Evoked Methane Photocatalytic Conversion to C2 Oxygenates over Ceria with Oxygen Vacancy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
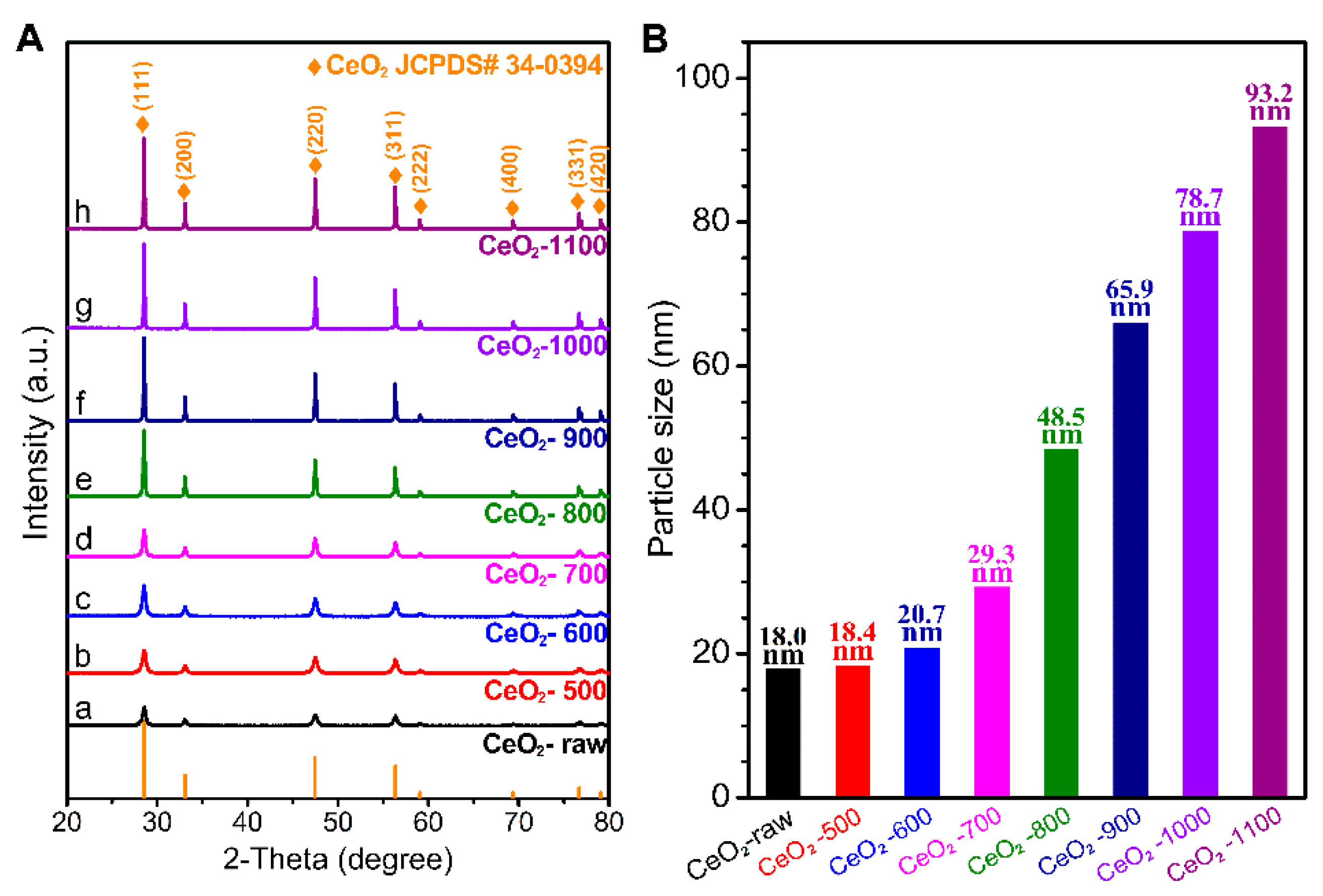
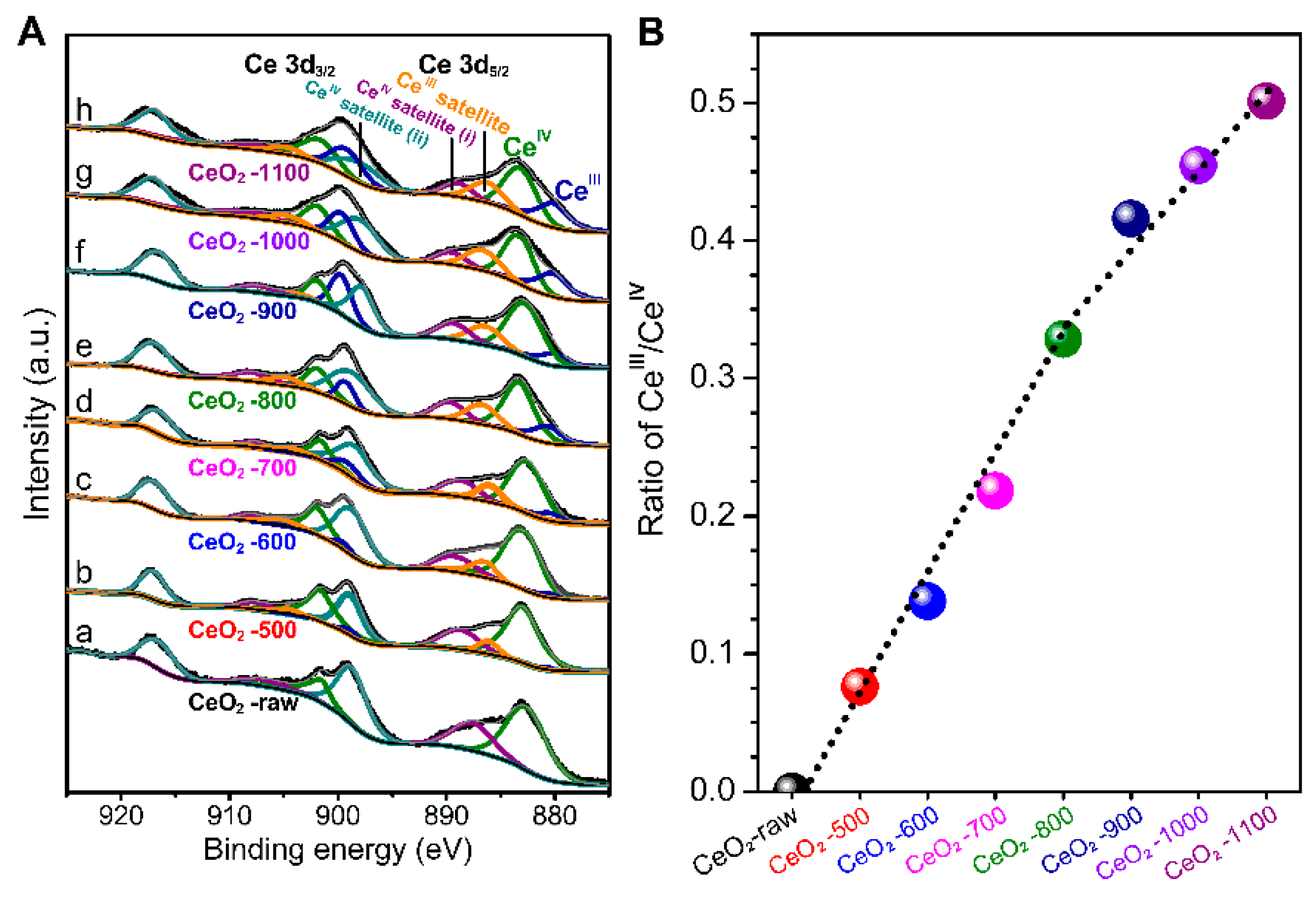
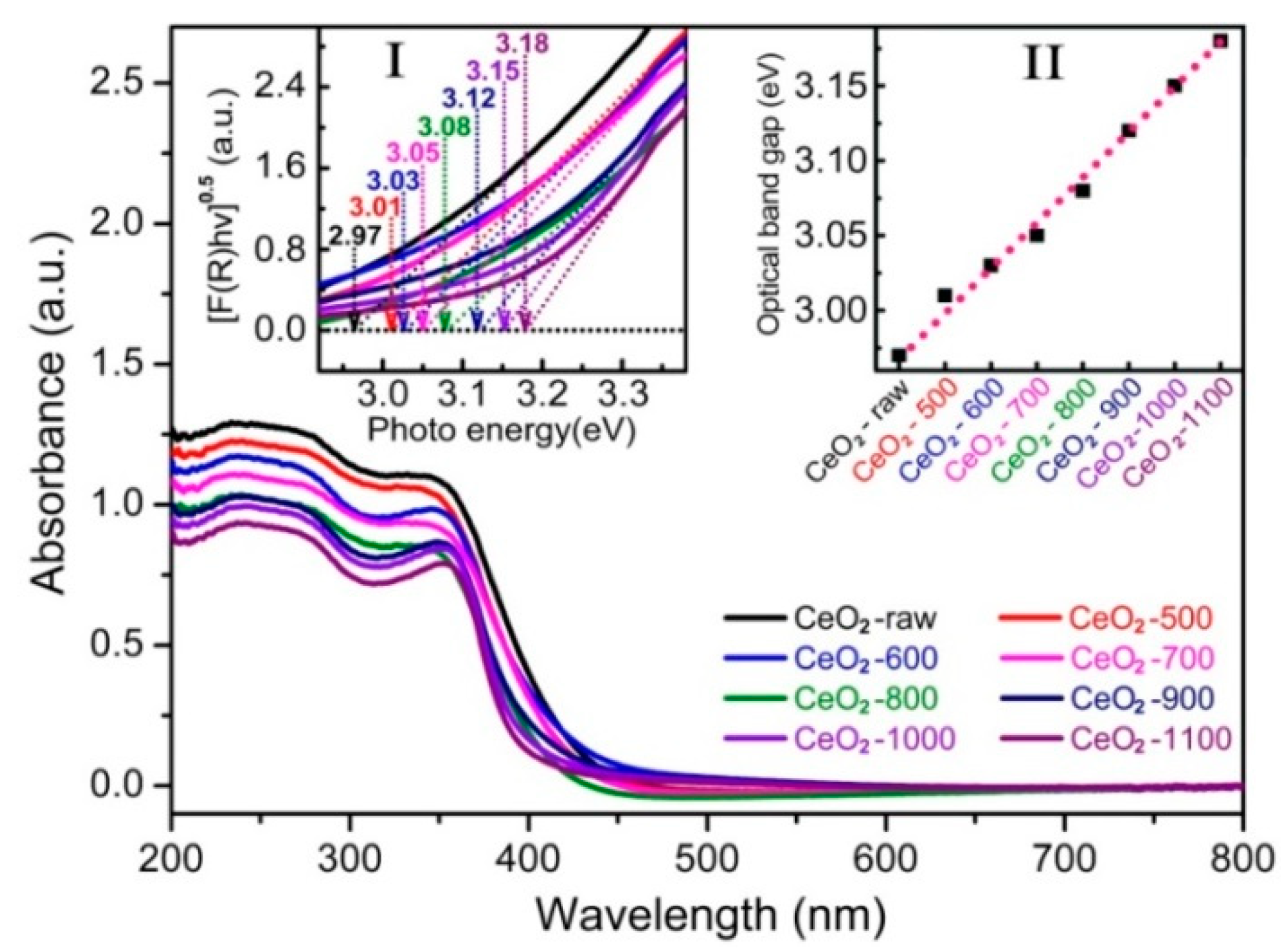
2.1. Characterizations of the Catalysts
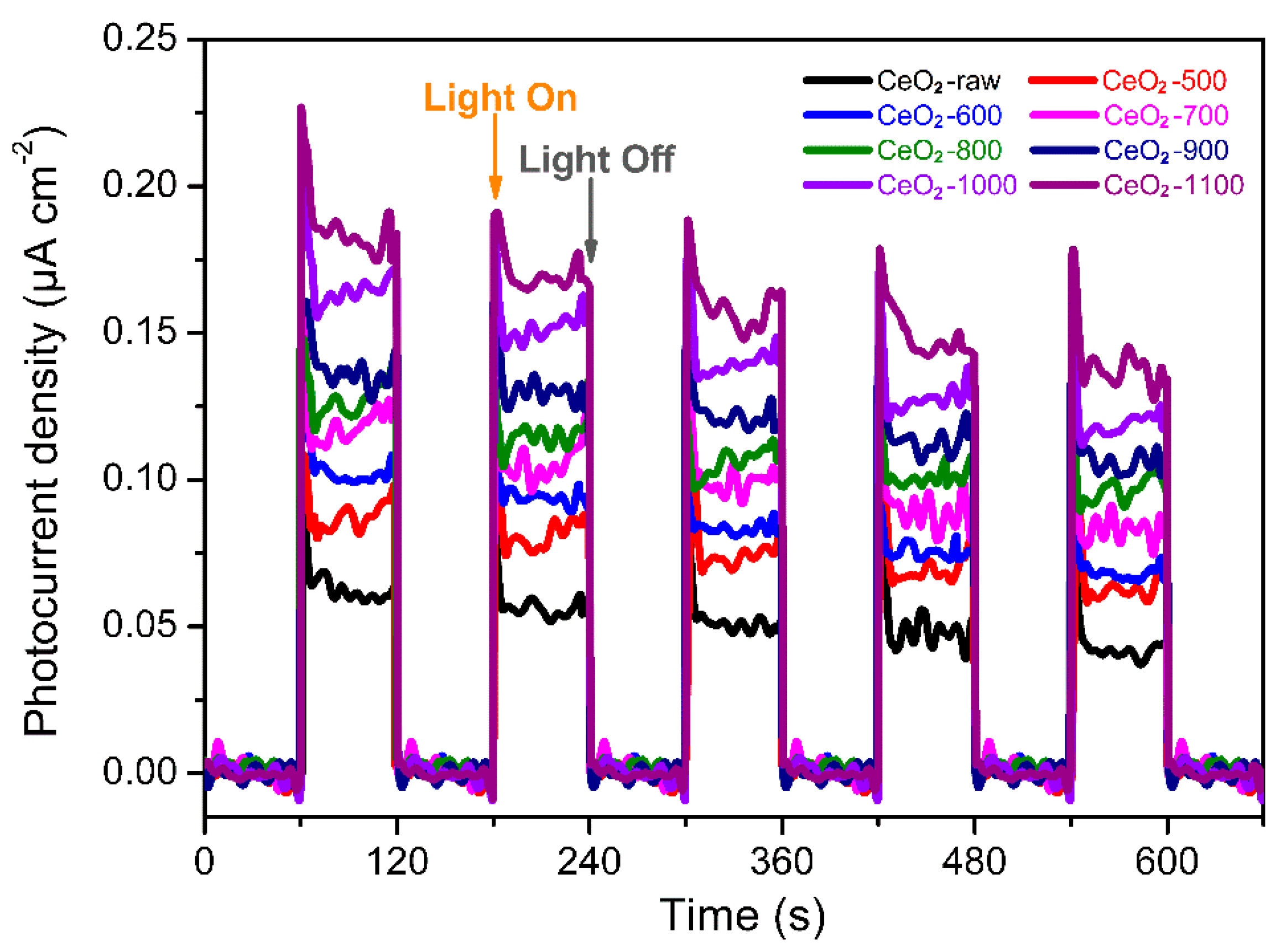
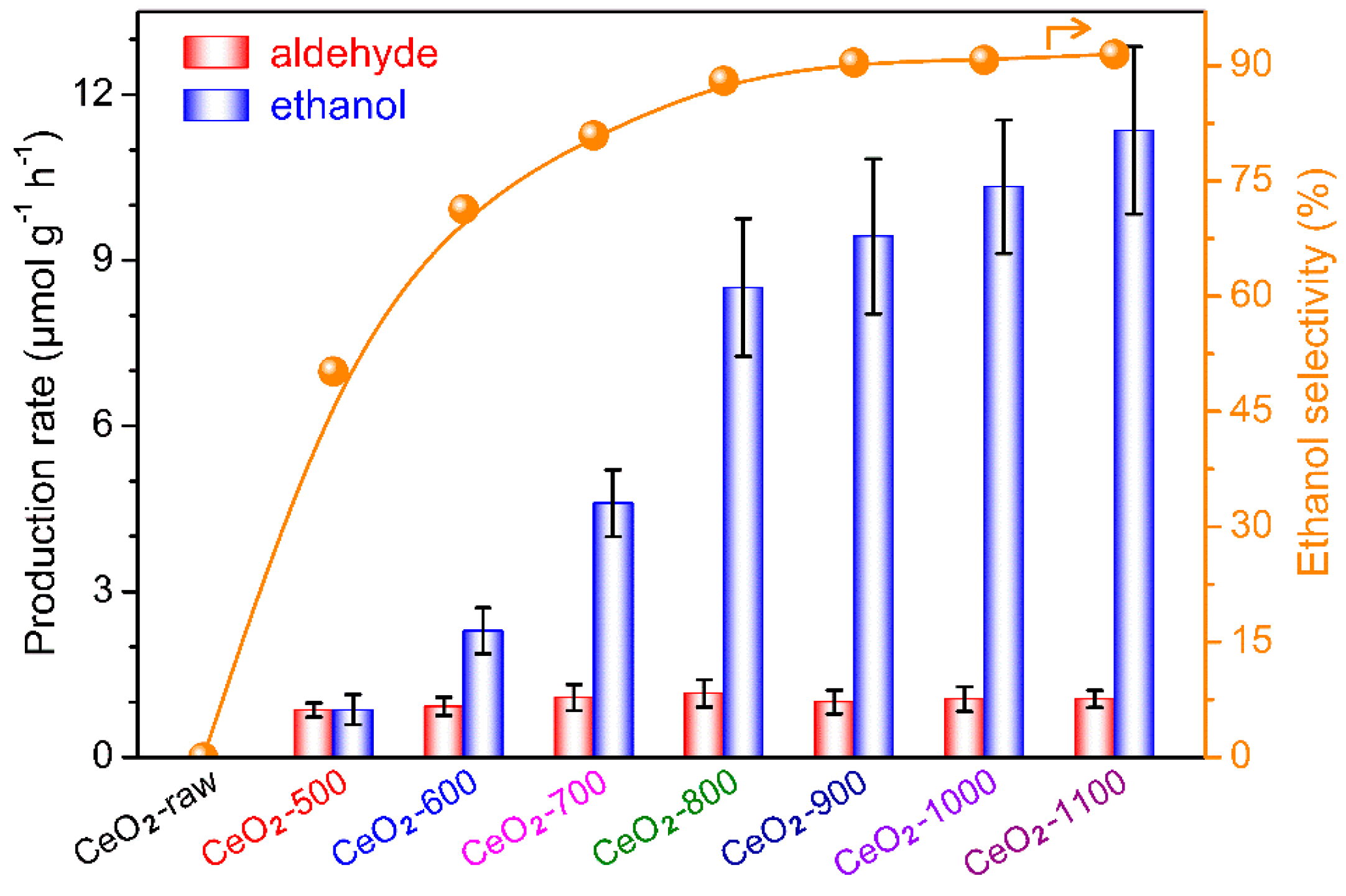
2.2. Photocatalytic Conversion of CH4
3. Materials and Methods
3.1. Preparation of Catalysts
3.2. Physical Characterizations
3.3. Photocatalytic Activity Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yuliati, L.; Yoshida, H. Photocatalytic conversion of methane. Chem. Soc. Rev. 2008, 37, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous functionalization of methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef]
- Xie, S.; Lin, S.; Zhang, Q.; Tian, Z.; Wang, Y. Selective electrocatalytic conversion of methane to fuels and chemicals. J. Energy Chem. 2018, 27, 1629–1636. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: Challenges and prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Tang, P.; Zhu, Q.; Wu, Z.; Ma, D. Methane activation: The past and future. Energy Environ. Sci. 2014, 7, 2580–2591. [Google Scholar] [CrossRef]
