Active Expression of Membrane-Bound L-Amino Acid Deaminase from Proteus mirabilis in Recombinant Escherichia coli by Fusion with Maltose-Binding Protein for Enhanced Catalytic Performance


Abstract
:1. Introduction
2. Results and Discussion
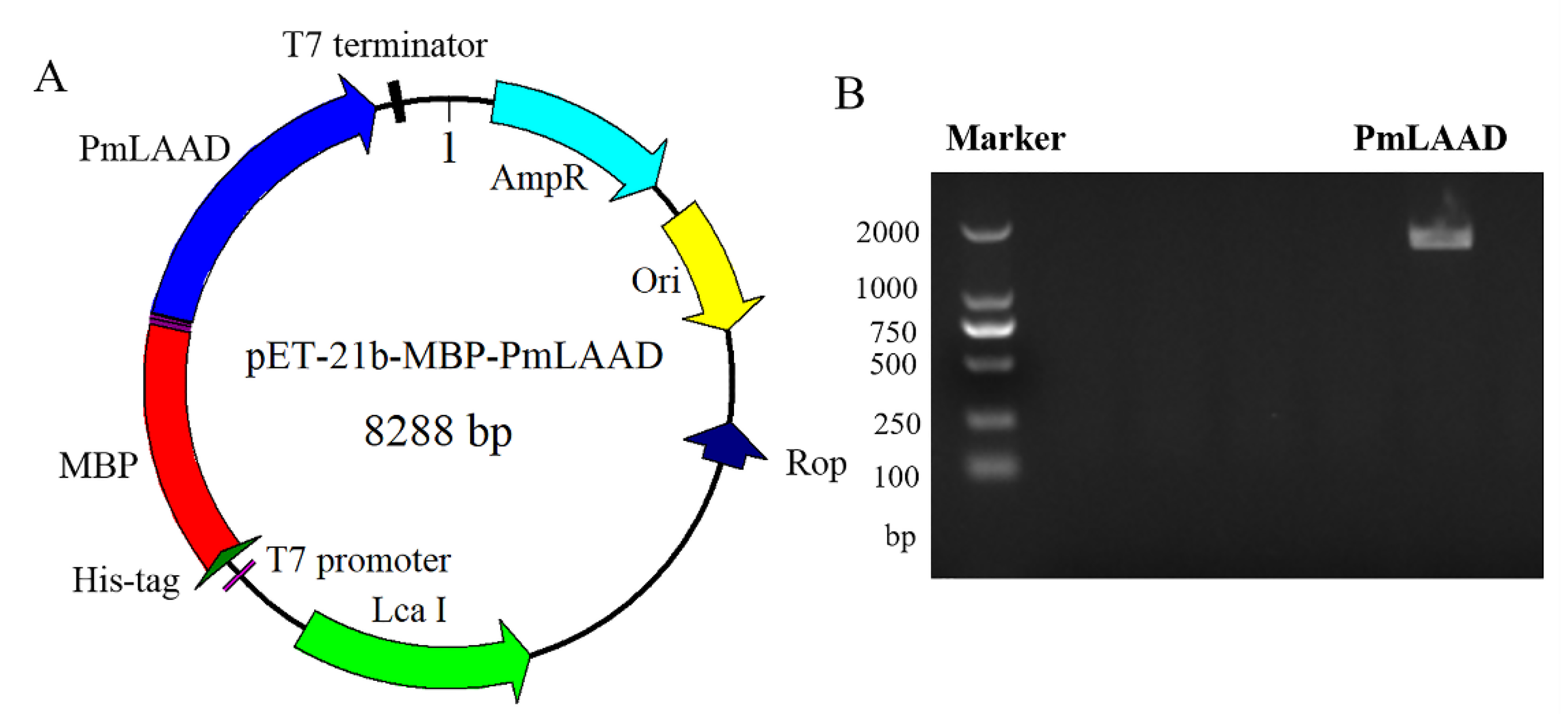
2.1. Construction of MBP-PmLAAD Recombinant Protein Expression Plasmids
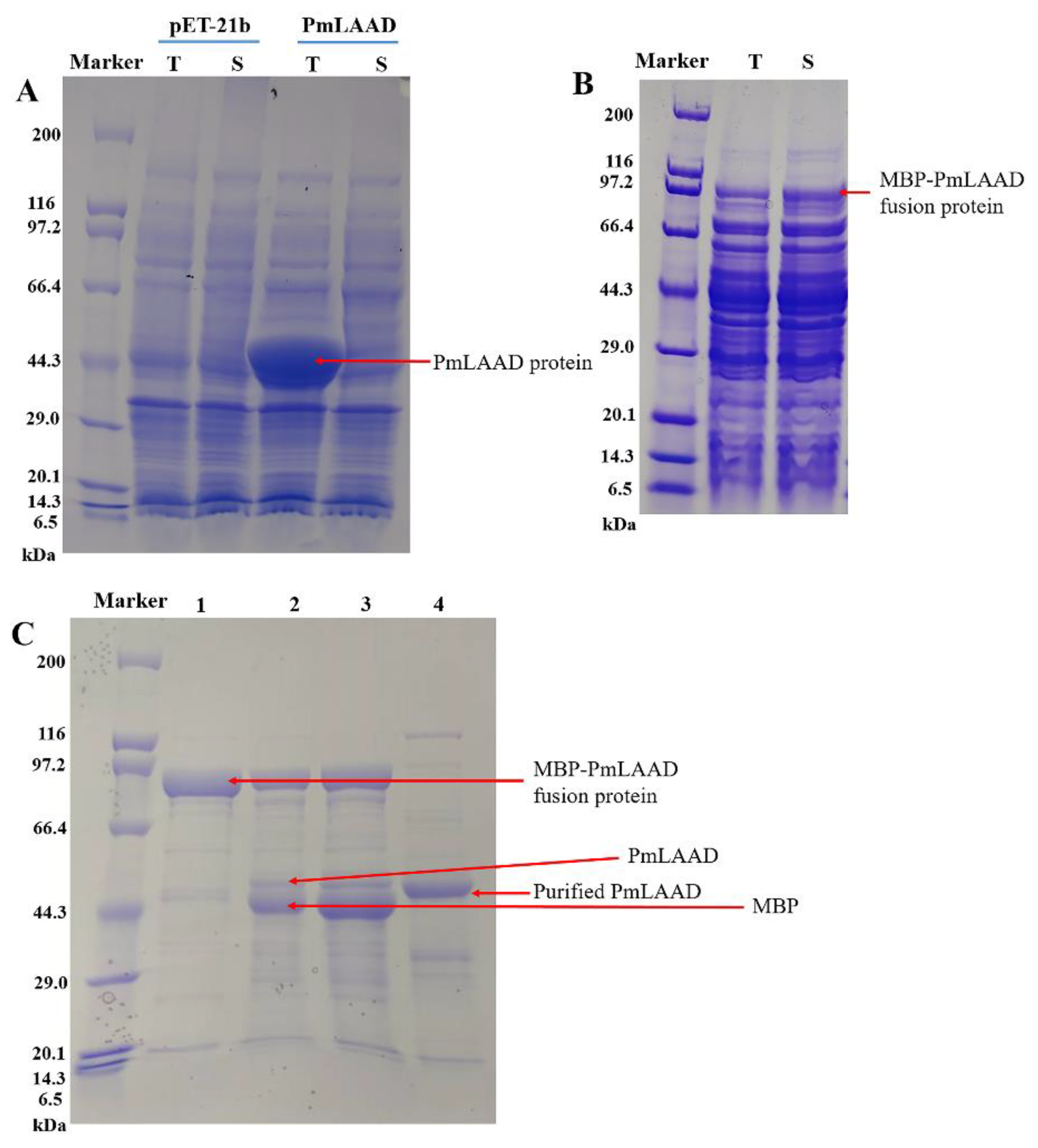
2.2. Expression and Purification of MBP-PmLAAD
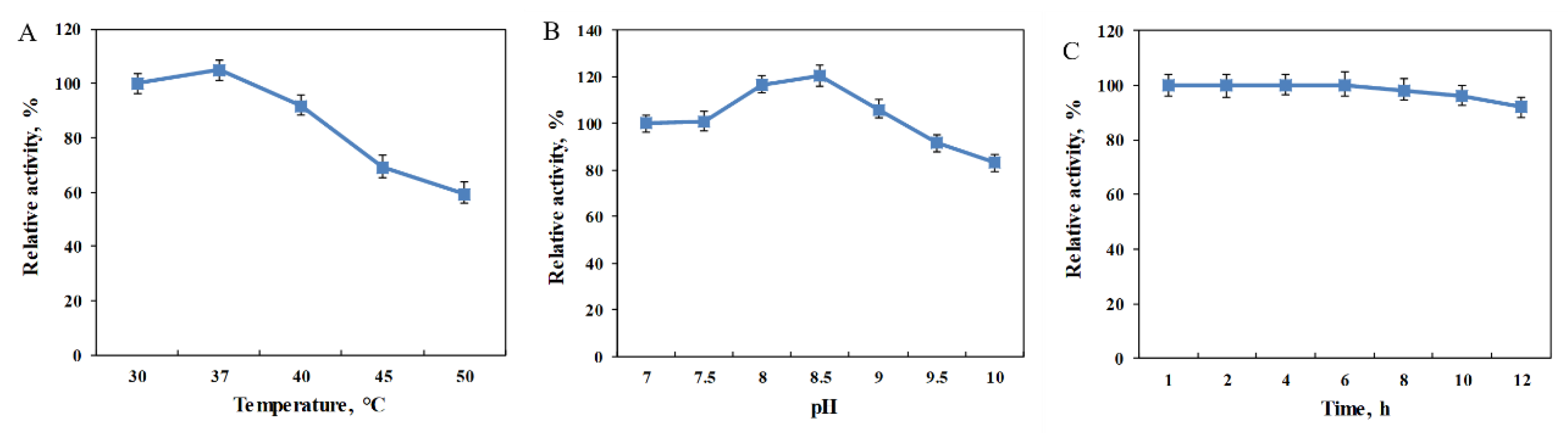
2.3. Biochemical Characterization of the MBP-PmLAAD Fusion Enzyme
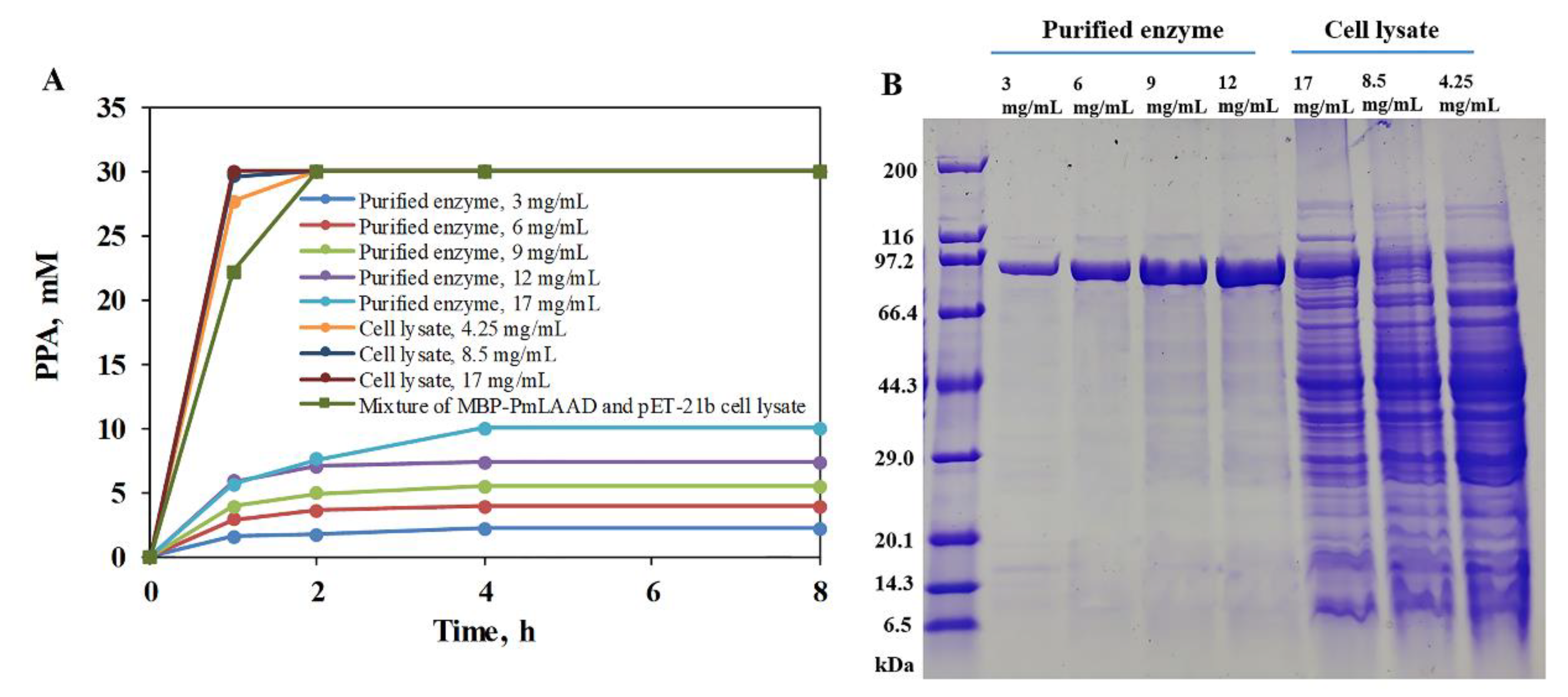
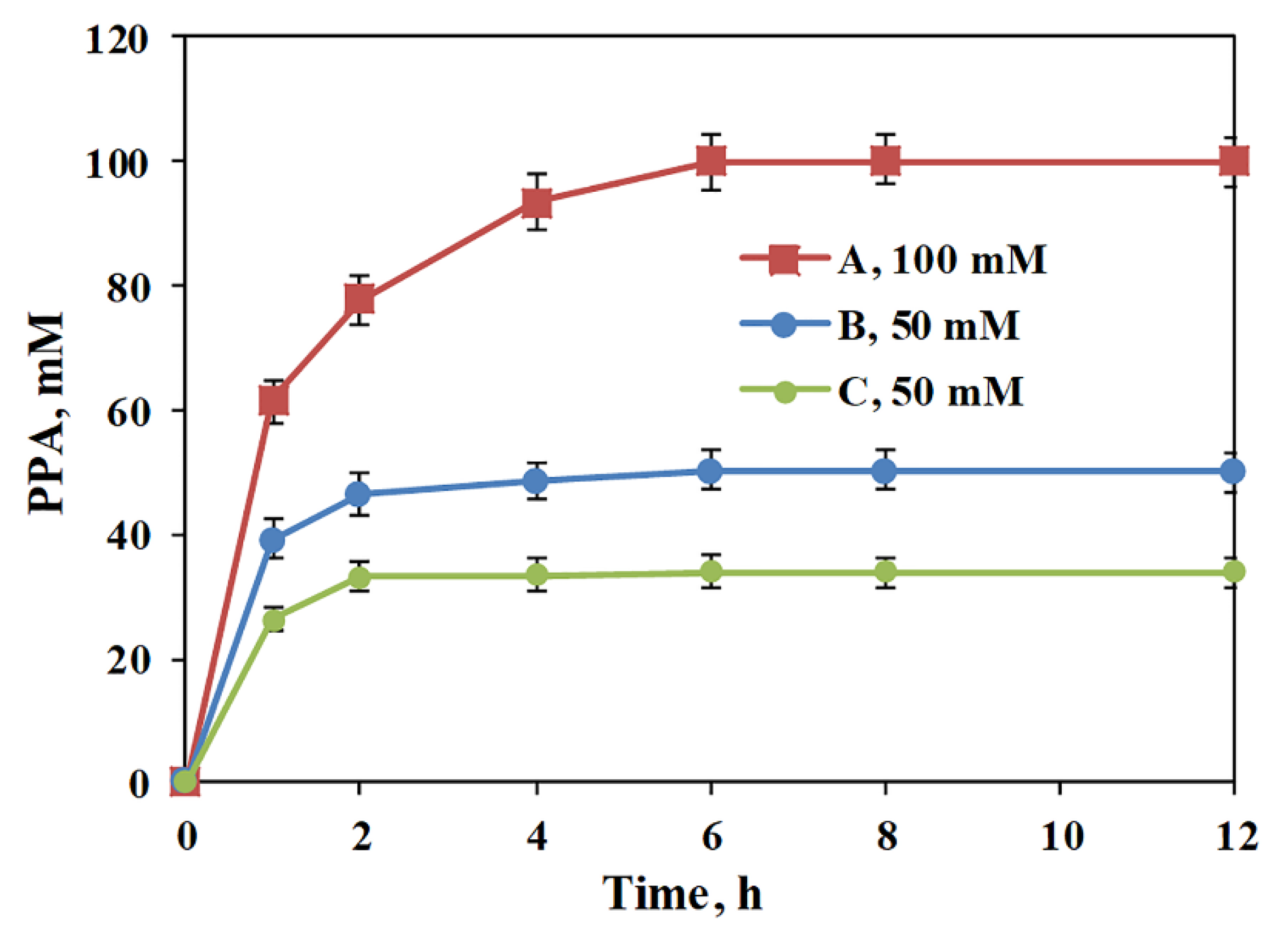
2.4. Conversion of L-Phe to PPA with MBP-PmLAAD Cell Lysate
3. Materials and Methods
3.1. Materials
3.2. Construction of the Expression Plasmid pET-21b-MBP-PmLAAD
3.3. Expression and Purification of MBP-PmLAAD Fusion Proteins
3.4. Activity Assay
3.5. Effect of pH, Temperature and Storage Time on the Activity of MBP-PmLAAD
3.6. Conversion of L-Phe to PPA Using the MBP-PmLAAD Fusion Protein
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pantaleone, D.P.; Geller, A.M.; Taylor, P.P. Purification and characterization of an L-amino acid deaminase used to prepare unnatural amino acids. J. Mol. Catal. B Enzym. 2001, 11, 795–803. [Google Scholar] [CrossRef]
- Baek, J.O.; Seo, J.W.; Kwon, O.; Seong, S.I.; Kim, I.H.; Kim, C.H. Expression and characterization of a second L-amino acid deaminase isolated from Proteus mirabilis in Escherichia coli. J. Basic Microbiol. 2011, 51, 129–135. [Google Scholar] [CrossRef]
