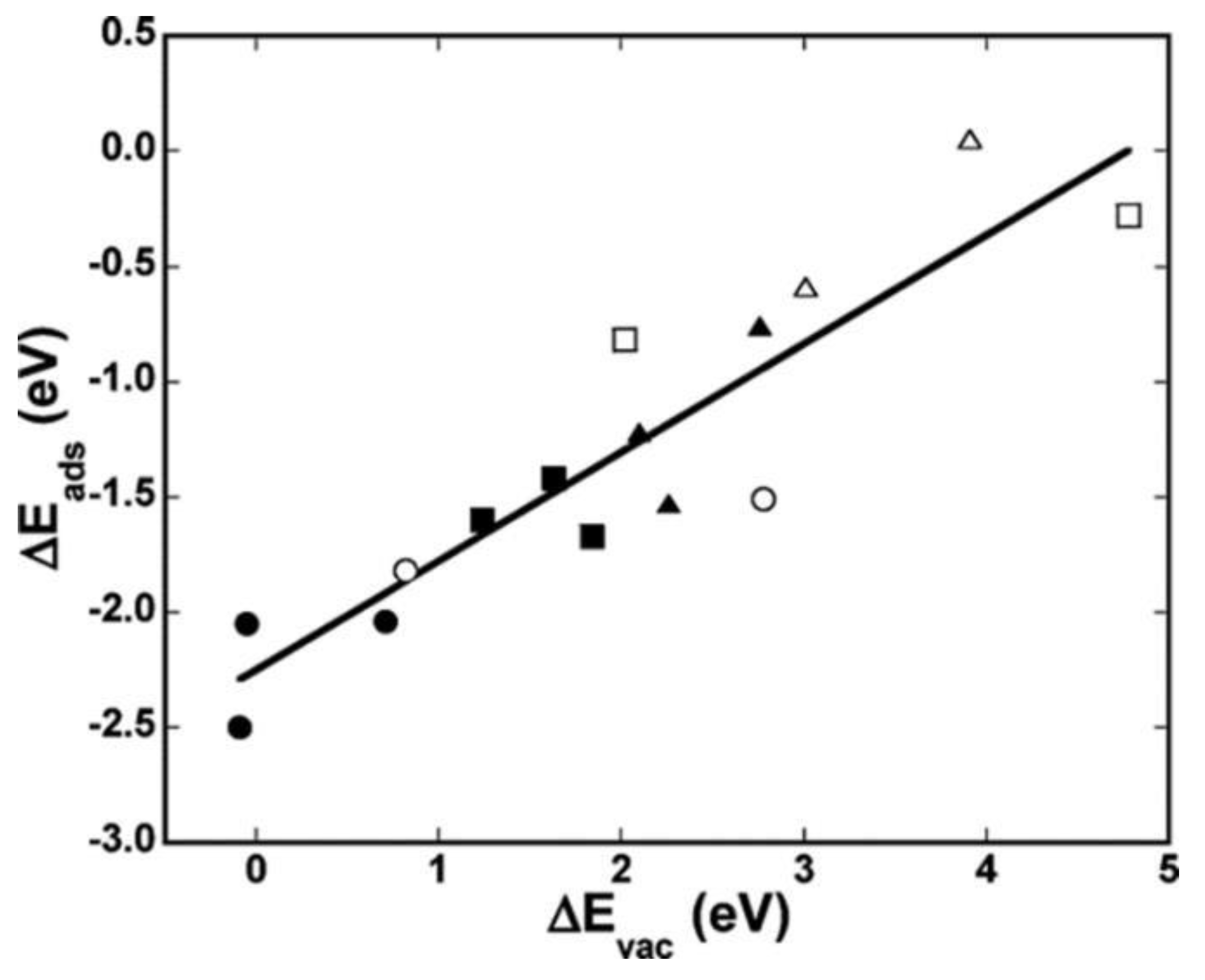
- Tian, D.; Li, K.; Wei, Y.; Zhu, X.; Zeng, C.; Cheng, X.; Zheng, Y.; Wang, H. DFT insights into oxygen vacancy formation and CH4 activation over CeO2 surfaces modified by transition metals (Fe, Co and Ni). Phys. Chem. Chem. Phys. 2018, 20, 11912–11929. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Puigdollers, A.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies. ACS Catal. 2017, 7, 6493–6513. [Google Scholar] [CrossRef] [Green Version]
- Menon, U.; Poelman, H.; Bliznuk, V.; Galvita, V.V.; Poelman, D.; Marin, G.B. Nature of the active sites for the total oxidation of toluene by CuOCeO2/Al2O3. J. Catal. 2012, 295, 91–103. [Google Scholar] [CrossRef]
- Pacchioni, G. Electronic interactions and charge transfers of metal atoms and clusters on oxide surfaces. Phys. Chem. Chem. Phys. 2013, 15, 1737–1757. [Google Scholar] [CrossRef] [PubMed]
- Kashfi-Sadabad, R.; Yazdani, S.; Huan, T.D.; Cai, Z.; Pettes, M.T. Role of Oxygen Vacancy Defects in the Electrocatalytic Activity of Substoichiometric Molybdenum Oxide. J. Phys. Chem. C 2018, 122, 18212–18222. [Google Scholar] [CrossRef]
- Singha, R.K.; Tsuji, Y.; Mahyuddin, M.H.; Yoshizawa, K. Methane Activation at the Metal–Support Interface of Ni4–CeO2(111) Catalyst: A Theoretical Study. J. Phys. Chem. C 2019, 123, 9788–9798. [Google Scholar] [CrossRef]
