In Situ Spectroscopic Methods for Electrocatalytic CO2 Reduction



Abstract
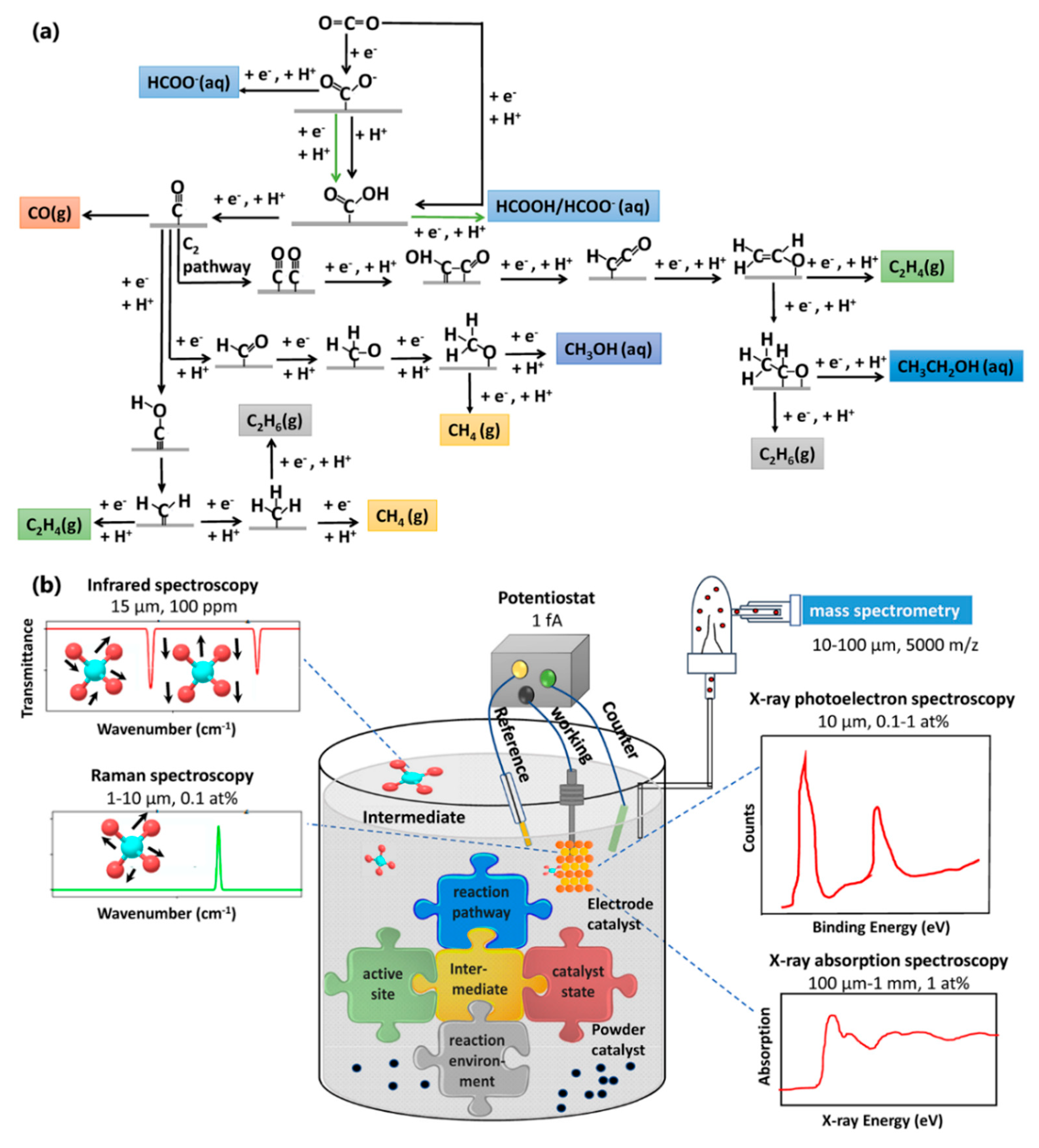
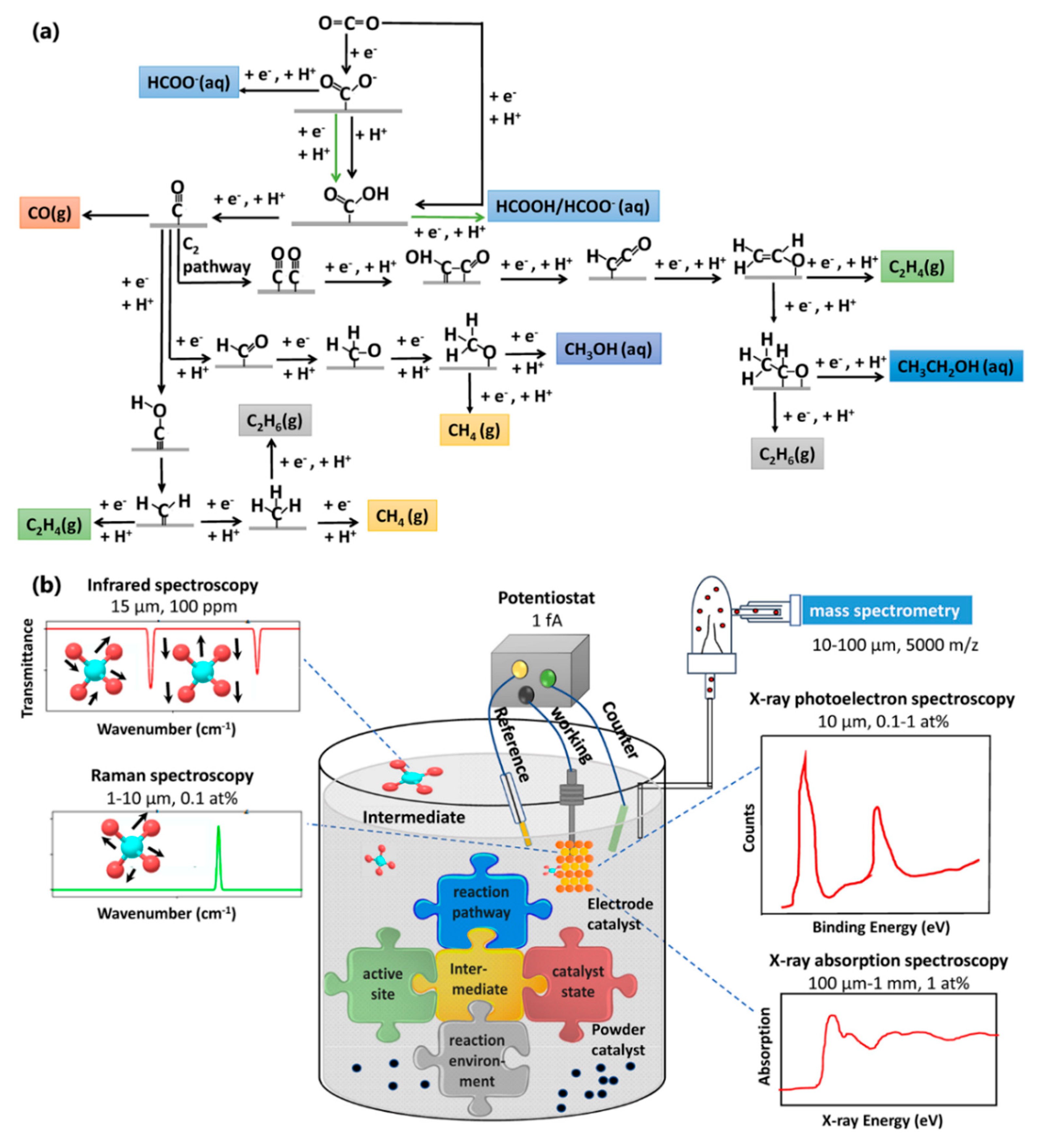
:1. Introduction
2. In Situ Spectroscopic Techniques
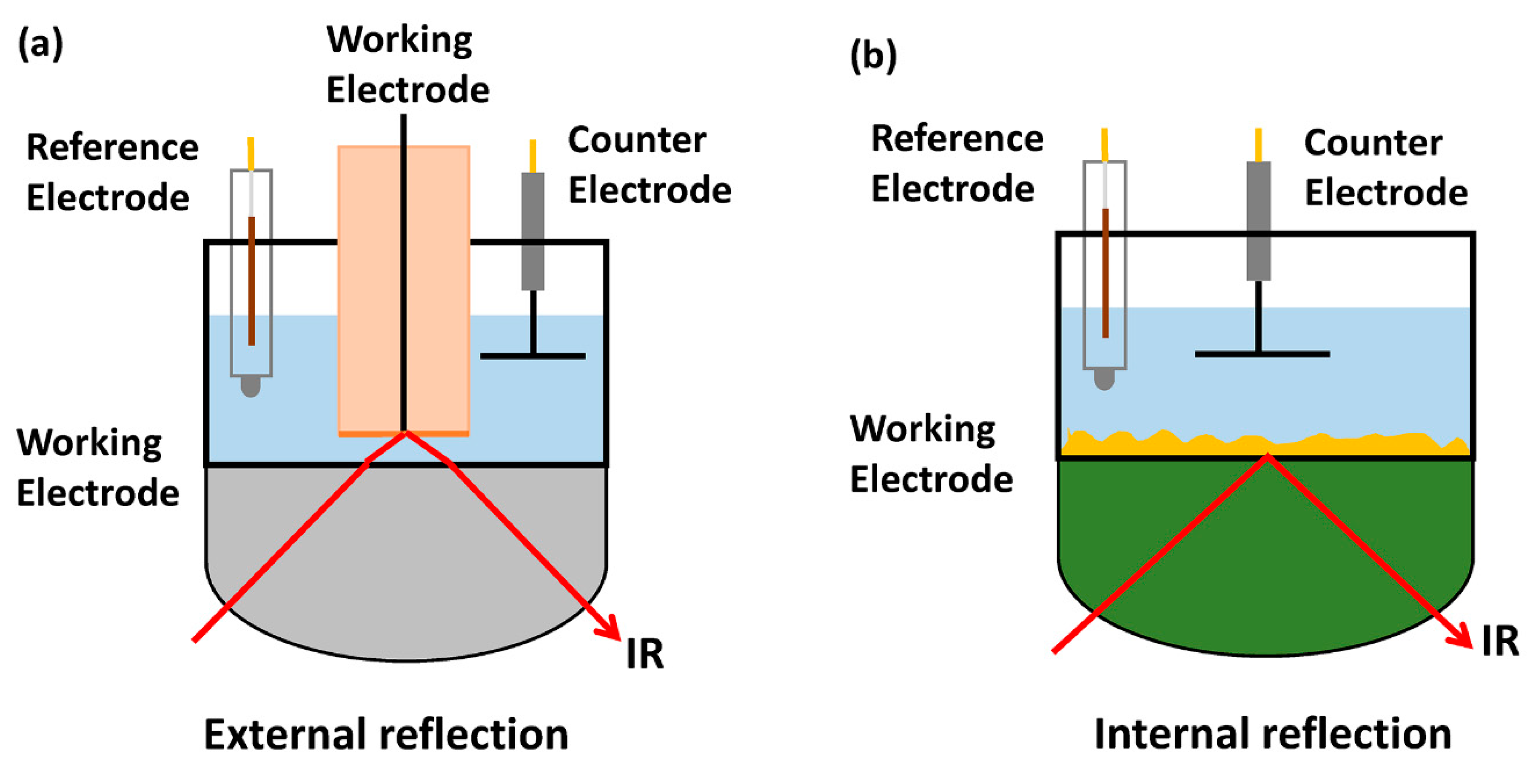
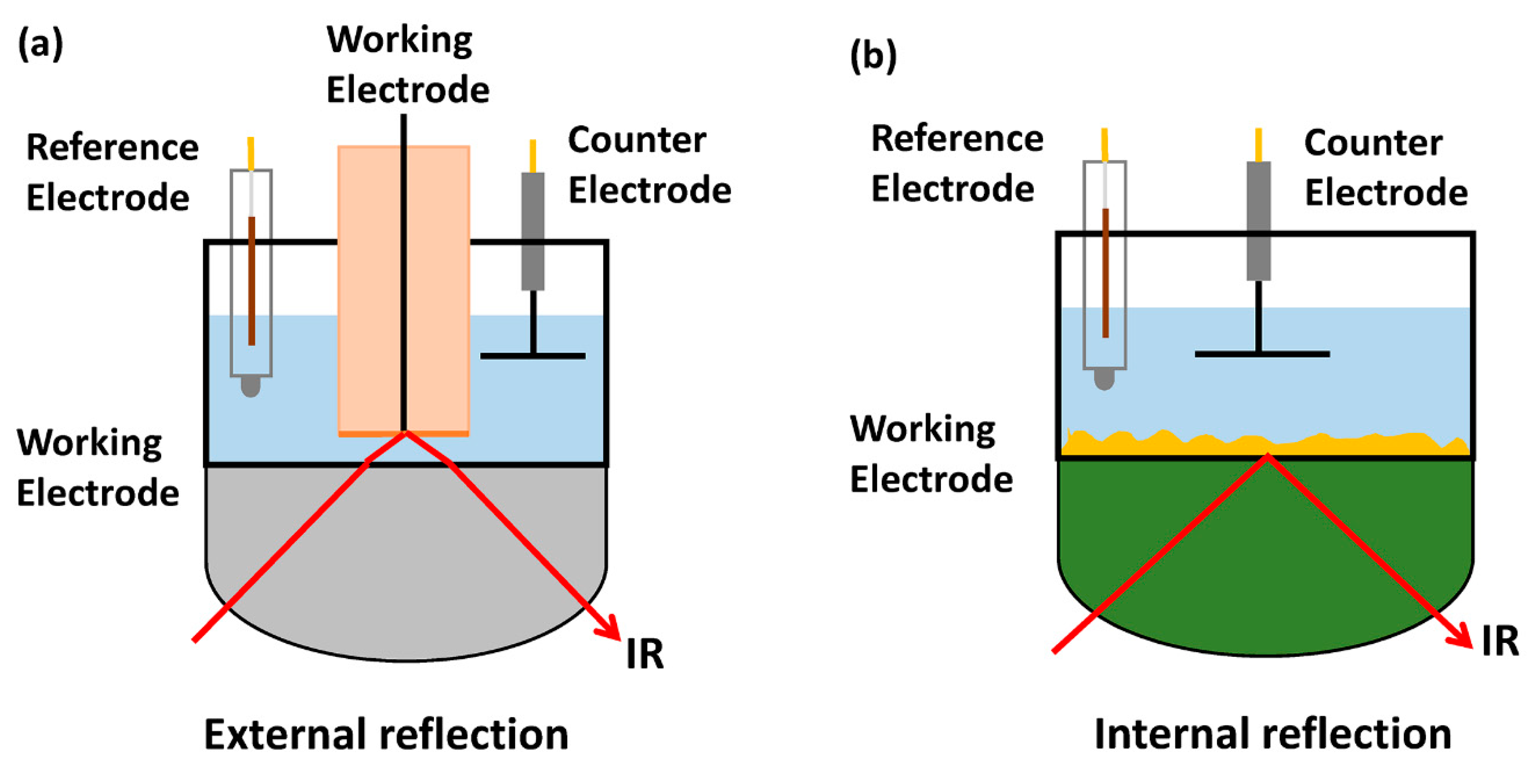
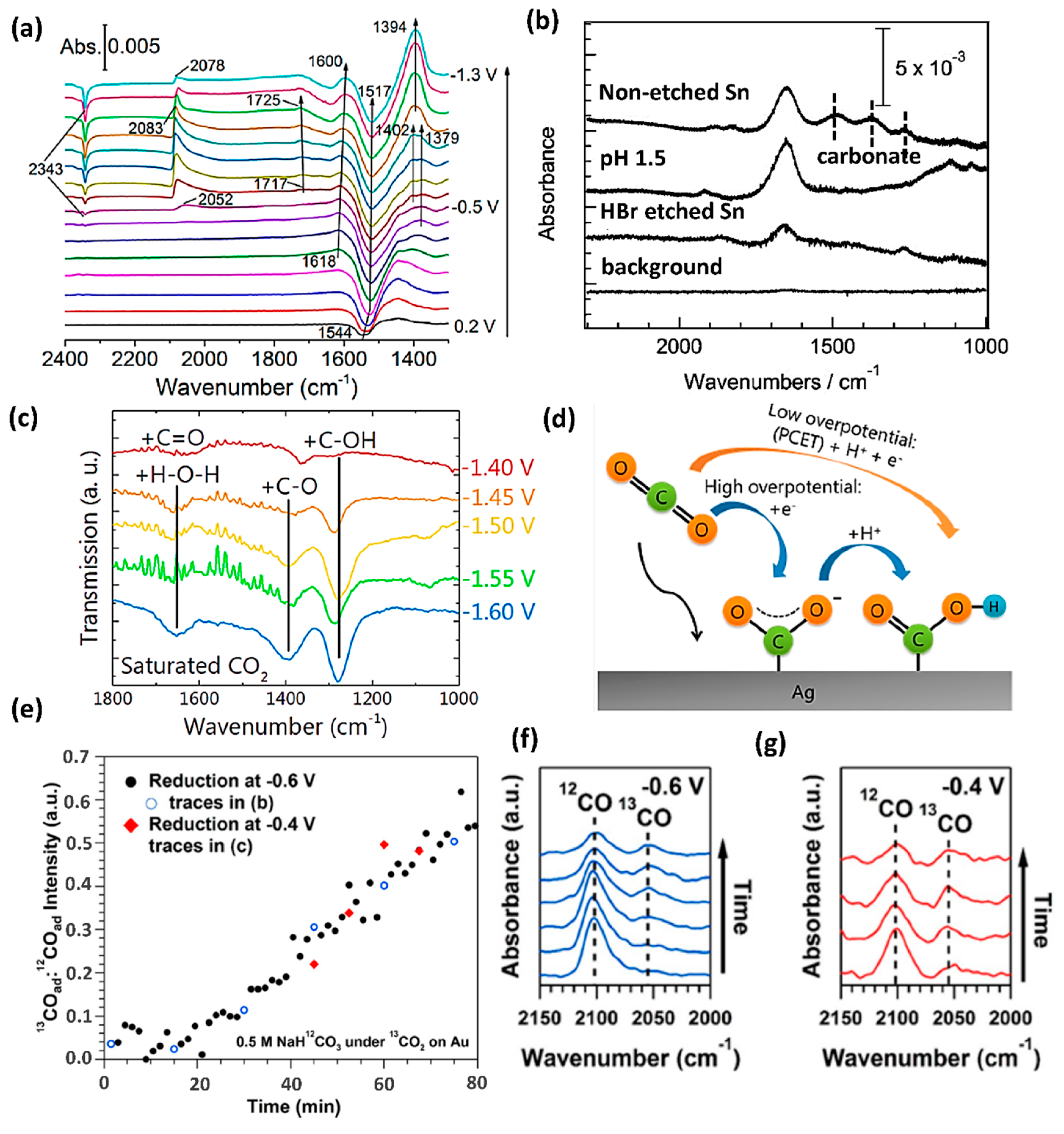
2.1. Infrared Spectroscopy (IR)
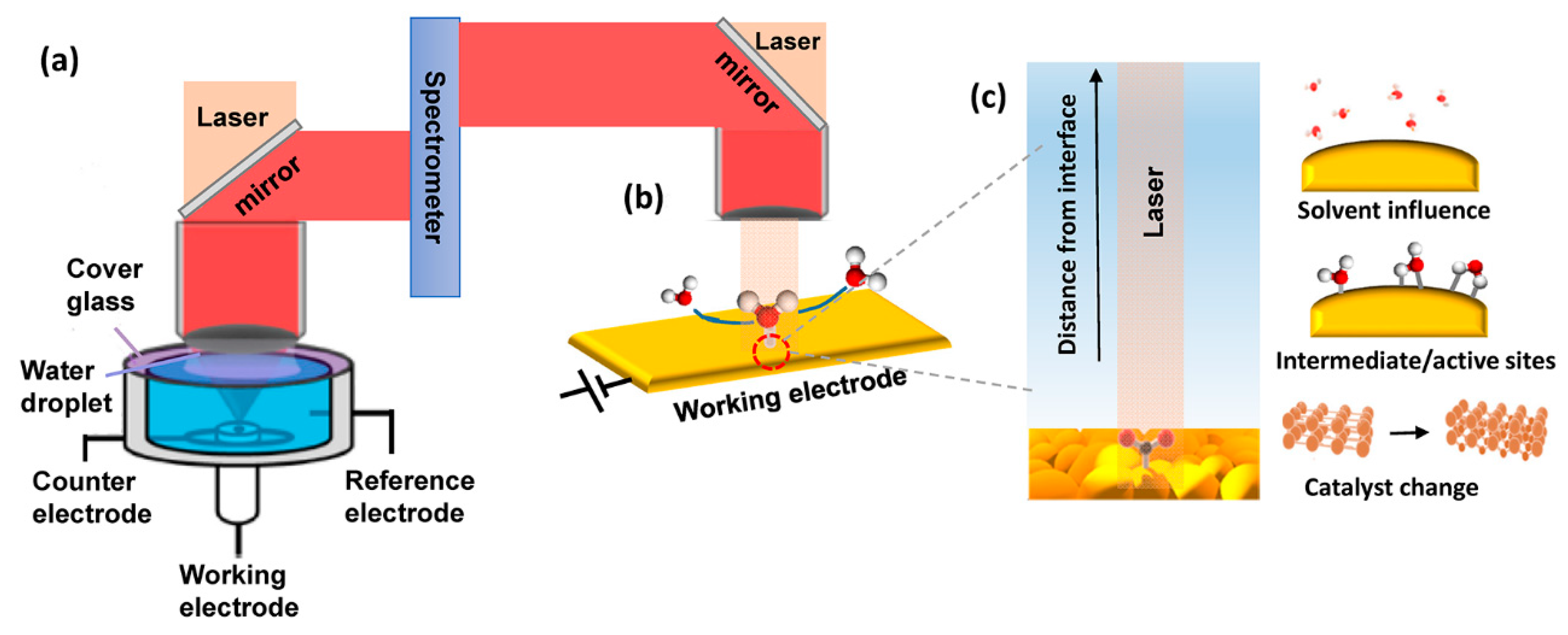
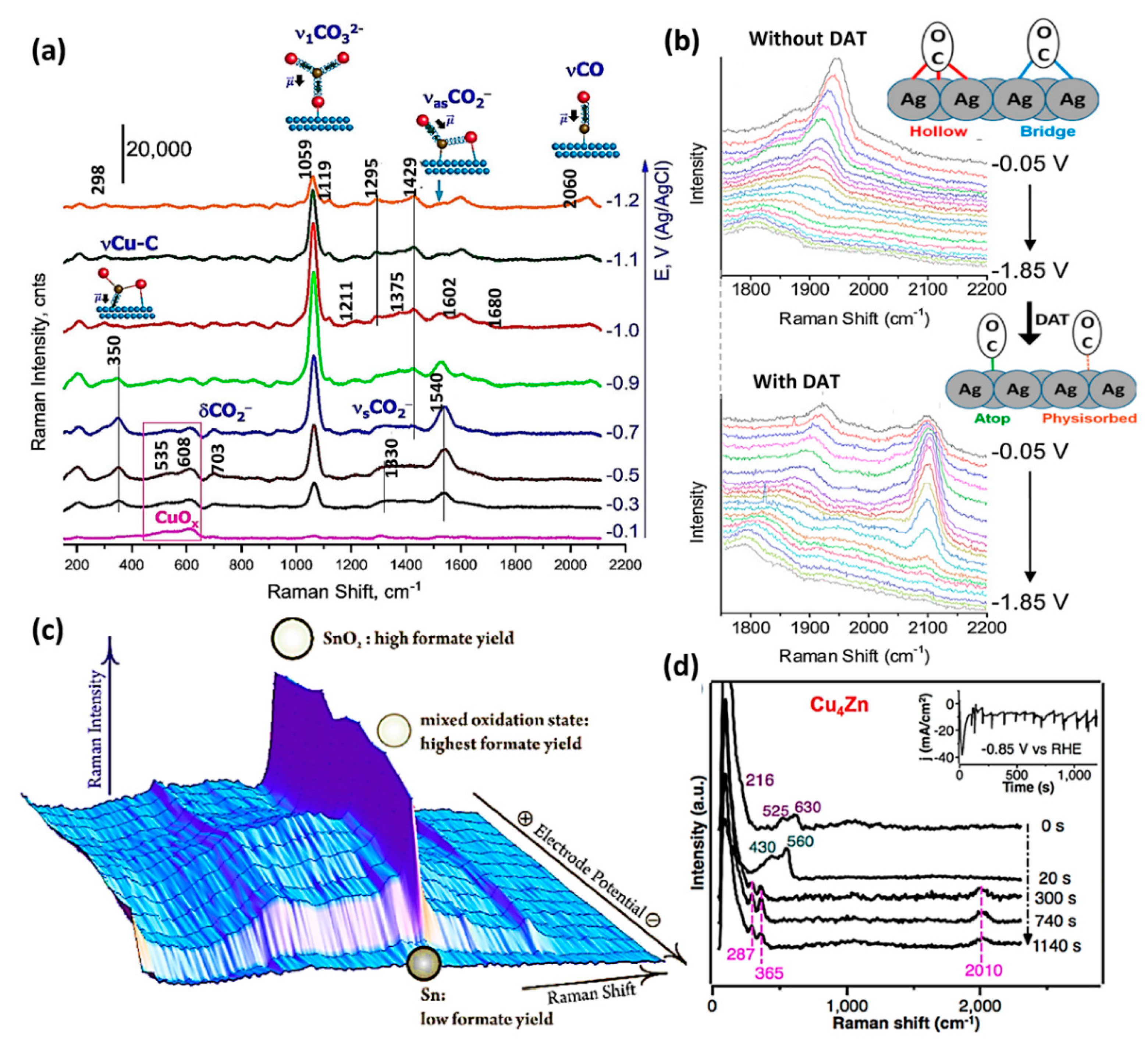
2.2. Raman Spectroscopy (RS)
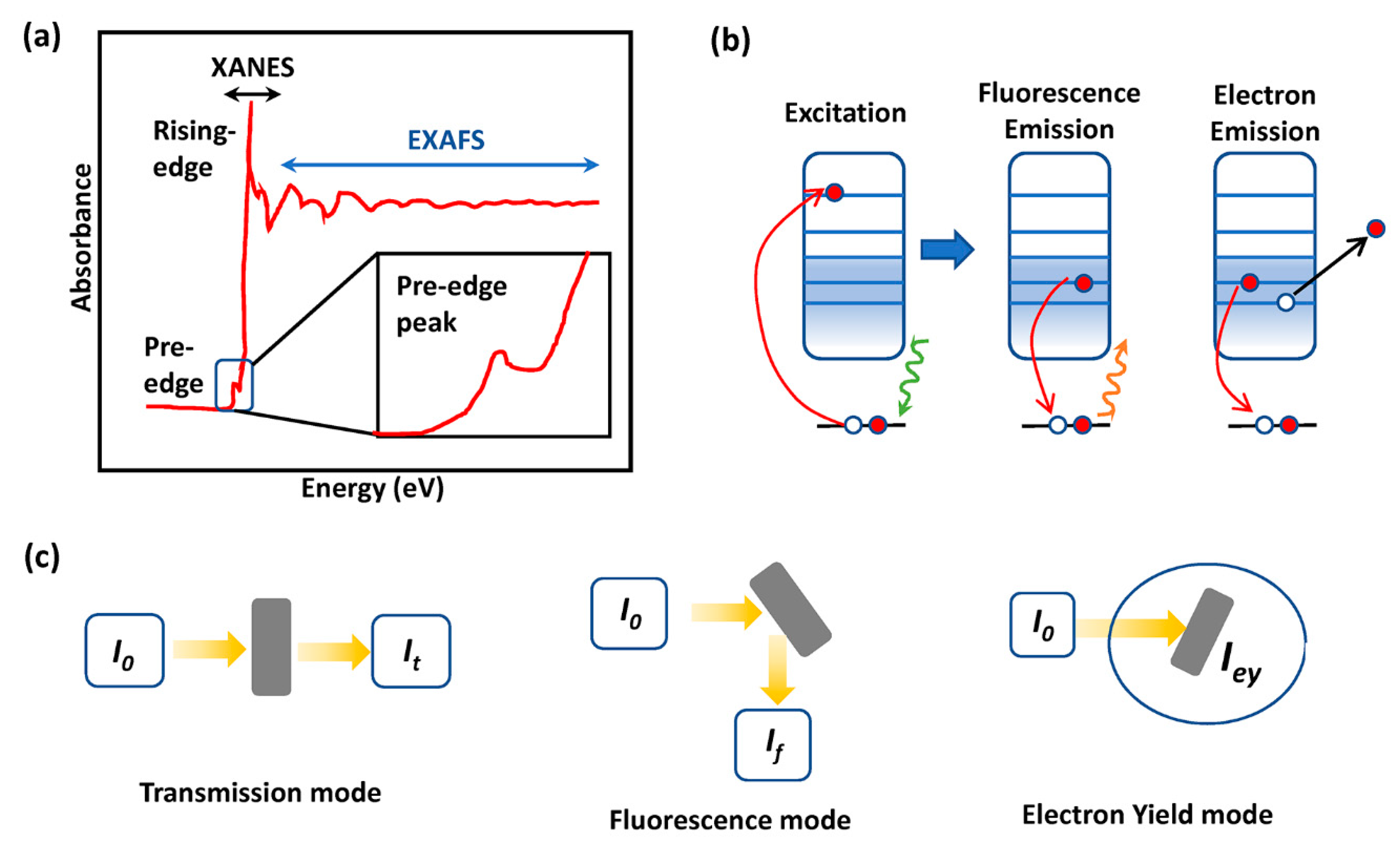
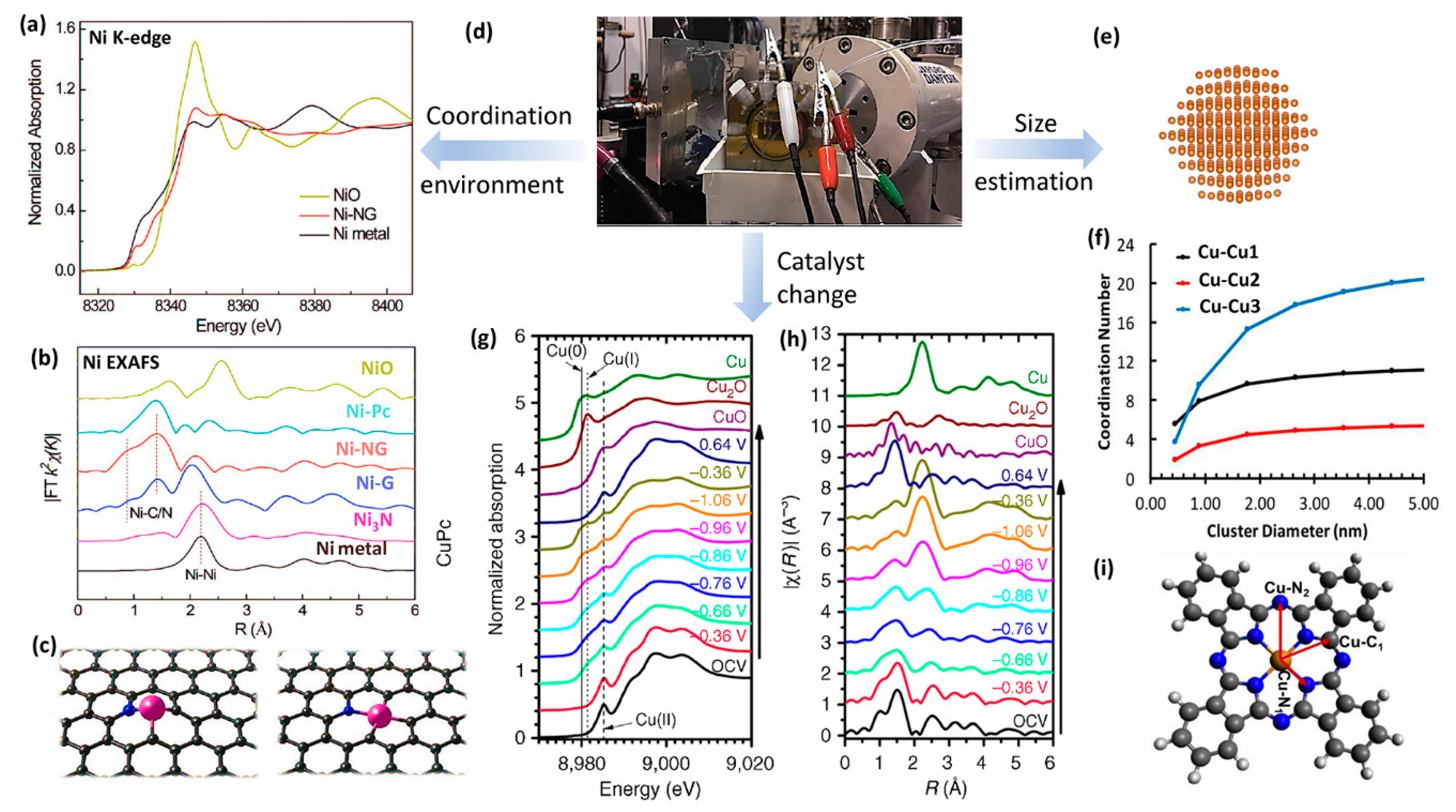
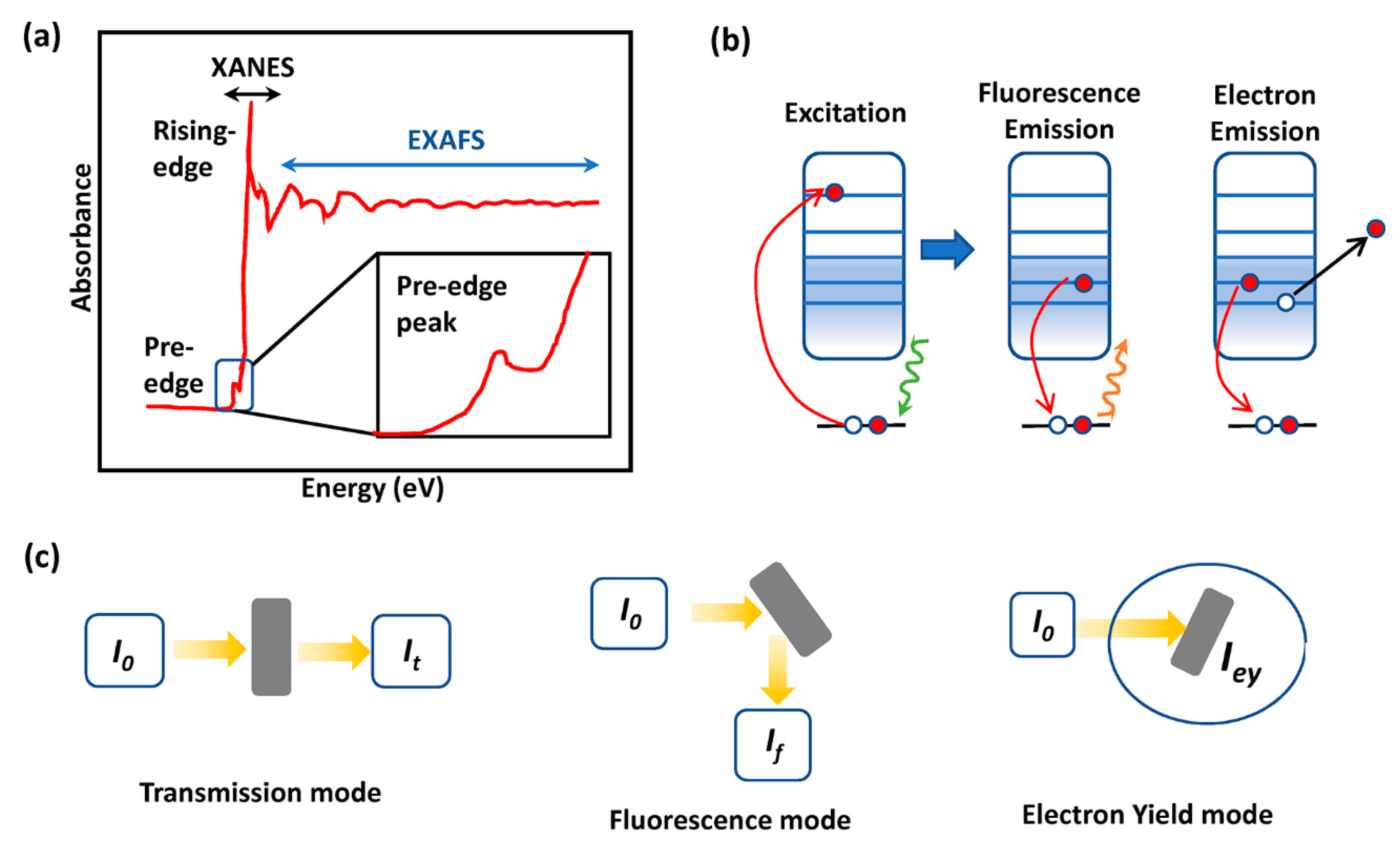
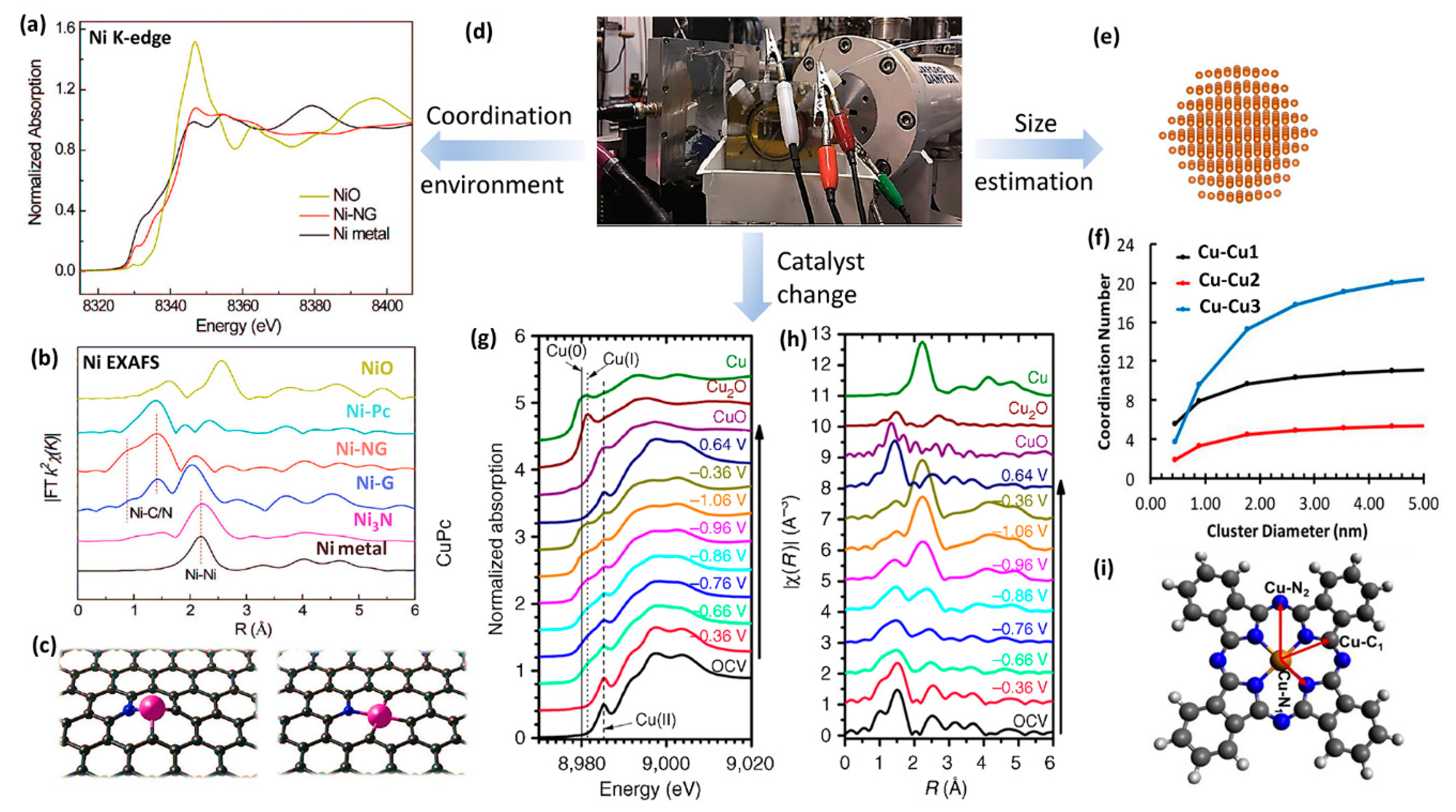
2.3. X-ray Absorption Spectroscopy (XAS)
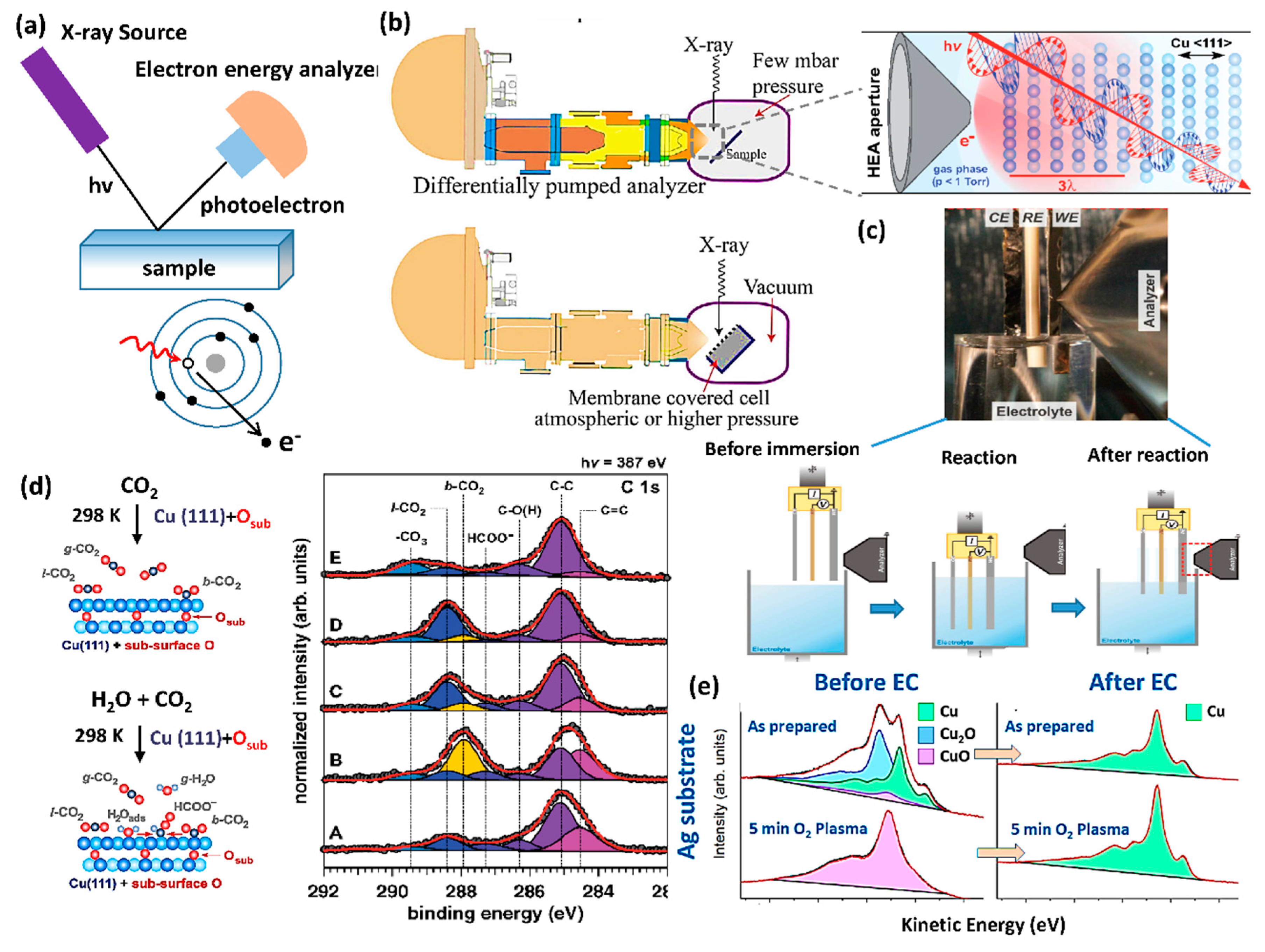
2.4. X-ray Photoelectron Spectroscopy (XPS)
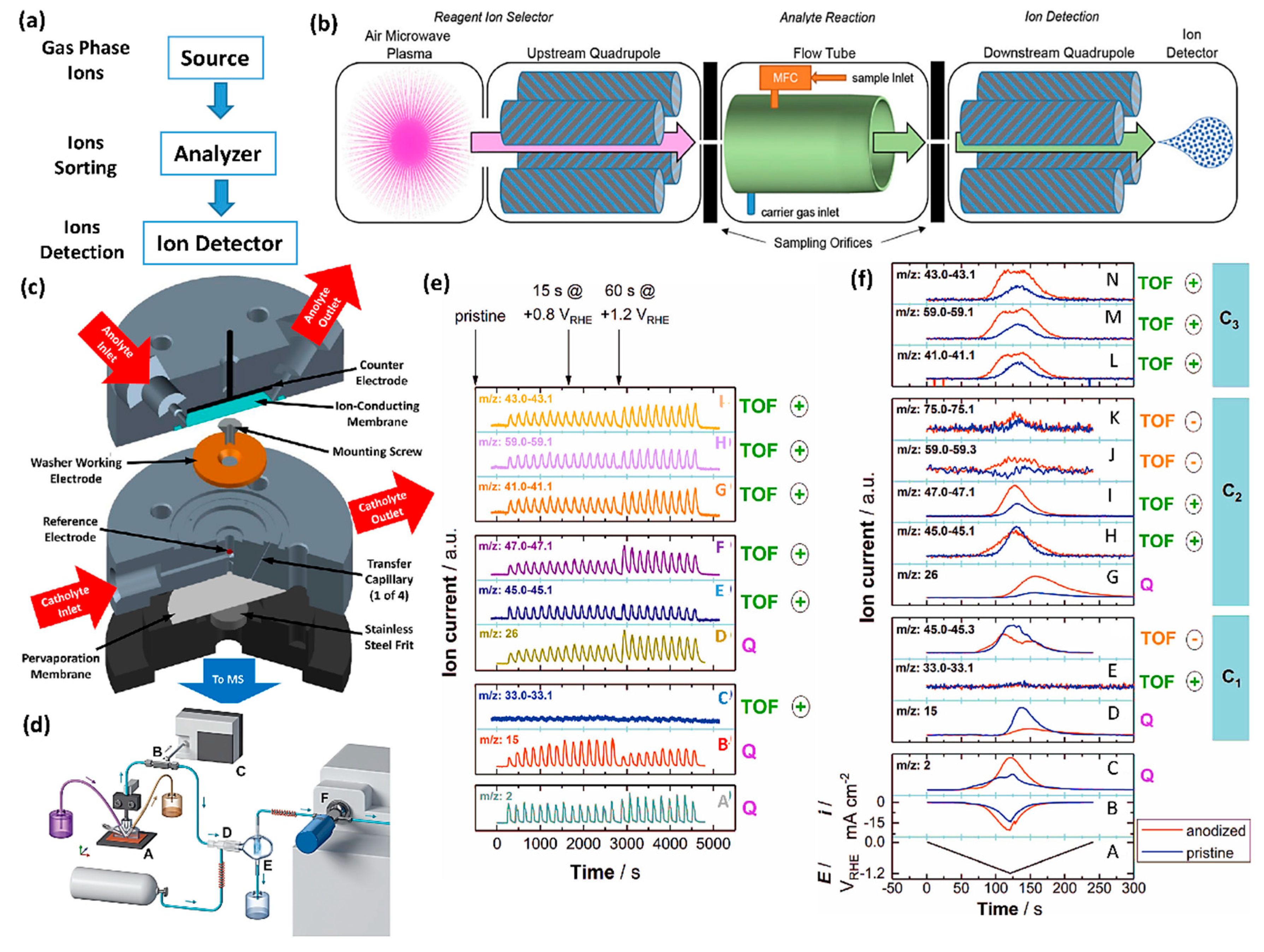
2.5. In Situ Mass Spectrometry (MS)
3. Summary and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATR | attenuated total reflection |
| ATR-SEIRAS | surface-enhanced ATR-IR spectroscopy |
| CO2 | carbon dioxide |
| CO2RR | CO2 reduction reaction |
| COF | covalent organic frameworks |
| CuPc | copper (II) phthalocyanine |
| CV | cyclic voltammetry |
| DEMS | differential electrochemical mass spectrometry |
| DFT | density functional theory |
| DRIFTS | diffuse reflectance infrared Fourier-transform spectroscopy |
| EC | electrochemical |
| EC-RTMS | electrochemical real-time mass spectrometry |
| EXAFS | extended X-ray absorption fine structure |
| FTIR | Fourier-transform infrared spectroscopy |
| FWHM | full width at half maximum |
| GC | gas chromatography |
| HPLC | high-performance liquid chromatography |
| HV | high vacuum |
| IR | infrared spectroscopy |
| MS | mass spectrometry |
| NAP-XPS | near-ambient-pressure XPS |
| Ni-G | Ni-coordinated graphene |
| NMR | nuclear magnetic resonance |
| Ni-NG | Ni-coordinated N-doped graphene |
| Ni NPs/G | Ni nanoparticles loaded graphene nanosheets |
| PCET | proton coupled electron transfer |
| RAS-IR | reflection-absorption infrared spectroscopy |
| SEIRA | surface-enhancement of the infrared absorption |
| SIFT | selective-ion flow-tube |
| SPEM | scanning photoelectron microscopes |
| XAS | X-ray absorption spectroscopy |
| XANES | X-ray absorption near edge structure |
| XPS | X-ray photoelectron spectroscopy |
References
- Connell, S.D.; Kroeker, K.J.; Fabricius, K.E.; Kline, D.I.; Russell, B.D. The other ocean acidification problem: CO2 as a resource among competitors for ecosystem dominance. Philos. Trans. R. Soc. B 2013, 368, 20120442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doney, S.C.; Fabry, V.J.; Feely, R.A.; Kleypas, J.A. Ocean acidification: The other CO2 problem. Annu. Rev. Mar. Sci. 2009, 1, 169–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; de Arquer, F.P.G.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibria, M.G.; Edwards, J.P.; Gabardo, C.M.; Dinh, C.T.; Seifitokaldani, A.; Sinton, D.; Sargent, E.H. Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design. Adv. Mater. 2019, 1807166. [Google Scholar] [CrossRef]
- Handoko, A.D.; Wei, F.; Yeo, B.S.; Seh, Z.W. Understanding heterogeneous electrocatalytic carbon dioxide reduction through operando techniques. Nat. Catal. 2018, 1, 922. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef]
- Koper, M.T. Structure sensitivity and nanoscale effects in electrocatalysis. Nanoscale 2011, 3, 2054–2073. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37. [Google Scholar] [CrossRef]
- Bandarenka, A.S.; Koper, M.T. Structural and electronic effects in heterogeneous electrocatalysis: Toward a rational design of electrocatalysts. J. Catal. 2013, 308, 11–24. [Google Scholar] [CrossRef]
- Stephens, I.E.; Bondarenko, A.S.; Grønbjerg, U.; Rossmeisl, J.; Chorkendorff, I. Understanding the electrocatalysis of oxygen reduction on platinum and its alloys. Energy Environ. Sci. 2012, 5, 6744–6762. [Google Scholar] [CrossRef] [Green Version]
- Bard, A.J.; Faulkner, L.R.; Leddy, J.; Zoski, C.G. Electrochemical Methods: Fundamentals and Applications; Wiley: New York, NY, USA, 1980; Volume 2. [Google Scholar]
- Kirk, C.; Chen, L.D.; Siahrostami, S.; Karamad, M.; Bajdich, M.; Voss, J.; Nørskov, J.K.; Chan, K. Theoretical investigations of the electrochemical reduction of CO on single metal atoms embedded in graphene. ACS Cent. Sci. 2017, 3, 1286–1293. [Google Scholar] [CrossRef] [Green Version]
- Bañares, M.A. Operando spectroscopy: The knowledge bridge to assessing structure–performance relationships in catalyst nanoparticles. Adv. Mater. 2011, 23, 5293–5301. [Google Scholar] [CrossRef]
- Zhu, K.; Zhu, X.; Yang, W. Application of In Situ Techniques for the Characterization of NiFe-Based Oxygen Evolution Reaction (OER) Electrocatalysts. Angew. Chem. Int. Ed. 2019, 58, 1252–1265. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, M.; Bai, Z.; Jiang, G.; Liu, B.; Wu, T.; Chen, Z.; Amine, K.; Lu, J. The Absence and Importance of Operando Techniques for Metal-Free Catalysts. Adv. Mater. 2019, 31, 1805609. [Google Scholar] [CrossRef]