- Paunović, V.; Zichittella, G.; Moser, M.; Amrute, A.P.; Pérez-Ramírez, J. Catalyst design for natural-gas upgrading through oxybromination chemistry. Nat. Chem. 2016, 8, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Dempsey, J.L. When electrochemistry met methane: Rapid catalyst oxidation fuels hydrocarbon functionalization. ACS Cent. Sci. 2017, 3, 1137–1139. [Google Scholar] [CrossRef] [PubMed]
- Nikolla, E.; Schwank, J.W.; Linic, S. Hydrocarbon steam reforming on Ni alloys at solid oxide fuel cell operating conditions. Catal. Today 2008, 136, 243–248. [Google Scholar] [CrossRef]
- Wang, B.; Albarracín-Suazo, S.; Pagán-Torres, Y.; Nikolla, E. Advances in methane conversion processes. Catal. Today 2017, 285, 147–158. [Google Scholar] [CrossRef]
- Enger, B.C.; Lødeng, R.; Holmen, A. A review of catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanisms over transition metal catalysts. Appl. Catal. A Gen. 2008, 346, 1–27. [Google Scholar] [CrossRef]
- Olivos-Suarez, A.I.; Szécsényi, A.; Hensen, E.J.; Ruiz-Martinez, J.; Pidko, E.A.; Gascon, J. Strategies for the direct catalytic valorization of methane using heterogeneous catalysis: Challenges and opportunities. ACS Catal. 2016, 6, 2965–2981. [Google Scholar] [CrossRef]