- Liu, L.; Hossain, G.S.; Shin, H.D.; Li, J.H.; Du, G.C.; Chen, J. One-step production of α-ketoglutaric acid from glutamic acid with an engineered L-amino acid deaminase from Proteus mirabilis. J. Biotechnol. 2013, 164, 97–104. [Google Scholar] [CrossRef]
- Zhang, D.P.; Jing, X.R.; Zhang, W.L.; Nie, Y.; Xu, Y. Highly selective synthesis of D-amino acids from readily available L-amino acids by a one-pot biocatalytic stereoinversion cascade. RSC Adv. 2019, 9, 29927–29935. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Hossain, G.S.; Li, J.H.; Shin, H.D.; Du, G.C.; Liu, L. Combination of phenylpyruvic acid (PPA) pathway engineering and molecular engineering of L-amino acid deaminase improves PPA production with an Escherichia coli whole-cell biocatalyst. Appl. Microbiol. Biotechnol. 2016, 100, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Hossain, G.S.; Li, J.; Shin, H.D.; Liu, L.; Du, G.C. Production of phenylpyruvic acid from L-phenylalanine using an L-amino acid deaminase from Proteus mirabilis: Comparison of enzymatic and whole-cell biotransformation approaches. Appl. Microbiol. Biotechnol. 2015, 99, 8391–8402. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, J.; Shin, H.D.; Du, G.C.; Liu, L.; Chen, J. One-step biosynthesis of α-ketoisocaproate from L-leucine by an Escherichia coli whole-cell biocatalyst expressing an L-amino acid deaminase from Proteus vulgaris. Sci. Rep. 2015, 5, 12614–12625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molla, G.; Melis, R.; Pollegioni, L. Breaking the mirror: L-Amino acid deaminase, a novel stereoselective biocatalyst. Biotechnol. Adv. 2017, 35, 657–668. [Google Scholar] [CrossRef]