- Lukashuk, L.; Foettinger, K. In Situ and Operando Spectroscopy: A Powerful Approach Towards Understanding Catalysts. Johnson Matthey Technol. Rev. 2018, 62, 316–331. [Google Scholar] [CrossRef]
- Mistry, H.; Varela, A.S.; Bonifacio, C.S.; Zegkinoglou, I.; Sinev, I.; Choi, Y.-W.; Kisslinger, K.; Stach, E.A.; Yang, J.C.; Strasser, P. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 2016, 7, 12123. [Google Scholar] [CrossRef]
- Gao, D.; Zegkinoglou, I.; Divins, N.J.; Scholten, F.; Sinev, I.; Grosse, P.; Roldan Cuenya, B. Plasma-activated copper nanocube catalysts for efficient carbon dioxide electroreduction to hydrocarbons and alcohols. ACS Nano 2017, 11, 4825–4831. [Google Scholar] [CrossRef]
- Grosse, P.; Gao, D.; Scholten, F.; Sinev, I.; Mistry, H.; Roldan Cuenya, B. Dynamic changes in the structure, chemical state and catalytic selectivity of Cu nanocubes during CO2 electroreduction: Size and support effects. Angew. Chem. Int. Ed. 2018, 57, 6192–6197. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Che, F.; Liu, M.; Zou, C.; Liang, Z.; De Luna, P.; Yuan, H.; Li, J.; Wang, Z.; Xie, H.; et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 2018, 10, 974. [Google Scholar] [CrossRef]
- Bernal, M.; Bagger, A.; Scholten, F.; Sinev, I.; Bergmann, A.; Ahmadi, M.; Rossmeisl, J.; Cuenya, B.R. CO2 electroreduction on copper-cobalt nanoparticles: Size and composition effect. Nano Energy 2018, 53, 27–36. [Google Scholar] [CrossRef]
- Pérez-Gallent, E.; Figueiredo, M.C.; Calle-Vallejo, F.; Koper, M.T. Spectroscopic observation of a hydrogenated CO dimer intermediate during CO reduction on Cu (100) electrodes. Angew. Chem. Int. Ed. 2017, 129, 3675–3678. [Google Scholar] [CrossRef]
- Dunwell, M.; Lu, Q.; Heyes, J.M.; Rosen, J.; Chen, J.G.; Yan, Y.; Jiao, F.; Xu, B. The central role of bicarbonate in the electrochemical reduction of carbon dioxide on gold. J. Am. Chem. Soc. 2017, 139, 3774–3783. [Google Scholar] [CrossRef]
- Klingan, K.; Kottakkat, T.; Jovanov, Z.P.; Jiang, S.; Pasquini, C.; Scholten, F.; Kubella, P.; Bergmann, A.; Roldan Cuenya, B.; Roth, C.; et al. Reactivity determinants in electrodeposited Cu foams for electrochemical CO2 reduction. ChemSusChem 2018, 11, 3449–3459. [Google Scholar] [CrossRef]
- Mandal, L.; Yang, K.R.; Motapothula, M.R.; Ren, D.; Lobaccaro, P.; Patra, A.; Sherburne, M.; Batista, V.S.; Yeo, B.S.; Ager, J.W.; et al. Investigating the role of copper oxide in electrochemical CO2 reduction in real time. ACS Appl. Mater. Interfaces 2018, 10, 8574–8584. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Yeo, B.S. Characterization of electrocatalytic water splitting and CO2 reduction reactions using in situ/operando Raman spectroscopy. ACS Catal. 2017, 7, 7873–7889. [Google Scholar] [CrossRef]
- Dutta, A.; Kuzume, A.; Rahaman, M.; Vesztergom, S.; Broekmann, P. Monitoring the chemical state of catalysts for CO2 electroreduction: An in operando study. ACS Catal. 2015, 5, 7498–7502. [Google Scholar] [CrossRef]
- Stuart, B. Infrared spectroscopy. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley: New York, NY, USA, 2000; pp. 1–18. [Google Scholar]
- Ryczkowski, J. IR spectroscopy in catalysis. Catal. Today 2001, 68, 263–381. [Google Scholar] [CrossRef]
- Reed, A.H.; Yeager, E. Infrared internal reflexion studies of the germanium/electrolyte interface. Electrochim. Acta 1970, 15, 1345–1354. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, Y.-W.; Cai, W.-B. Recent applications of in situ ATR-IR spectroscopy in interfacial electrochemistry. Curr. Opin. Electrochem. 2017, 1, 73–79. [Google Scholar] [CrossRef]
- Eischens, R.; Pliskin, W. The infrared spectra of adsorbed molecules. In Advances in Catalysis; Elsevier: Amsterdam, The Netherlands, 1958; Volume 10, pp. 1–56. [Google Scholar]
- Drochner, A.; Fehlings, M.; Krauß, K.; Vogel, H. A New DRIFTS Cell for the In-Situ Investigation of Heterogeneously Catalyzed Reactions. Chem. Eng. Technol. 2000, 23, 319–322. [Google Scholar] [CrossRef]
- Osawa, M.; Ikeda, M. Surface-enhanced infrared absorption of p-nitrobenzoic acid deposited on silver island films: Contributions of electromagnetic and chemical mechanisms. J. Phys. Chem. Lett. 1991, 95, 9914–9919. [Google Scholar] [CrossRef]
- Osawa, M.; Ataka, K.-I.; Yoshii, K.; Yotsuyanagi, T. Surface-enhanced infrared ATR spectroscopy for in situ studies of electrode/electrolyte interfaces. J. Electron. Spectros. Relat. Phenomena 1993, 64, 371–379. [Google Scholar] [CrossRef]
- Osawa, M.; Ataka, K.-I.; Yoshii, K.; Nishikawa, Y. Surface-enhanced infrared spectroscopy: The origin of the absorption enhancement and band selection rule in the infrared spectra of molecules adsorbed on fine metal particles. Appl. Spectrosc. 1993, 47, 1497–1502. [Google Scholar] [CrossRef]
- Osawa, M. Dynamic processes in electrochemical reactions studied by surface-enhanced infrared absorption spectroscopy (SEIRAS). Bull. Chem. Soc. Jpn. 1997, 70, 2861–2880. [Google Scholar] [CrossRef]
- Kan, B.-C.; Boo, J.-H.; Lee, I.; Zaera, F. Thermal chemistry of tetrakis (ethylmethylamido) titanium on Si (100) surfaces. J. Phys. Chem. A 2009, 113, 3946–3954. [Google Scholar] [CrossRef]