- Karakaya, C.; Kee, R.J. Progress in the direct catalytic conversion of methane to fuels and chemicals. Prog. Energy Combust. Sci. 2016, 55, 60–97. [Google Scholar] [CrossRef]
- Mesters, C. A selection of recent advances in C1 chemistry. Annu. Rev. Chem. Biomol. Eng 2016, 7, 223–238. [Google Scholar] [CrossRef]
- Song, J.; Sun, Y.; Ba, R.; Huang, S.; Zhao, Y.; Zhang, J.; Zhu, Y. Monodisperse Sr–La2O3 hybrid nanofibers for oxidative coupling of methane to synthesize C2 hydrocarbons. Nanoscale 2015, 7, 2260–2264. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Tan, D. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef]
- Soulivong, D.; Norsic, S.; Taoufik, M.; Coperet, C.; Thivolle-Cazat, J.; Chakka, S.; Basset, J.M. Non-Oxidative Coupling Reaction of Methane to Ethane and Hydrogen Catalyzed by the Silica-Supported Tantalum Hydride: (≡SiO)2Ta−H. J. Am. Chem. Soc. 2008, 130, 5044–5045. [Google Scholar] [CrossRef]
- Murcia-López, S.; Villa, K.; Andreu, T.; Morante, J.R. Partial oxidation of methane to methanol using bismuth-based photocatalysts. ACS Catal. 2014, 4, 3013–3019. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Pan, X.; Cortie, D.; Huang, X.; Yi, Z. Photocatalytic oxidation of methane over silver decorated zinc oxide nanocatalysts. Nat. Commun. 