- Ju, Y.; Tong, S.; Gao, Y.; Zhao, W.; Liu, Q.; Gu, Q.; Xu, J.; Niu, L.; Teng, M.; Zhou, H. Crystal structure of a membrane-bound L-amino acid deaminase from Proteus vulgaris. J. Struct. Biol. 2016, 195, 306–315. [Google Scholar] [CrossRef]
- Motta, P.; Molla, G.; Pollegioni, L.; Nardini, M. Structure-function relationships in L-amino acid deaminase, a flavoprotein belonging to a novel class of biotechnologically relevant enzymes. J. Biol. Chem. 2016, 291, 10457–10475. [Google Scholar] [CrossRef] [Green Version]
- Hossain, G.S.; Li, J.; Shin, H.D.; Du, G.C.; Wang, M.; Liu, L.; Chen, J. One-step biosynthesis of α-keto-γ-methylthiobutyric acid from L-methionine by an Escherichia coli whole-cell biocatalyst expressing an engineered L-amino acid deaminase from Proteus vulgaris. PLoS ONE 2014, 9, 114291–114307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, G.S.; Li, J.; Shin, H.D.; Chen, R.R.; Du, G.C.; Liu, L.; Chen, J. Bioconversion of L-glutamic acid to α-ketoglutaric acid by an immobilized whole-cell biocatalyst expressing L-amino acid deaminase from Proteus mirabilis. J. Biotechnol. 2014, 169, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Nshimiyimana, P.; Liu, L.; Du, G.C. Engineering of L-amino acid deaminases for the production of α-keto acids from L-amino acids. Bioengineered 2019, 10, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, G.S.; Shin, H.D.; Li, J.H.; Du, G.C.; Chen, J.; Liu, L. Transporter engineering and enzyme evolution for pyruvate production from d/L-alanine with a whole-cell biocatalyst expressing L-amino acid deaminase from Proteus mirabilis. RSC Adv. 2016, 6, 82676–82684. [Google Scholar] [CrossRef]
- Hossain, G.S.; Shin, H.D.; Li, J.; Wang, M.; Du, G.C.; Liu, L.; Chen, J. Integrating error-prone PCR and DNA shuffling as an effective molecular evolution strategy for the production of α-ketoglutaric acid by L-amino acid deaminase. RSC Adv. 2016, 6, 46149–46158. [Google Scholar] [CrossRef]
- Li, R.; Sakir, H.G.; Li, J.; Shin, H.D.; Du, G.C.; Chen, J.; Liu, L. Rational molecular engineering of L-amino acid deaminase for production of α-ketoisovaleric acid from L-valine by Escherichia coli. RSC Adv. 2017, 7, 6615–6621. [Google Scholar] [CrossRef] [Green Version]
- Okino, S.; Inui, M.; Yukawa, H. Production of organic acids by Corynebacterium glutamicum under oxygen deprivation. Appl. Microbiol. Biotechnol. 2005, 68, 475–480. [Google Scholar] [CrossRef]
- Hanson, R.L.; Davis, B.L.; Goldberg, S.L.; Johnston, R.M.; Parker, W.L.; Tully, T.P.; Montana, M.A.; Patel, R.N. Enzymatic preparation of a D-amino acid from a racemic amino acid or keto acid. Org. Process. Res. Dev. 2008, 12, 1119–1129. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Lovelock, S.L.; Weise, N.J.; Ahmed, S.T.; Turner, N.J. Synthesis of D- and L-phenylalanine derivatives by phenylalanine ammonia lyases: A multienzymatic cascade process. Angew. Chem. Int. Ed. 2015, 54, 4608–4611. [Google Scholar] [CrossRef] [Green Version]