- Kubota, J.; Ma, Z.; Zaera, F. In situ characterization of adsorbates in solid−liquid interfaces by reflection−absorption infrared spectroscopy. Langmuir 2003, 19, 3371–3376. [Google Scholar] [CrossRef]
- Greenler, R.G. Infrared study of adsorbed molecules on metal surfaces by reflection techniques. J. Chem. Phys. 1966, 44, 310–315. [Google Scholar] [CrossRef]
- Hoffmann, F.M. Infrared reflection-absorption spectroscopy of adsorbed molecules. Surf. Sci. Rep. 1983, 3, 107–192. [Google Scholar] [CrossRef]
- Zaera, F. Infrared and molecular beam studies of chemical reactions on solid surfaces. Int. Rev. Phys. Chem. 2002, 21, 433–471. [Google Scholar] [CrossRef]
- Ye, S.; Kondo, T.; Hoshi, N.; Inukai, J.; Yoshimoto, S.; Osawa, M.; Itaya, K. Recent progress in electrochemical surface science with atomic and molecular levels. Electrochemistry 2009, 77, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Osawa, M. Surface enhanced vibrational spectroscopy. Handb. Vib. Spectrosc. 2002, 1, 785–815. [Google Scholar]
- Osawa, M. In-situ surface-enhanced infrared spectroscopy of the electrode/solution interface. Adv. Electrochem. Sci. Eng. 2006, 9, 269. [Google Scholar]
- Kawata, S.; Ohtsu, M.; Irie, M. Near-Field Optics and Surface Plasmon Polaritons; Springer Science & Business Media: Berlin, Germany, 2001; Volume 81. [Google Scholar]
- Hartstein, A.; Kirtley, J.; Tsang, J. Enhancement of the infrared absorption from molecular monolayers with thin metal overlayers. Phys. Rev. Lett. 1980, 45, 201. [Google Scholar] [CrossRef]
- Vimont, A.; Thibault-Starzyk, F.; Daturi, M. Analysing and understanding the active site by IR spectroscopy. Chem. Soc. Rev. 2010, 39, 4928–4950. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Ford, M.E.; Gregory, D.; Hu, R.; Keturakis, C.J.; Lwin, S.; Tang, Y.; Yang, Z.; Zhu, M.; Banares, M.A. A decade+ of operando spectroscopy studies. Catal. Today 2017, 283, 27–53. [Google Scholar] [CrossRef]
- Dou, J.; Sun, Z.; Opalade, A.A.; Wang, N.; Fu, W.; Tao, F.F. Operando chemistry of catalyst surfaces during catalysis. Chem. Soc. Rev. 2017, 46, 2001–2027. [Google Scholar] [CrossRef]
- Karim, W.; Kleibert, A.; Hartfelder, U.; Balan, A.; Gobrecht, J.; Van Bokhoven, J.A.; Ekinci, Y. Size-dependent redox behavior of iron observed by in-situ single nanoparticle spectro-microscopy on well-defined model systems. Sci. Rep. 2016, 6, 18818. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.; Whittle, D.; Rhodes, C.; Ward, A.; Rohan, D.; Shannon, M.; Hutchings, G.; Kiely, C. Microstructural studies of the copper promoted iron oxide/chromia water-gas shift catalyst. Phys. Chem. Chem. Phys. 2002, 4, 3902–3908. [Google Scholar] [CrossRef]
- Firet, N.J.; Smith, W.A. Probing the reaction mechanism of CO2 electroreduction over Ag films via operando infrared spectroscopy. ACS Catal. 2016, 7, 606–612. [Google Scholar] [CrossRef]
- Kraack, J.P.; Kaech, A.; Hamm, P. Surface enhancement in ultrafast 2D ATR IR spectroscopy at the metal-liquid interface. J. Phys. Chem. C 2016, 120, 3350–3359. [Google Scholar] [CrossRef]
- Gunathunge, C.M.; Li, X.; Li, J.; Hicks, R.P.; Ovalle, V.J.; Waegele, M.M. Spectroscopic observation of reversible surface reconstruction of copper electrodes under CO2 reduction. J. Phys. Chem. C 2017, 121, 12337–12344. [Google Scholar] [CrossRef]
- Heyes, J.; Dunwell, M.; Xu, B. CO2 reduction on Cu at low overpotentials with surface-enhanced in situ spectroscopy. J. Phys. Chem. C 2016, 120, 17334–17341. [Google Scholar] [CrossRef]
- Wuttig, A.; Liu, C.; Peng, Q.; Yaguchi, M.; Hendon, C.H.; Motobayashi, K.; Ye, S.; Osawa, M.; Surendranath, Y. Tracking a common surface-bound intermediate during CO2-to-fuels catalysis. ACS Cent. Sci. 2016, 2, 522–528. [Google Scholar] [CrossRef]
- Wain, A.J.; O’Connell, M.A. Advances in surface-enhanced vibrational spectroscopy at electrochemical interfaces. Adv. Phys. X 2017, 2, 188–209. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.; Koga, O.; Yamazaki, H.; Matsuo, T. Infrared spectroscopy of adsorbed CO and intermediate species in electrochemical reduction of CO2 to hydrocarbons on a Cu electrode. Electrochim. Acta 1995, 40, 2617–2622. [Google Scholar] [CrossRef]
- Oda, I.; Ogasawara, H.; Ito, M. Carbon monoxide adsorption on copper and silver electrodes during carbon dioxide electroreduction studied by infrared reflection absorption spectroscopy and surface-enhanced raman spectroscopy. Langmuir 1996, 12, 1094–1097. [Google Scholar] [CrossRef]
- Hori, Y.; Koga, O.; Watanabe, Y.; Matsuo, T. FTIR measurements of charge displacement adsorption of CO on poly-and single crystal (100) of Cu electrodes. Electrochim. Acta 1998, 44, 1389–1395. [Google Scholar] [CrossRef]
- Zhu, S.; Jiang, B.; Cai, W.-B.; Shao, M. Direct observation on reaction intermediates and the role of bicarbonate anions in CO2 electrochemical reduction reaction on Cu surfaces. J. Am. Chem. Soc. 2017, 139, 15664–15667. [Google Scholar] [CrossRef]
- Gunathunge, C.M.; Ovalle, V.J.; Li, Y.; Janik, M.J.; Waegele, M.M. Existence of an electrochemically inert CO population on Cu electrodes in alkaline pH. ACS Catal. 2018, 8, 7507–7516. [Google Scholar] [CrossRef]