2016, 7, 12273. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Jin, R.; Li, A.; Bi, Y.; Ruan, Q.; Deng, Y.; Zhang, Y.; Yao, S.; Sankar, G.; Ma, D.; et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 2018, 1, 889–896. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, L.; Wang, W. Direct functionalization of methane into ethanol over copper modified polymeric carbon nitride via photocatalysis. Nat. Commun. 2019, 10, 506. [Google Scholar] [CrossRef]
- Ji, P.; Zhang, J.; Chen, F.; Anpo, M. Study of Adsorption and Degradation of Acid Orange 7 on the Surface of CeO2 under Visible Light Irradiation. Appl. Catal. B 2009, 85, 148–154. [Google Scholar] [CrossRef]
- Feng, T.; Wang, X.; Feng, G. Synthesis of Novel CeO2 Microspheres with Enhanced Solar Light Photocatalyic Properties. Mater. Lett. 2013, 100, 36–39. [Google Scholar] [CrossRef]
- Chen, F.; Cao, Y.; Jia, D. Preparation and Photocatalytic Property of CeO2 Lamellar. Appl. Surf. Sci. 2011, 257, 9226–9231. [Google Scholar] [CrossRef]
- Sabari Arul, N.; Mangalaraj, D.; Kim, T.W.; Chen, P.C.; Ponpandian, N.; Meena, P.; Masuda, Y. Synthesis of CeO2 Nanorods with Improved Photocatalytic Activity: Comparison between Precipitation and Hydrothermal Process. J. Mater. Sci. Mater. Electron. 2013, 24, 1644–1650. [Google Scholar] [CrossRef]