- Busto, E.; Richter, N.; Grischek, B.; Kroutil, W. Biocontrolled formal inversion or retention of L-α-amino acids to enantiopure (R)- or (S)-hydroxyacids. Chem. Eur. J. 2014, 20, 11225–11228. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Ahmed, S.T.; Thompson, M.P.; Weise, N.J.; Galman, J.L.; Gahloth, D.; Dunstan, M.S.; Leys, D.; Turner, N.J. Single-biocatalyst synthesis of enantiopured-arylalanines exploiting an engineered D-amino acid dehydrogenase. Adv. Synth. Catal. 2016, 358, 3298–3306. [Google Scholar] [CrossRef]
- Zhao, W.; Ding, H.; Lv, C.; Hu, S.; Huang, J.; Zheng, X.; Yao, S.; Mei, L. Two-step biocatalytic reaction using recombinant Escherichia coli cells for efficient production of phenyllactic acid from L-phenylalanine. Process Biochem. 2018, 64, 31–37. [Google Scholar] [CrossRef]
- Zhu, L.; Feng, G.; Ge, F.; Song, P.; Wang, T.; Liu, Y.; Tao, Y.; Zhou, Z. One-pot enzymatic synthesis of D-arylalanines using phenylalanine ammonia lyase and L-amino acid deaminase. Appl. Biochem. Biotechnol. 2018, 187, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Melis, R.; Rosini, E.; Pirillo, V.; Pollegioni, L.; Molla, G. In vitro evolution of an L-amino acid deaminase active on L-1-naphthylalanine. Catal. Sci. Technol. 2018, 8, 5359–5367. [Google Scholar] [CrossRef]