- Liu, Z.; Lustemberg, P.; Gutierrez, R.A.; Carey, J.J.; Palomino, R.M.; Vorokhta, M.; Grinter, D.C.; Ramirez, P.J.; Matolin, V.; Nolan, M.; et al. In Situ Investigation of Methane Dry Reforming on Metal/Ceria(111) Surfaces: Metal-Support Interactions and C-H Bond Activation at Low Temperature. Angew. Chem. Int. Ed. 2017, 56, 13041–13046. [Google Scholar] [CrossRef]
- Sadykov, V.A.; Kuznetsova, T.G.; Alikina, G.M.; Frolova, Y.V.; Lukashevich, A.I.; Potapova, Y.V.; Muzykantov, V.S.; Rogov, V.A.; Kriventsov, V.V.; Kochubei, D.I.; et al. Ceria- based fluorite-like oxide solid solutions as catalysts of methane selective oxidation into syngas by the lattice oxygen: Synthesis, characterization and performance. Catal. Today 2004, 93–95, 45–53. [Google Scholar] [CrossRef]
- Zeng, L.; Cheng, Z.; Fan, J.A.; Fan, L.-S.; Gong, J. Metal oxide redox chemistry for chemical looping processes. Nat. Rev. Chem. 2018, 2, 349–364. [Google Scholar] [CrossRef]
- Menon, U.; Galvita, V.V.; Marin, G.B. Reaction network for the total oxidation of toluene over CuO–CeO2/Al2O3. J. Catal. 2011, 283, 1–9. [Google Scholar] [CrossRef]
- Rodríguez, J.A.; Hrbek, J. Inverse oxide/metal catalysts: A versatile approach for activity tests and mechanistic studies. Surf. Sci. 2010, 604, 241–244. [Google Scholar] [CrossRef]
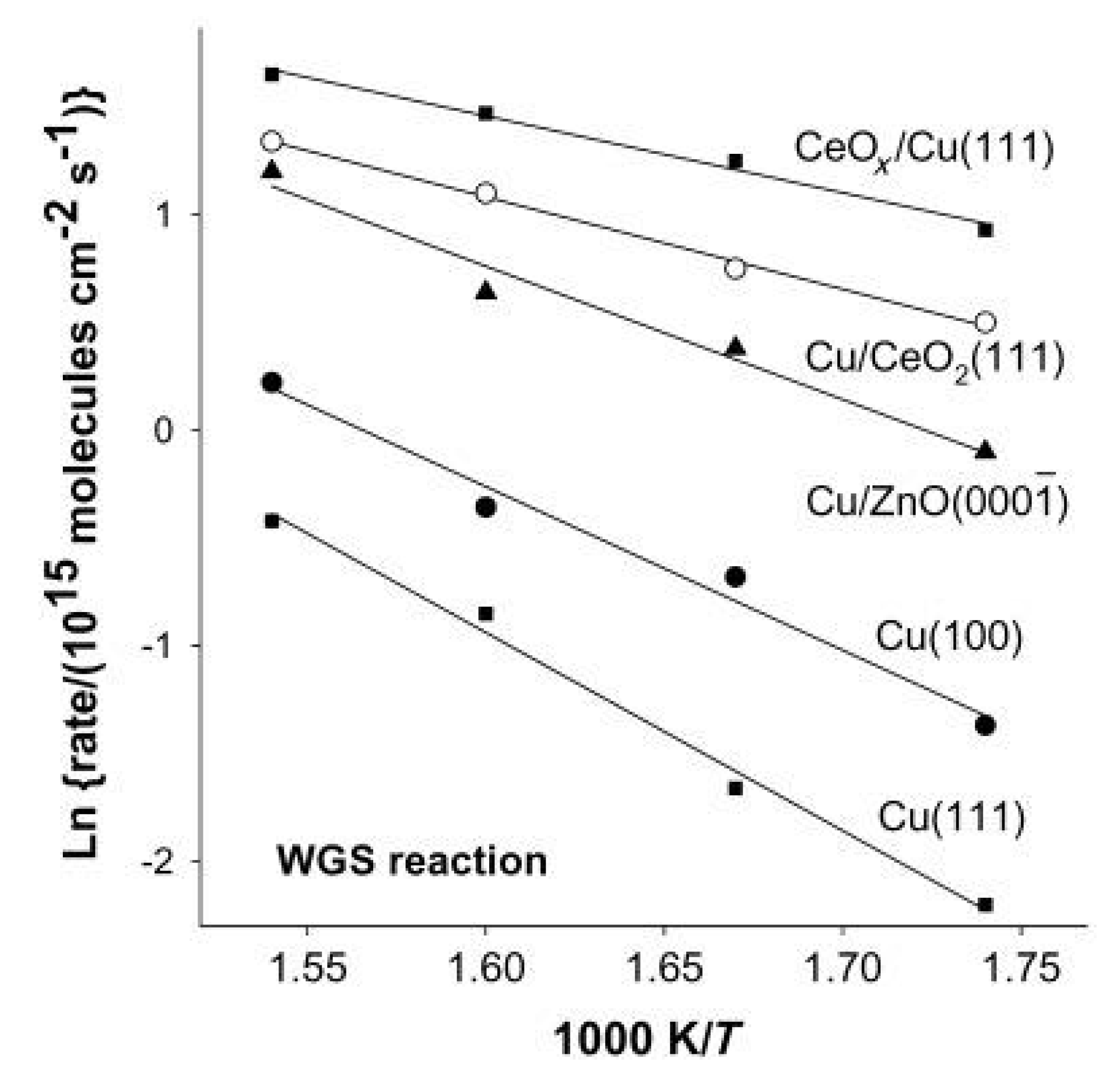
- Rodriguez, J.A.; Graciani, J.; Evans, J.; Park, J.B.; Yang, F.; Stacchiola, D.; Senanayake, S.D.; Ma, S.; Pérez, M.; Liu, P.; et al. Water-Gas Shift Reaction on a Highly Active Inverse CeOx/Cu(111) Catalyst: Unique Role of Ceria Nanoparticles. Angew. Chem. Int. Ed. 2009, 48, 8047–8050. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Stacchiola, D.; Rodriguez, J.A. Unique Properties of Ceria Nanoparticles Supported on Metals: Novel Inverse Ceria/Copper Catalysts for CO Oxidation and the Water-Gas Shift Reaction. Acc. Chem. Res. 2013, 46, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Loschen, C.; Migani, A.; Bromley, S.T.; Illas, F.; Neyman, K.M. Density functional studies of model cerium oxide nanoparticles. Phys. Chem. Chem. Phys. 2008, 10, 5730–5738. [Google Scholar] [CrossRef] [PubMed]
- Vayssilov, G.N.; Mihaylov, M.; Petkov, P.S.; Hadjiivanov, K.I.; Neyman, K.M. Reassignment of the Vibrational Spectra of Carbonates, Formates, and Related Surface Species on Ceria: A Combined Density Functional and Infrared Spectroscopy Investigation. J. Phys. Chem. C 2011, 115, 23435–23454. [Google Scholar] [CrossRef]
- Paier, J.; Penschke, C.; Sauer, J. Oxygen Defects and Surface Chemistry of Ceria: Quantum Chemical Studies Compared to Experiment. Chem. Rev. 2013, 113, 3949–3985. [Google Scholar] [CrossRef]
- Yang, Z.; Woo, T.K.; Baudin, M.; Hermansson, K. Atomic and electronic structure of unreduced and reduced CeO2 surfaces: A first-principles study. J. Chem. Phys. 2004, 120, 7741–7749. [Google Scholar] [CrossRef]
- Matko, V.; Milanović, M. Temperature-compensated capacitance–frequency converter with high resolution. Sens. Actuators A Phys. 2014, 220, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Fan, R.; Li, C.; Yang, X.; Li, H.; Lin, J.; Zhou, X.; Lv, R. A fast-response and highly linear humidity sensor based on quartz crystal microbalance. Sens. Actuators B Chem. 2019, 283, 659–665. [Google Scholar] [CrossRef]