- Chan, S.H.S.; Yeong Wu, T.; Juan, J.C.; Teh, C.Y. Recent Developments of Metal Oxide Semiconductors as Photocatalysts in Advanced Oxidation Processes (AOPs) for Treatment of Dye Waste-Water. J. Chem. Technol. Biotechnol. 2011, 86, 1130–1158. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Arakawa, H. Cerium Dioxide as a Photocatalyst for Water Decomposition to O2 in the Presence of Ce aq4+ and Fe aq3+ Species. J. Mol. Catal. A Chem. 2000, 161, 105–113. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Uesigi, T.; Abe, Y.; Sayama, K.; Arakawa, H. The Photocatalytic Oxidation of Water to O2 over Pure CeO2, WO3 and TiO2 Using Fe3+ and Ce4+ as Electron Acceptors. Appl. Catal. A 2001, 205, 117–128. [Google Scholar] [CrossRef]
- Primo, A.; Marino, T.; Corma, A.; Molinari, R.; García, H. Efficient Visible-Light Photocatalytic Water Splitting by Minute Amounts of Gold Supported on Nanoparticulate CeO2 Obtained by a Biopolymer Templating Method. J. Am. Chem. Soc. 2011, 133, 6930–6933. [Google Scholar] [CrossRef]
- Abanades, S.; Flamant, G. Thermochemical hydrogen production from a two-step solar-driven water-splitting cycle based on cerium oxides. Sol. Energy 2006, 80, 1611–1623. [Google Scholar] [CrossRef]