- Korepanova, A.; Moore, J.D.; Nguyen, H.B.; Hua, Y.; Cross, T.A.; Gao, F. Expression of membrane proteins from Mycobacterium tuberculosis in Escherichia coli as fusions with maltose binding protein. Prot. Expr. Purif. 2007, 53, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Cabanne, C.; Pezzini, J.; Joucla, G.; Hocquellet, A.; Barbot, C.; Garbay, B.; Santarelli, X. Efficient purification of recombinant proteins fused to maltose-binding protein by mixed-mode chromatography. J. Chromatogr. A 2009, 1216, 4451–4456. [Google Scholar] [CrossRef]
- Bokhove, M.; Sadat Al Hosseini, H.; Saito, T.; Dioguardi, E.; Gegenschatz-Schmid, K.; Nishimura, K.; Raj, I.; de Sanctis, D.; Han, L.; Jovine, L. Easy mammalian expression and crystallography of maltose-binding protein-fused human proteins. J. Struct. Biol. 2016, 194, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Moua, P.S.; Gonzalez, A.; Oshiro, K.T.; Tam, V.; Li, Z.H.; Chang, J.; Leung, W.; Yon, A.; Thor, D.; Venkatram, S.; et al. Differential secretion pathways of proteins fused to the Escherichia coli maltose binding protein (MBP) in Pichia pastoris. Prot. Expr. Purif. 2016, 124, 1–9. [Google Scholar] [CrossRef]
- Hahn, K.; Neumeister, K.; Mix, A.; Kottke, T.; Groger, H.; Fischer von Mollard, G. Recombinant expression and characterization of a L-amino acid oxidase from the fungus Rhizoctonia solani. Appl. Microbiol. Biotechnol. 2017, 101, 2853–2864. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, M.; Jing, N.; Zhang, S. Production of soluble bioactive mouse leukemia inhibitory factor from Escherichia coli using MBP tag. Prot. Expr. Purif. 2018, 150, 86–91. [Google Scholar] [CrossRef]