- Baruch, M.F.; Pander, J.E., III; White, J.L.; Bocarsly, A.B. Mechanistic insights into the reduction of CO2 on tin electrodes using in situ ATR-IR spectroscopy. ACS Catal. 2015, 5, 3148–3156. [Google Scholar] [CrossRef]
- Pander, J.E., III; Baruch, M.F.; Bocarsly, A.B. Probing the mechanism of aqueous CO2 reduction on post-transition-metal electrodes using ATR-IR spectroelectrochemistry. ACS Catal. 2016, 6, 7824–7833. [Google Scholar] [CrossRef]
- Feaster, J.T.; Shi, C.; Cave, E.R.; Hatsukade, T.; Abram, D.N.; Kuhl, K.P.; Hahn, C.; Nørskov, J.K.; Jaramillo, T.F. Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes. ACS Catal. 2017, 7, 4822–4827. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Quan, X.; Yu, H. Efficient electrochemical reduction of carbon dioxide to acetate on nitrogen-doped nanodiamond. J. Am. Chem. Soc. 2015, 137, 11631–11636. [Google Scholar] [CrossRef]
- Hori, Y.I. Electrochemical CO2 reduction on metal electrodes. In Modern Aspects of Electrochemistry; Springer: Berlin, Germany, 2008; pp. 89–189. [Google Scholar]
- Chen, Y.; Li, C.W.; Kanan, M.W. Aqueous CO2 reduction at very low overpotential on oxide-derived Au nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. [Google Scholar] [CrossRef]
- Morhart, T.A.; Unni, B.; Lardner, M.J.; Burgess, I.J. Electrochemical ATR-SEIRAS Using Low-Cost, Micromachined Si Wafers. Anal. Chem. 2017, 89, 11818–11824. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.-K.; Wang, J.-Y.; Li, Q.-X.; Yan, Y.-G.; Liu, J.-H.; Cai, W.-B. Practically modified attenuated total reflection surface-enhanced IR absorption spectroscopy for high-quality frequency-extended detection of surface species at electrodes. Anal. Chem. 2008, 80, 166–171. [Google Scholar] [CrossRef]
- Shao, M.-h.; Liu, P.; Adzic, R.R. Superoxide anion is the intermediate in the oxygen reduction reaction on platinum electrodes. J. Am. Chem. Soc. 2006, 128, 7408–7409. [Google Scholar] [CrossRef]
- Neubrech, F.; Pucci, A.; Cornelius, T.W.; Karim, S.; García-Etxarri, A.; Aizpurua, J. Resonant plasmonic and vibrational coupling in a tailored nanoantenna for infrared detection. Phys. Rev. Lett. 2008, 101, 157403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubrech, F.; Huck, C.; Weber, K.; Pucci, A.; Giessen, H. Surface-enhanced infrared spectroscopy using resonant nanoantennas. Chem. Rev. 2017, 117, 5110–5145. [Google Scholar] [CrossRef] [PubMed]
- Huck, C.; Vogt, J.; Sendner, M.; Hengstler, D.; Neubrech, F.; Pucci, A. Plasmonic enhancement of infrared vibrational signals: Nanoslits versus nanorods. ACS Photonics 2015, 2, 1489–1497. [Google Scholar] [CrossRef]
- Zeng, Z.-C.; Hu, S.; Huang, S.-C.; Zhang, Y.-J.; Zhao, W.-X.; Li, J.-F.; Jiang, C.; Ren, B. Novel electrochemical Raman spectroscopy enabled by water immersion objective. Anal. Chem. 2016, 88, 9381–9385. [Google Scholar] [CrossRef]
- Fleischmann, M.; Hendra, P.J.; McQuillan, A.J. Raman spectra of pyridine adsorbed at a silver electrode. Chem. Phys. Lett. 1974, 26, 163–166. [Google Scholar] [CrossRef]
- Ren, B.; Li, X.; She, C.; Wu, D.; Tian, Z. Surface Raman spectroscopy as a versatile technique to study methanol oxidation on rough Pt electrodes. Electrochim. Acta 2000, 46, 193–205. [Google Scholar] [CrossRef]
- Yeo, B.S.; Klaus, S.L.; Ross, P.N.; Mathies, R.A.; Bell, A.T. Identification of hydroperoxy species as reaction intermediates in the electrochemical evolution of oxygen on gold. ChemPhysChem 2010, 11, 1854–1857. [Google Scholar] [CrossRef]
- Deng, Y.; Handoko, A.D.; Du, Y.; Xi, S.; Yeo, B.S. In situ Raman spectroscopy of copper and copper oxide surfaces during electrochemical oxygen evolution reaction: Identification of CuIII oxides as catalytically active species. ACS Catal. 2016, 6, 2473–2481. [Google Scholar] [CrossRef]
- Stiles, P.L.; Dieringer, J.A.; Shah, N.C.; Van Duyne, R.P. Surface-enhanced Raman spectroscopy. Annu. Rev. Anal. Chem. 2008, 1, 601–626. [Google Scholar] [CrossRef] [Green Version]
- Ichinohe, Y.; Wadayama, T.; Hatta, A. Electrochemical reduction of CO2 on silver as probed by surface-enhanced Raman scattering. J. Raman Spectrosc. 1995, 26, 335–340. [Google Scholar] [CrossRef]
- Mahojey, M.R.; Howard, M.W.; Cooney, R.P. Carbon dioxide conversion to hydrocarbons at silver electrode surfaces: Raman Spectroscpic evidence for surface carbon intermediates. Chem. Phys. Lett. 1980, 71, 59–63. [Google Scholar] [CrossRef]
- Deng, Y.; Huang, Y.; Ren, D.; Handoko, A.D.; Seh, Z.W.; Hirunsit, P.; Yeo, B.S. On the Role of Sulfur for the Selective Electrochemical Reduction of CO2 to Formate on CuS x Catalysts. ACS Appl. Mater. Interfaces 2018, 10, 28572–28581. [Google Scholar] [CrossRef]
- Smith, B.; Irish, D.; Kedzierzawski, P.; Augustynski, J. A Surface Enhanced Roman Scattering Study of the Intermediate and Poisoning Species Formed during the Electrochemical Reduction of CO2 on Copper. J. Electrochem. Soc. 1997, 144, 4288–4296. [Google Scholar] [CrossRef]