- Meiqing, S.; Xinquan, W.; Yuan, A.; Duan, W.; Minwei, Z.; Jun, W. Dynamic Oxygen Storage Capacity Measurements on Ceria-Based Material. J. Rare Earths 2007, 25, 48–52. [Google Scholar] [CrossRef]
- Melchionna, M.; Fornasiero, P. The role of ceria-based nanostructured materials in energy applications. Mater. Today 2014, 17, 349–357. [Google Scholar] [CrossRef]
- Karran Woan, Y.-Y.T.W.S. Synthesis and characterization of luminescent cerium oxide nanoparticles. Nanomedicine 2010, 5, 233–242. [Google Scholar]
- Muduli, S.K.; Wang, S.; Chen, S.; Ng, C.F.; Huan, C.H.A.; Sum, T.C.; Soo, H.S. Mesoporous cerium oxide nanospheres for the visible-light driven photocatalytic degradation of dyes. Beilstein J. Nanotechnol. 2014, 5, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, B.; Chetri, P.; Choudhury, A. Oxygen defects and formation of Ce3+ affecting the photocatalytic performance of CeO2 nanoparticles. RSC Adv. 2014, 4, 4663–4671. [Google Scholar] [CrossRef]
- Gao, H.; Yang, H.; Yang, G.; Wang, S. Effects of oxygen vacancy and sintering temperature on the photoluminescence properties and photocatalytic activity of CeO2 nanoparticles with high uniformity. Mater. Technol. 2018, 33, 321–332. [Google Scholar] [CrossRef]