- Lustemberg, P.G.; Palomino, R.M.; Gutiérrez, R.A.; Grinter, D.C.; Vorokhta, M.; Liu, Z.; Ramírez, P.J.; Matolín, V.; Ganduglia-Pirovano, M.V.; Senanayake, S.D.; et al. Direct Conversion of Methane to Methanol on Ni-Ceria Surfaces: Metal–Support Interactions and Water-Enabled Catalytic Conversion by Site Blocking. J. Am. Chem. Soc. 2018, 140, 7681–7687. [Google Scholar] [CrossRef]
- De Vrieze, J.E.; Thybaut, J.W.; Saeys, M. Role of Surface Hydroxyl Species in Copper-Catalyzed Hydrogenation of Ketones. ACS Catal. 2018, 8, 7539–7548. [Google Scholar] [CrossRef]
- Gunasooriya, G.T.K.K.; van Bavel, A.P.; Kuipers, H.P.C.E.; Saeys, M. Key Role of Surface Hydroxyl Groups in C–O Activation during Fischer–Tropsch Synthesis. ACS Catal. 2016, 6, 3660–3664. [Google Scholar] [CrossRef]
- Henderson, M.A. The interaction of water with solid surfaces: Fundamental aspects revisited. Surf. Sci. Rep. 2002, 46, 1–308. [Google Scholar] [CrossRef]
- Kauppi, E.I.; Honkala, K.; Krause, A.O.I.; Kanervo, J.M.; Lefferts, L. ZrO2 Acting as a Redox Catalyst. Top. Catal. 2016, 59, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.; Lau, S.L.J.; Krcha, M.D.; Janik, M.J. Correlation of Methane Activation and Oxide Catalyst Reducibility and Its Implications for Oxidative Coupling. ACS Catal. 2016, 6, 1812–1821. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Bliznuk, V.; Detavernier, C.; Marin, G.B. CeO2-Modified Fe2O3 for CO2 Utilization via Chemical Looping. Ind. Eng. Chem. Res. 2013, 52, 8416–8426. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. Catalyst-assisted chemical looping for CO2 conversion to CO. Appl. Catal. B 2015, 164, 184–191. [Google Scholar] [CrossRef]
- Siriwardane, R.; Tian, H.; Richards, G.; Simonyi, T.; Poston, J. Chemical-Looping Combustion of Coal with Metal Oxide Oxygen Carriers. Energy Fuels 2009, 23, 3885–3892. [Google Scholar] [CrossRef]
- Hu, J.; Galvita, V.V.; Poelman, H.; Marin, B.G. Advanced Chemical Looping Materials for CO2 Utilization: A Review. Materials 2018, 11, 1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Du, X.; Yu, W.; Zhang, J.; Wei, J. Coproduction of Hydrogen and Methanol from Methane by Chemical Looping Reforming. Ind. Eng. Chem. Res. 2019, 58, 10296–10306. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Marin, G.B. Hydrogen Production from Methane and Carbon Dioxide 1007 by Catalyst-Assisted Chemical Looping. Top. Catal. 2011, 54, 907. [Google Scholar] [CrossRef]
- Mihai, O.; Chen, D.; Holmen, A. Chemical looping methane partial oxidation: The effect of the crystal size and O content of LaFeO3. J. Catal. 2012, 293, 175–185. [Google Scholar] [CrossRef]
- Park, M.B.; Park, E.D.; Ahn, W.-S. Recent Progress in Direct Conversion of Methane to Methanol Over Copper-Exchanged Zeolites. Front. Chem. 2019, 7, 514. [Google Scholar] [CrossRef] [PubMed]


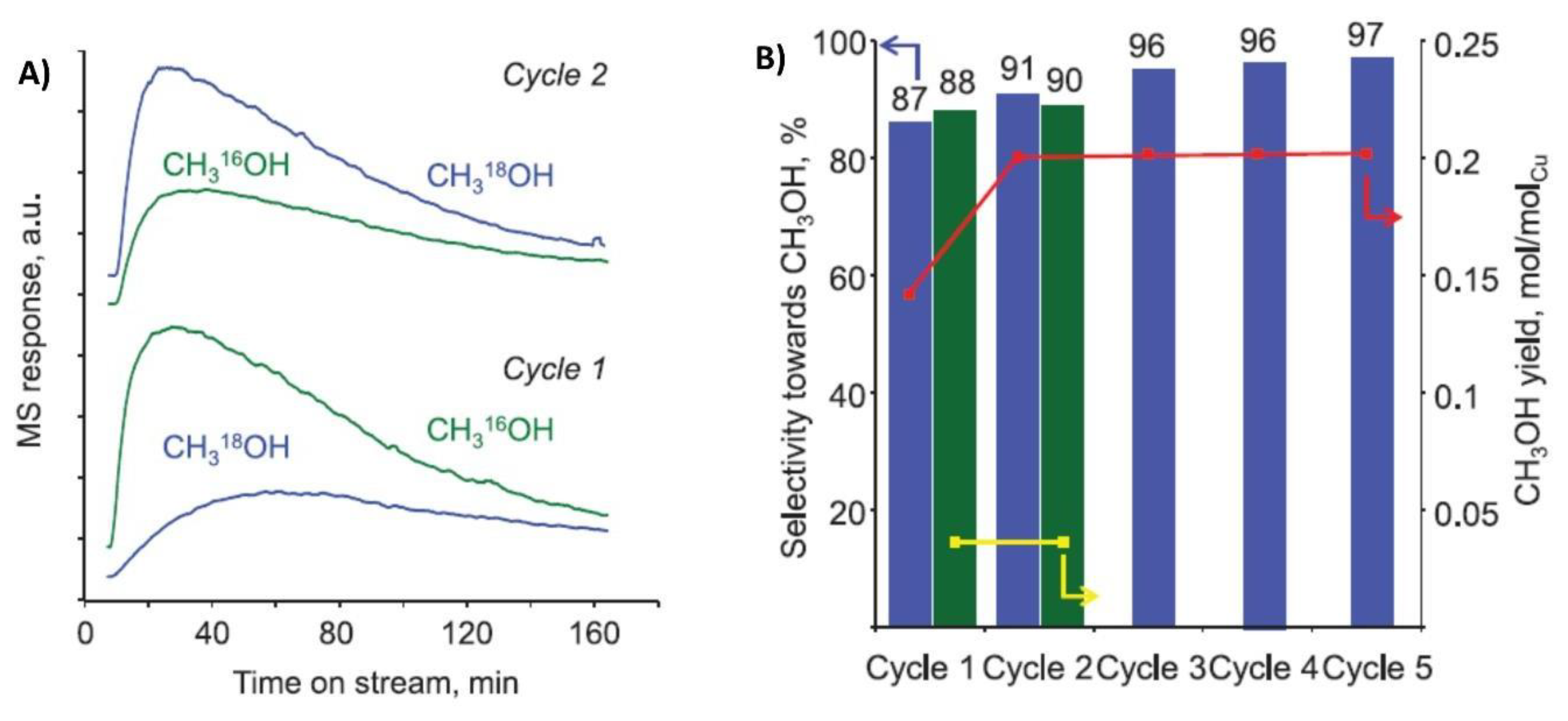
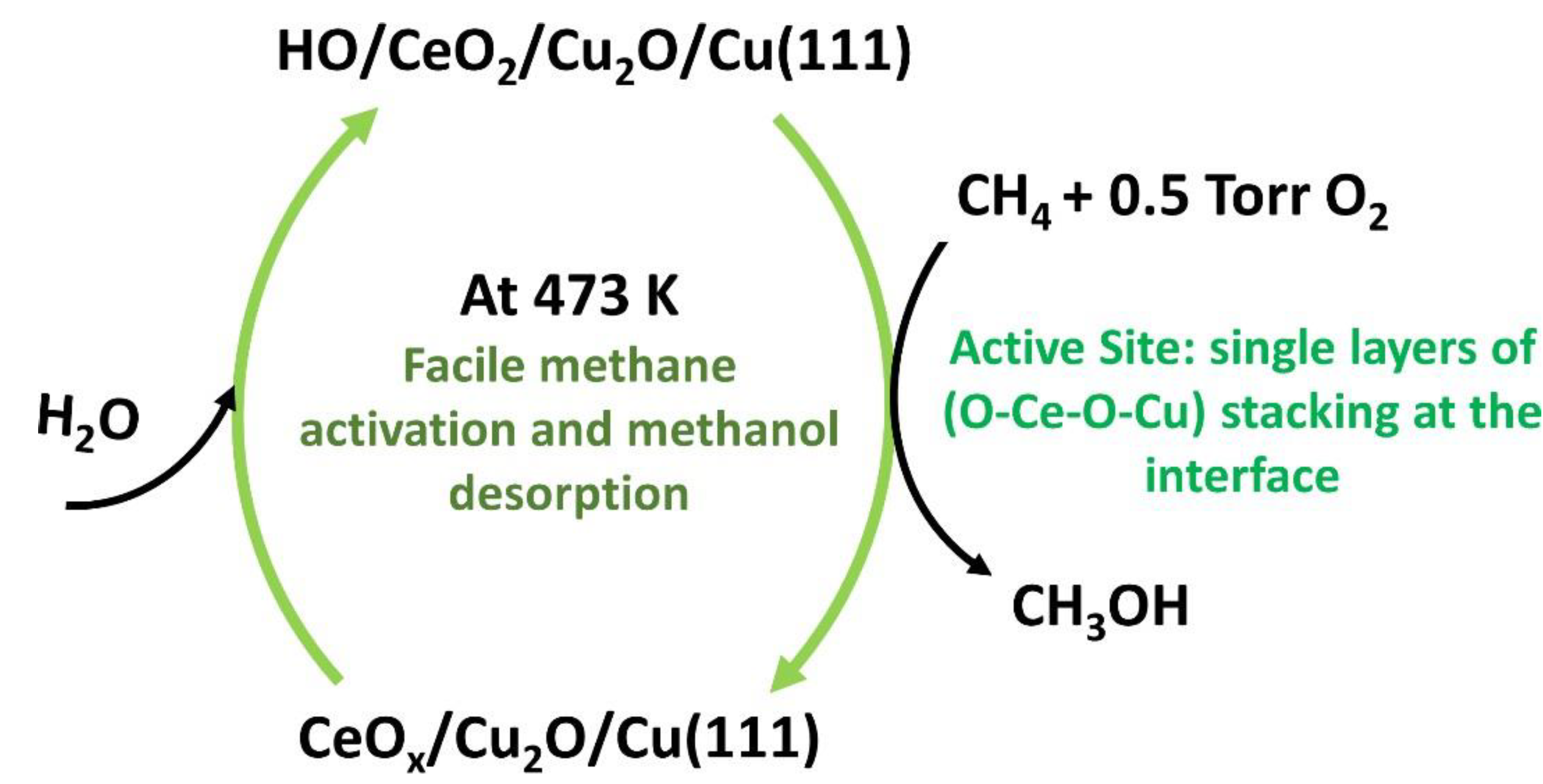