- Ju, Y.; Liu, Z.; Zhang, Z.; Duan, L.; Liu, Q.; Gu, Q.; Zhang, C.; Xu, J.; Zhou, H. Membrane binding of the insertion sequence of Proteus vulgaris L-amino acid deaminase stabilizes protein structure and increases catalytic activity. Sci. Rep. 2017, 7, 13719–13730. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PmLAAD Cell Lysate | MBP-PmLAAD Cell Lysate | MBP-PmLAAD Purified Enzyme | PmLAAD Purified Enzyme | E. coli pET-21b Cell Lysate | |
|---|---|---|---|---|---|
| Specific activity (μmol/mg PmLAAD min) | 0.076 | 0.17 | 0.025 | ND b | ND b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.-P.; Jing, X.-R.; Fan, A.-W.; Liu, H.; Nie, Y.; Xu, Y. Active Expression of Membrane-Bound L-Amino Acid Deaminase from Proteus mirabilis in Recombinant Escherichia coli by Fusion with Maltose-Binding Protein for Enhanced Catalytic Performance. Catalysts 2020, 10, 215. https://doi.org/10.3390/catal10020215
Zhang D-P, Jing X-R, Fan A-W, Liu H, Nie Y, Xu Y. Active Expression of Membrane-Bound L-Amino Acid Deaminase from Proteus mirabilis in Recombinant Escherichia coli by Fusion with Maltose-Binding Protein for Enhanced Catalytic Performance. Catalysts. 2020; 10(2):215. https://doi.org/10.3390/catal10020215
Chicago/Turabian StyleZhang, Dan-Ping, Xiao-Ran Jing, An-Wen Fan, Huan Liu, Yao Nie, and Yan Xu. 2020. "Active Expression of Membrane-Bound L-Amino Acid Deaminase from Proteus mirabilis in Recombinant Escherichia coli by Fusion with Maltose-Binding Protein for Enhanced Catalytic Performance" Catalysts 10, no. 2: 215. https://doi.org/10.3390/catal10020215