- Zhou, X.D.; Huebner, W. Size-induced lattice relaxation in CeO2 nanoparticles. Appl. Phys. Lett. 2001, 79, 3512–3514. [Google Scholar] [CrossRef]
- Choudhury, B.; Choudhury, A. Ce3+ and oxygen vacancy mediated tuning of structural and optical properties of CeO2 nanoparticles. Mater. Chem. Phys. 2012, 131, 666–671. [Google Scholar] [CrossRef]
- Choudhury, B.; Chetri, P.; Choudhury, A. Annealing temperature and oxygen-vacancy-dependent variation of lattice strain, band gap and luminescence properties of CeO2 nanoparticles. J. Exp. Nanosci. 2015, 10, 103–114. [Google Scholar] [CrossRef]
- Mogensen, M.; Sammes, N.M.; Tompsett, G.A. Physical, chemical and electrochemical properties of pure and doped ceria. Solid State Ion. 2000, 129, 63–94. [Google Scholar] [CrossRef]
- Goubin, F.; Rocquefelte, X.; Whangbo, M.H.; Montardi, Y.; Brec, R.; Jobic, S. Experimental and theoretical characterization of the optical properties of CeO2, SrCeO3, and Sr2CeO4 containing Ce4+ (f0) ions. Chem. Mater. 2004, 16, 662–669. [Google Scholar] [CrossRef]
- Shi, S.; Ke, X.; Ouyang, C.; Zhang, H.; Ding, H.; Tang, Y.; Tang, W. First-principles investigation of the bonding, optical and lattice dynamical properties of CeO2. J. Power Sources 2009, 194, 830–834. [Google Scholar] [CrossRef]