- Bohra, D.; Ledezma-Yanez, I.; Li, G.; de Jong, W.; Pidko, E.A.; Smith, W.A. Lateral Adsorbate Interactions Inhibit HCOO− while Promoting CO Selectivity for CO2 Electrocatalysis on Silver. Angew. Chem. Int. Ed. 2019, 131, 1359–1363. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, K.G.; Gewirth, A.A. In situ surface-enhanced Raman spectroscopy of the electrochemical reduction of carbon dioxide on silver with 3, 5-diamino-1, 2, 4-triazole. J. Phys. Chem. C 2014, 118, 17567–17576. [Google Scholar] [CrossRef]
- Mahoney, M.R.; Howard, M.W.; Cooney, R.P. Raman spectra of carbon monoxide adsorbed on silver electrodes. J. Electroanal. Chem. 1984, 161, 163–167. [Google Scholar] [CrossRef]
- Orozco, G.; Pérez, M.C.; Rincon, A.; Gutiérrez, C. Adsorption and electrooxidation of carbon monoxide on silver. Langmuir 1998, 14, 6297–6306. [Google Scholar] [CrossRef]
- Gajdoš, M.; Eichler, A.; Hafner, J. CO adsorption on close-packed transition and noble metal surfaces: Trends from ab initio calculations. J. Phys. Condens. Matter 2004, 16, 1141. [Google Scholar] [CrossRef] [Green Version]
- Ikezawa, Y.; Saito, H.; Matsubayashi, H.; Toda, G. Comparative study of CO adsorbed on Pt, Pd, Au and Ag electrodes in neutral solution by IR reflection absorption spectroscopy. J. Electroanal. Chem. 1988, 252, 395–402. [Google Scholar] [CrossRef]
- Ren, D.; Ang, B.S.-H.; Yeo, B.S. Tuning the selectivity of carbon dioxide electroreduction toward ethanol on oxide-derived CuxZn catalysts. ACS Catal. 2016, 6, 8239–8247. [Google Scholar] [CrossRef]
- Singhal, A.; Pai, M.R.; Rao, R.; Pillai, K.T.; Lieberwirth, I.; Tyagi, A.K. Copper (I) oxide nanocrystals–one step synthesis, characterization, formation mechanism, and photocatalytic properties. Eur. J. Inorg. Chem. 2013, 2013, 2640–2651. [Google Scholar] [CrossRef]
- Das, D.; Mondal, P. Photoluminescence phenomena prevailing in c-axis oriented intrinsic ZnO thin films prepared by RF magnetron sputtering. RSC Adv. 2014, 4, 35735–35743. [Google Scholar] [CrossRef]
- Jiang, S.; Klingan, K.; Pasquini, C.; Dau, H. New aspects of operando Raman spectroscopy applied to electrochemical CO2 reduction on Cu foams. J. Chem. Phys. 2019, 150, 041718. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Deng, Y.; Handoko, A.D.; Chen, C.S.; Malkhandi, S.; Yeo, B.S. Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper (I) oxide catalysts. ACS Catal. 2015, 5, 2814–2821. [Google Scholar] [CrossRef]
- Zhang, Z.; Sheng, S.; Wang, R.; Sun, M. Tip-Enhanced Raman Spectroscopy; ACS Publications: Washington, DC, USA, 2016. [Google Scholar]
- Zaleski, S.; Wilson, A.J.; Mattei, M.; Chen, X.; Goubert, G.; Cardinal, M.F.; Willets, K.A.; Van Duyne, R.P. Investigating nanoscale electrochemistry with surface-and tip-enhanced Raman spectroscopy. Acc. Chem. Res. 2016, 49, 2023–2030. [Google Scholar] [CrossRef]
- Bressler, C.; Chergui, M. Ultrafast X-ray absorption spectroscopy. Chem. Rev. 2004, 104, 1781–1812. [Google Scholar] [CrossRef]
- Koningsberger, D.; Prins, R. X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS, and XANES; Wiley: New York, NY, USA, 1988. [Google Scholar]
- Stötzel, J.; Lützenkirchen-Hecht, D.; Fonda, E.; De Oliveira, N.; Briois, V.; Frahm, R. Novel angular encoder for a quick-extended x-ray absorption fine structure monochromator. Rev. Sci. Instrum. 2008, 79, 083107. [Google Scholar] [CrossRef]
- McBreen, J.; Ogrady, W.E.; Pandya, K.I. EXAFS: New tool for study of battery and fuel cell materials. J. Power Sour. 1987, 22, 323–340. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, M. A review on the structural studies of batteries and host materials by X-ray absorption spectroscopy. ISRN Mater. Sci. 2013, 2013, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Bunker, G. Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy; Cambridge University Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Beer, A. Bestimmung der absorption des rothen lichts in farbigen flussigkeiten. Ann. Phys. 1852, 162, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Jiang, J.; Weng, Z.; Wang, M.; Broere, D.L.; Zhong, Y.; Brudvig, G.W.; Feng, Z.; Wang, H. Electroreduction of CO2 catalyzed by a heterogenized Zn–porphyrin complex with a redox-innocent metal center. ACS Cent. Sci. 2017, 3, 847–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García de Arquer, F.P.; Bushuyev, O.S.; De Luna, P.; Dinh, C.T.; Seifitokaldani, A.; Saidaminov, M.I.; Tan, C.S.; Quan, L.N.; Proppe, A.; Kibria, M.G.; et al. 2D Metal Oxyhalide-Derived Catalysts for Efficient CO2 Electroreduction. Adv. Mater. 2018, 30, 1802858. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Diercks, C.S.; Zhang, Y.-B.; Kornienko, N.; Nichols, E.M.; Zhao, Y.; Paris, A.R.; Kim, D.; Yang, P.; Yaghi, O.M. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 2015, 349, 1208–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Siahrostami, S.; Zheng, T.; Hu, Y.; Hwang, S.; Stavitski, E.; Peng, Y.; Dynes, J.; Gangisetty, M.; Su, D. Isolated Ni single atoms in graphene nanosheets for high-performance CO2 reduction. Energy Environ. Sci. 2018, 11, 893–903. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, J.; Shi, J.; Tan, D.; Liu, L.; Zhang, F.; Lu, C.; Su, Z.; Tan, X.; Cheng, X.; et al. Manganese acting as a high-performance heterogeneous electrocatalyst in carbon dioxide reduction. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.-H.; Bushuyev, O.S.; Li, J.; De Luna, P.; Seifitokaldani, A.; Dinh, C.-T.; Garcia de Arquer, F.P.; Wang, Y.; Liang, Z.; Proppe, A.H.; et al. Metal–Organic Frameworks Mediate Cu Coordination for Selective CO2 Electroreduction. J. Am. Chem. Soc. 2018, 140, 11378–11386. [Google Scholar] [CrossRef]