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Process | Preparation | Methanol Selectivity (%) | Methanol Productivity (mol/g/s) | Ref. |
|---|---|---|---|---|---|
| pMMO | Biocatalytic with O2 as oxidant | Embedding pMMO in a polymer hydrogel | 100 | 10−8 | [38] |
| Pt (II) salts | Liquid phase with H2SO4 as oxidant | Homogenous phase | 81 | 10−7 mol/cm3/s | [44] |
| Cu-Fe/silicalite- 1 | Aqueous hydrogen peroxide | Mixed homogenous phase | >90 | 10−7 | [45] |
| AuPdCu/TiO2 | Aqueous hydrogen peroxide | Incipient wetness impregnation | >80 | NA | [52] |
| Fe-ZSM5 | N2O as oxidant | Hydrothermal synthesis | >90 | NA | [54,55] |
| Cu-MOR | Water as oxidant | Conventional ion exchange | >90 | 10−8 | [22,40] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, R.; Poelman, H.; Marin, G.B.; Galvita, V.V. Approaches for Selective Oxidation of Methane to Methanol. Catalysts 2020, 10, 194. https://doi.org/10.3390/catal10020194
Sharma R, Poelman H, Marin GB, Galvita VV. Approaches for Selective Oxidation of Methane to Methanol. Catalysts. 2020; 10(2):194. https://doi.org/10.3390/catal10020194
Chicago/Turabian StyleSharma, Richa, Hilde Poelman, Guy B. Marin, and Vladimir V. Galvita. 2020. "Approaches for Selective Oxidation of Methane to Methanol" Catalysts 10, no. 2: 194. https://doi.org/10.3390/catal10020194
APA StyleSharma, R., Poelman, H., Marin, G. B., & Galvita, V. V. (2020). Approaches for Selective Oxidation of Methane to Methanol. Catalysts, 10(2), 194. https://doi.org/10.3390/catal10020194