- Murali, A.; Lan, Y.P.; Sohn, H.Y. Effect of oxygen vacancies in non-stoichiometric ceria on its photocatalytic properties. Nano Struct. Nano Objects 2019, 18, 100257. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.; Chen, W.; Wu, G.; Song, Y.; Dong, X.; Li, G.; Fang, J.; Wei, W.; Sun, Y. Evoked Methane Photocatalytic Conversion to C2 Oxygenates over Ceria with Oxygen Vacancy. Catalysts 2020, 10, 196. https://doi.org/10.3390/catal10020196
Du J, Chen W, Wu G, Song Y, Dong X, Li G, Fang J, Wei W, Sun Y. Evoked Methane Photocatalytic Conversion to C2 Oxygenates over Ceria with Oxygen Vacancy. Catalysts. 2020; 10(2):196. https://doi.org/10.3390/catal10020196
Chicago/Turabian StyleDu, Jin, Wei Chen, Gangfeng Wu, Yanfang Song, Xiao Dong, Guihua Li, Jianhui Fang, Wei Wei, and Yuhan Sun. 2020. "Evoked Methane Photocatalytic Conversion to C2 Oxygenates over Ceria with Oxygen Vacancy" Catalysts 10, no. 2: 196. https://doi.org/10.3390/catal10020196
APA StyleDu, J., Chen, W., Wu, G., Song, Y., Dong, X., Li, G., Fang, J., Wei, W., & Sun, Y. (2020). Evoked Methane Photocatalytic Conversion to C2 Oxygenates over Ceria with Oxygen Vacancy. Catalysts, 10(2), 196. https://doi.org/10.3390/catal10020196