- Weng, Z.; Wu, Y.; Wang, M.; Jiang, J.; Yang, K.; Huo, S.; Wang, X.-F.; Ma, Q.; Brudvig, G.W.; Batista, V.S.; et al. Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction. Nat. Commun. 2018, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- Clausen, B.; Grabaek, L.; Topsoe, H.; Hansen, L.; Stoltze, P.; Norskov, J.; Nielsen, O. A new procedure for particle size determination by EXAFS based on molecular dynamics simulations. J. Catal. 1993, 141, 368–379. [Google Scholar] [CrossRef]
- Seifitokaldani, A.; Gabardo, C.M.; Burdyny, T.; Dinh, C.-T.; Edwards, J.P.; Kibria, M.G.; Bushuyev, O.S.; Kelley, S.O.; Sinton, D.; Sargent, E.H. Hydronium-induced switching between CO2 electroreduction pathways. J. Am. Chem. Soc. 2018, 140, 3833–3837. [Google Scholar] [CrossRef]
- Gabardo, C.M.; Seifitokaldani, A.; Edwards, J.P.; Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; O’Brien, C.P.; Sargent, E.H.; Sinton, D. Combined high alkalinity and pressurization enable efficient CO 2 electroreduction to CO. Energy Environ. Sci. 2018, 11, 2531–2539. [Google Scholar] [CrossRef]
- Kibria, M.G.; Dinh, C.T.; Seifitokaldani, A.; De Luna, P.; Burdyny, T.; Quintero-Bermudez, R.; Ross, M.B.; Bushuyev, O.S.; García de Arquer, F.P.; Yang, P. A surface reconstruction route to high productivity and selectivity in CO2 electroreduction toward C2+ hydrocarbons. Adv. Mater. 2018, 30, 1804867. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.-Q.; Zhuang, T.-T.; Seifitokaldani, A.; Li, J.; Huang, C.-W.; Tan, C.-S.; Li, Y.; De Luna, P.; Dinh, C.T.; Hu, Y.; et al. Copper-on-nitride enhances the stable electrosynthesis of multi-carbon products from CO2. Nat. Commun. 2018, 9, 3828. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.-T.; Liang, Z.-Q.; Seifitokaldani, A.; Li, Y.; De Luna, P.; Burdyny, T.; Che, F.; Meng, F.; Min, Y.; Quintero-Bermudez, R.; et al. Steering post-C–C coupling selectivity enables high efficiency electroreduction of carbon dioxide to multi-carbon alcohols. Nat. Catal. 2018, 1, 421. [Google Scholar] [CrossRef]
- Braun, A.; Sivula, K.; Bora, D.K.; Zhu, J.; Zhang, L.; Gratzel, M.; Guo, J.; Constable, E.C. Direct observation of two electron holes in a hematite photoanode during photoelectrochemical water splitting. J. Phys. Chem. C 2012, 116, 16870–16875. [Google Scholar] [CrossRef]
- Hämäläinen, K.; Siddons, D.; Hastings, J.; Berman, L. Elimination of the inner-shell lifetime broadening in X-ray-absorption spectroscopy. Phys. Rev. Lett. 1991, 67, 2850. [Google Scholar] [CrossRef] [PubMed]
- Lelong, G.R.; Radtke, G.; Cormier, L.; Bricha, H.; Rueff, J.-P.; Ablett, J.M.; Cabaret, D.; Gélébart, F.D.R.; Shukla, A. Detecting non-bridging oxygens: Non-resonant inelastic X-ray scattering in crystalline lithium borates. Inorg. Chem. 2014, 53, 10903–10908. [Google Scholar] [CrossRef]
- Al Samarai, M.; Delgado-Jaime, M.U.; Ishii, H.; Hiraoka, N.; Tsuei, K.-D.; Rueff, J.P.; Lassale-Kaiser, B.; Weckhuysen, B.M.; De Groot, F.M. 1s3p resonant inelastic X-ray scattering of cobalt oxides and sulfides. J. Phys. Chem. C 2016, 120, 24063–24069. [Google Scholar] [CrossRef] [Green Version]
- Glatzel, P.; Singh, J.; Kvashnina, K.O.; van Bokhoven, J.A. In situ characterization of the 5d density of states of Pt nanoparticles upon adsorption of CO. J. Am. Chem. Soc. 2010, 132, 2555–2557. [Google Scholar] [CrossRef]
- Yan, Y.J.; Zhang, W.; Che, J. Time–frequency theory of pump-probe absorption spectroscopy. J. Chem. Phys. 1997, 106, 2212–2224. [Google Scholar] [CrossRef]
- Goulielmakis, E.; Loh, Z.-H.; Wirth, A.; Santra, R.; Rohringer, N.; Yakovlev, V.S.; Zherebtsov, S.; Pfeifer, T.; Azzeer, A.M.; Kling, M.F.; et al. Real-time observation of valence electron motion. Nature 2010, 466, 739. [Google Scholar] [CrossRef]
- Wang, M.; Árnadóttir, L.; Xu, Z.J.; Feng, Z. In situ X-ray absorption spectroscopy studies of nanoscale electrocatalysts. Nanomicro Lett. 2019, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Knop-Gericke, A.; Kleimenov, E.; Hävecker, M.; Blume, R.; Teschner, D.; Zafeiratos, S.; Schlögl, R.; Bukhtiyarov, V.I.; Kaichev, V.V.; Prosvirin, I.P. X-ray photoelectron spectroscopy for investigation of heterogeneous catalytic processes. Adv. Catal. 2009, 52, 213–272. [Google Scholar]
- Daulton, T.L.; Little, B.J.; Lowe, K.; Jones-Meehan, J. In situ environmental cell–transmission electron microscopy study of microbial reduction of chromium (VI) using electron energy loss spectroscopy. Microsc. Microanal. 2001, 7, 470–485. [Google Scholar] [PubMed]
- Nishijima, K.; Yamasaki, J.; Orihara, H.; Tanaka, N. Development of microcapsules for electron microscopy and their application to dynamical observation of liquid crystals in transmission electron microscopy. Nanotechnology 2004, 15, S329. [Google Scholar] [CrossRef]
- Mohanty, N.; Fahrenholtz, M.; Nagaraja, A.; Boyle, D.; Berry, V. Impermeable graphenic encasement of bacteria. Nano Lett. 2011, 11, 1270–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, M.; Tromp, R.; Vereecken, P.; Hull, R.; Ross, F. Dynamic microscopy of nanoscale cluster growth at the solid–liquid interface. Nat. Mater. 2003, 2, 532. [Google Scholar] [CrossRef]
- Kolmakov, A.; Dikin, D.A.; Cote, L.J.; Huang, J.; Abyaneh, M.K.; Amati, M.; Gregoratti, L.; Günther, S.; Kiskinova, M. Graphene oxide windows for in situ environmental cell photoelectron spectroscopy. Nat. Nanotechnol. 2011, 6, 651. [Google Scholar] [CrossRef]
- Kraus, J.; Reichelt, R.; Günther, S.; Gregoratti, L.; Amati, M.; Kiskinova, M.; Yulaev, A.; Vlassiouk, I.; Kolmakov, A. Photoelectron spectroscopy of wet and gaseous samples through graphene membranes. Nanoscale 2014, 6, 14394–14403. [Google Scholar] [CrossRef]
- Kolmakov, A.; Gregoratti, L.; Kiskinova, M.; Günther, S. Recent approaches for bridging the pressure gap in photoelectron microspectroscopy. Top. Catal. 2016, 59, 448–468. [Google Scholar] [CrossRef] [Green Version]
- Velasco-Velez, J.J.; Pfeifer, V.; Hävecker, M.; Weatherup, R.S.; Arrigo, R.; Chuang, C.H.; Stotz, E.; Weinberg, G.; Salmeron, M.; Schlögl, R. Photoelectron spectroscopy at the graphene-liquid interface reveals the electronic structure of an electrodeposited cobalt/graphene electrocatalyst. Angew. Chem. Int. Ed. 2015, 54, 14554–14558. [Google Scholar] [CrossRef] [Green Version]
- Axnanda, S.; Crumlin, E.J.; Mao, B.; Rani, S.; Chang, R.; Karlsson, P.G.; Edwards, M.O.; Lundqvist, M.; Moberg, R.; Ross, P. Using “tender” X-ray ambient pressure X-ray photoelectron spectroscopy as a direct probe of solid-liquid interface. Sci. Rep. 2015, 5, 9788. [Google Scholar] [CrossRef] [PubMed]
- Favaro, M.; Xiao, H.; Cheng, T.; Goddard, W.A.; Yano, J.; Crumlin, E.J. Subsurface oxide plays a critical role in CO2 activation by Cu (111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl. Acad. Sci. USA 2017, 114, 6706–6711. [Google Scholar] [PubMed] [Green Version]
- Scholten, F.; Sinev, I.; Bernal, D.M.; Roldan Cuenya, B. Plasma-modified dendritic Cu catalyst for CO2 electroreduction. ACS Catal. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reller, C.; Krause, R.; Volkova, E.; Schmid, B.; Neubauer, S.; Rucki, A.; Schuster, M.; Schmid, G. Selective electroreduction of CO2 toward ethylene on nano dendritic copper catalysts at high current density. Adv. Energy Mater. 2017, 7, 1602114. [Google Scholar] [CrossRef]
- Dutta, A.; Rahaman, M.; Luedi, N.C.; Mohos, M.; Broekmann, P. Morphology matters: Tuning the product distribution of CO2 electroreduction on oxide-derived Cu foam catalysts. ACS Catal. 2016, 6, 3804–3814. [Google Scholar] [CrossRef]
- Rosen, J.; Hutchings, G.S.; Lu, Q.; Forest, R.V.; Moore, A.; Jiao, F. Electrodeposited Zn dendrites with enhanced CO selectivity for electrocatalytic CO2 reduction. ACS Catal. 2015, 5, 4586–4591. [Google Scholar] [CrossRef]
- Ade, H.; Kirz, J.; Hulbert, S.; Johnson, E.; Anderson, E.; Kern, D. Scanning photoelectron microscope with a zone plate generated microprobe. Nucl. Instrum. Methods Phys. Res. Sec. A Acceler. Soectrom. Detectors Assoc. Equip. 1990, 291, 126–131. [Google Scholar] [CrossRef]
- Sezen, H.; Alemán, B.; Amati, M.; Dalmiglio, M.; Gregoratti, L. Spatially resolved chemical characterization with scanning photoemission spectromicroscopy: Towards near-ambient-pressure experiments. ChemCatChem 2015, 7, 3665–3673. [Google Scholar] [CrossRef]
- Amati, M.; Abyaneh, M.K.; Gregoratti, L. Dynamic High Pressure: A novel approach toward near ambient pressure photoemission spectroscopy and spectromicroscopy. J. Instrum. 2013, 8, T05001. [Google Scholar] [CrossRef]
- Roy, K.; Artiglia, L.; van Bokhoven, J.A. Ambient pressure photoelectron spectroscopy: Opportunities in catalysis from solids to liquids and introducing time resolution. ChemCatChem 2018, 10, 666–682. [Google Scholar] [CrossRef]
- Wolter, O.; Heitbaum, J. Differential electrochemical mass spectroscopy (DEMS)—A new method for the study of electrode processes. Berichte der Bunsengesellschaft für Physikalische Chemie 1984, 88, 2–6. [Google Scholar] [CrossRef]
- Clark, E.L.; Singh, M.R.; Kwon, Y.; Bell, A.T. Differential electrochemical mass spectrometer cell design for online quantification of products produced during electrochemical reduction of CO2. Anal. Chem. 2015, 87, 8013–8020. [Google Scholar] [CrossRef] [Green Version]
- Lobaccaro, P.; Mandal, L.; Motapothula, M.R.; Sherburne, M.; Martin, J.; Venkatesan, T.; Ager, J.W. Initial Application of Selected-Ion Flow-Tube Mass Spectrometry to Real-Time Product Detection in Electrochemical CO2 Reduction. Energy Technol. 2018, 6, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Khanipour, P.; Löffler, M.; Reichert, A.M.; Haase, F.T.; Mayrhofer, K.J.; Katsounaros, I. Electrochemical Real-Time Mass Spectrometry (EC-RTMS): Monitoring Electrochemical Reaction Products in Real Time. Angew. Chem. Int. Ed. 2019, 131, 7351–7355. [Google Scholar]
- Wonders, A.H.; Housmans, T.H.; Rosca, V.; Koper, M.T. On-line mass spectrometry system for measurements at single-crystal electrodes in hanging meniscus configuration. J. Appl. Electrochem. 2006, 36, 1215–1221. [Google Scholar] [CrossRef]
- Grote, J.-P.; Zeradjanin, A.R.; Cherevko, S.; Mayrhofer, K.J. Coupling of a scanning flow cell with online electrochemical mass spectrometry for screening of reaction selectivity. Rev. Sci. Instrum. 2014, 85, 104101. [Google Scholar] [CrossRef] [PubMed]
- Karabinas, P.; Wolter, O.; Heitbaum, J. Mechanistic studies with mass spectroscopic cyclic voltammetry: Anodic oxidation of hydroxylamine on Pt. Berichte der Bunsengesellschaft für Physikalische Chemie 1984, 88, 1191–1196. [Google Scholar] [CrossRef]
- Baltruschat, H. Differential electrochemical mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1693–1706. [Google Scholar] [CrossRef] [Green Version]
- Trimarco, D.B.; Pedersen, T.; Hansen, O.; Chorkendorff, I.; Vesborg, P.C. Fast and sensitive method for detecting volatile species in liquids. Rev. Sci. Instrum. 2015, 86, 075006. [Google Scholar] [CrossRef]
- Annesley, T.M. Ion suppression in mass spectrometry. Clin. Chem. 2003, 49, 1041–1044. [Google Scholar] [CrossRef] [Green Version]
- Oberacher, H.; Pitterl, F.; Erb, R.; Plattner, S. Mass spectrometric methods for monitoring redox processes in electrochemical cells. Mass Spectrom. Rev. 2015, 34, 64–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, T.A.; Chen, H.; Zare, R.N. Identification of fleeting electrochemical reaction intermediates using desorption electrospray ionization mass spectrometry. J. Am. Chem. Soc. 2015, 137, 7274–7277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jusys, Z.; Massong, H.; Baltruschat, H. A New Approach for Simultaneous DEMS, and EQCM: Electro-oxidation of Adsorbed CO on Pt and Pt-Ru. J. Electrochem. Soc. 1999, 146, 1093–1098. [Google Scholar] [CrossRef]
- Schuppert, A.K.; Topalov, A.A.; Katsounaros, I.; Klemm, S.O.; Mayrhofer, K.J. A scanning flow cell system for fully automated screening of electrocatalyst materials. J. Electrochem. Soc. 2012, 159, F670–F675. [Google Scholar] [CrossRef]
- Clark, E.L.; Bell, A.T. Direct observation of the local reaction environment during the electrochemical reduction of CO2. J. Am. Chem. Soc. 2018, 140, 7012–7020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, S.J. Design, Construction and Research Application of a Differential Electrochemical Mass Spectrometer (DEMS); Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Bravo-Suárez, J.J.; Srinivasan, P.D. Design characteristics of in situ and operando ultraviolet-visible and vibrational spectroscopic reaction cells for heterogeneous catalysis. Catal. Rev. 2017, 59, 1–151. [Google Scholar] [CrossRef]
- McNeil, B.W.; Thompson, N.R. X-ray free-electron lasers. Nat. Photonics 2010, 4, 814. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Probed Information | Merits | Limitation |
|---|---|---|---|
| IR | Absorption of molecular vibrations | Non-destructive sampling Useable from ultrahigh vacuum to atmospheric pressures Useable in vacuum, gaseous, liquid and even solid phases | Limited IR transmission Indirect information Macroscopic information |
| RS | Inelastically scattered light | Non-destructive sampling Easy sample preparation Fast spectra acquisition within seconds Not interfered by water Wide spectra region (4000–50 cm−1) | Low spatial resolution Macroscopic information Fluorescence interference Unable to detect the intermediates with low scattering cross-sections |
| XAS | Absorption coefficient | Element specific Metal site sensitive Not limited by the sample state (powder, solution, frozen solution) | Average information Hard to detect light elements |
| XPS | Kinetic energy and number of electrons that escape from the material | Nondestructive Surface sensitive (10–200 Å) Almost all elements (except H, He) High information content | Low atomic resolution Pressure gap Limited time resolution Limited spatial resolution |
| MS | Mass-to-charge ratio of ions | High accuracy (<1 ppm) Consume little sample | Limited product collection |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, L.; Seifitokaldani, A. In Situ Spectroscopic Methods for Electrocatalytic CO2 Reduction. Catalysts 2020, 10, 481. https://doi.org/10.3390/catal10050481
Jin L, Seifitokaldani A. In Situ Spectroscopic Methods for Electrocatalytic CO2 Reduction. Catalysts. 2020; 10(5):481. https://doi.org/10.3390/catal10050481
Chicago/Turabian StyleJin, Lei, and Ali Seifitokaldani. 2020. "In Situ Spectroscopic Methods for Electrocatalytic CO2 Reduction" Catalysts 10, no. 5: 481. https://doi.org/10.3390/catal10050481
APA StyleJin, L., & Seifitokaldani, A. (2020). In Situ Spectroscopic Methods for Electrocatalytic CO2 Reduction. Catalysts, 10(5), 481. https://doi.org/10.3390/catal10050481