1. Introduction
Layered double hydroxides (LDHs), also known as anionic-clays or hydrotalcites, are a group of inorganic lamellar compounds of basic nature. In the last decades, much attention is being paid to the study of these inorganic materials due to their chemical and structural properties, which make them useful in interesting applications, such as adsorbents, catalysts, anion exchangers or flame retardants, among others [
1,
2,
3]. These materials exhibit high chemical stability, good biocompatibility and pH-dependent solubility [
4]. The first LDH reported in the literature, discovered in 1842, was the mineral hydrotalcite [Mg
6Al
2(OH)
16]CO
3·4 H
2O. This inorganic structure results from stacked brucite layers, Mg(OH)
2, where some Mg
2+ ions can be replaced by Al
3+ ions, thus generating an excess of positive charge in layers, which must be counterbalanced by the presence of anions, mainly carbonates (CO
32−), in the interlayer spacing [
1,
2,
3].
The general formula of LDH is [M(II)
1−xM(III)
x(OH)
2]
x + [A
n − x/n]
x−·mH
2O, where M(II) are divalent cations (Mg
2+, Zn
2+, Cu
2+, Ni
2+) and M(III) are trivalent cations (Al
3+, Fe
3+, Cr
3+). A
n− is an anion of charge
n, and
m is the stoichiometric value of co-intercalated water [
1].
LDHs can be synthesized through several synthesis methods, such as co-precipitation, sol-gel, hydrothermal or urea hydrolysis, although the most usual method is co-precipitation, which can be performed at variable or constant pH [
3].
Focusing only on the catalytic properties of LDHs themselves, these inorganic compounds can display both acid and basic sites [
5,
6]. However, the amount and strength of these acid/basic sites can be modified by thermal treatment, where dehydroxylation and decarbonation processes cause the collapse of the layered structure, leading to the formation of their respective metal oxides [
6]. In addition, if any of M(II) or M(III) cations is easily reducible, it is feasible to obtain catalysts with metallic, acid and basic sites, in such a way that they can find a large spectrum of catalytic applications.
Among the great variety of catalytic processes that have emerged in the last century, the biomass valorization to produce high value-added chemicals is attracting the interest of many research groups, as a sustainable alternative to the use of fossil-based raw materials. This great interest has been prompted by the depletion of fossil fuels, which has led to the search and development of alternative sources that can satisfy both chemical and energy demands [
7,
8]. Biomass is the only feedstock that can replace fossil fuels, but many efforts are still required for the implementation and integration of these processes in the forthcoming biorefineries. However, the selection of a biomass source must be carried out with care and responsibility, since this biomass could interfere with the food chain, causing serious speculation problems and social imbalances. Taking into account these premises, lignocellulosic biomass, coming from agricultural waste, has emerged as an abundant, sustainable and non-edible source of energy, biofuels and chemicals [
9]. Lignocellulose is composed by cellulose (40–50%), hemicellulose (20–35%) and lignin (15–25%), which can be extracted selectively by using several thermal, physical and chemical treatments [
10].
Focusing only on the hemicellulose fraction, this can be isolated under mild hydrolysis treatment, leading, after depolymerization and hydrolysis, to the respective monosaccharides, mainly xylose [
11,
12]. Xylose, in turn, can also be dehydrated through homogeneous and/or heterogeneous catalysts to obtain furfural (FUR) as the main product [
12,
13]. After bioethanol, furfural is the second most produced compound in the sugar platform. The great interest in this organic compound lies in its chemical structure (an aldehyde group and a furan ring with α,β-unsaturations), which confer it a high reactivity [
12]. Thus, furfural can be used as feedstock to give rise to a wide range of products with applications in fields, such as polymers, pharmacy, cosmetics, among others, through hydrogenation, oxidation, dehydration, decarbonylation or condensation reactions [
12,
14,
15]. Thus, for instance, different environmentally friendly processes have been reported aimed at the furfural derivatization for the synthesis of valuable chemicals in aqueous media [
16,
17], as well as in in eco-sustainable removable media as deep eutectic solvents, by using homogeneous [
18] or heterogeneous catalysts [
19,
20].
Among the products that can be derived from FUR, furfuryl alcohol (FOL) is the most important. It has been estimated that about 62% of FUR production is employed for the synthesis of FOL due to its importance in resin manufacture for the foundry industry and for chemicals [
15]. Industrially, FOL has been synthesized through furfural hydrogenation using a copper chromite catalyst [
21,
22,
23,
24]. Another product obtained in high proportions during FUR hydrogenation is 2-methylfuran (MF) [
12], which is also considered a valuable product as a biofuel additive and for the synthesis of heterocycles [
15].
Despite the good performance of the commercial copper chromite catalyst, in the last decade, environmental awareness has led to the search for chromium-free catalysts, which can be more sustainable. As alternative, transition metal-based catalysts, e.g., Cu [
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35], Ni [
28,
35,
36,
37,
38,
39,
40,
41] or Pd [
28,
42,
43] have been proposed to replace copper chromite. Generally, these metals have been dispersed on different supports. Both the hydrogenating character of the metal and the acid/base/redox properties of the support play a determining role in the activity and the product pattern, mainly for those processes where FUR hydrogenation takes place in the gas phase.
In this context, the design of LDHs as catalyst precursors, where some of their cations can be easily reducible, is a suitable approach for the development of active catalysts for FUR hydrogenation to obtain high value-added chemicals. Several authors have reported the synthesis of Ni-based hydrotalcites as efficient catalysts for this reaction, although the high hydrogenating nature of Ni sites [
28] leads to a wide range of products, including a high proportion of carbonaceous deposits in many cases [
35,
36]. A suitable alternative could be the use of Cu-based hydrotalcites, since Cu active centers display a lower hydrogenating capacity, in such a way that the selectivity pattern can be easily controlled. In this sense, previous research has evaluated the role of Al source in the synthesis of Cu/ZnO/Al
2O
3 from their respective hydroxides, after calcination and reduction [
44]. These authors achieved well dispersed Cu centers, which were selective towards FOL [
44]. In the present work, a series of [CuZn
1−xAl
x(OH)
2]
x + [A
n − x/n]
x−·mH
2O hydrotalcites have been synthesized by coprecipitation method, with different Cu/Zn molar ratio, although the M(II)/M(III) molar ratio (M(II) is Cu(II) + Zn(II)) was 3 in all cases, since this value favors the formation of ordered LDH structures [
1,
2,
3]. Moreover, the effect of other parameters, like aging time, was also evaluated for the synthesis of LDHs. Precursors were calcined and reduced, and then characterized and tested in the FUR hydrogenation, with special emphasis on the correlation between the amount of available metal sites and their catalytic behavior.
2. Characterization of the Catalysts
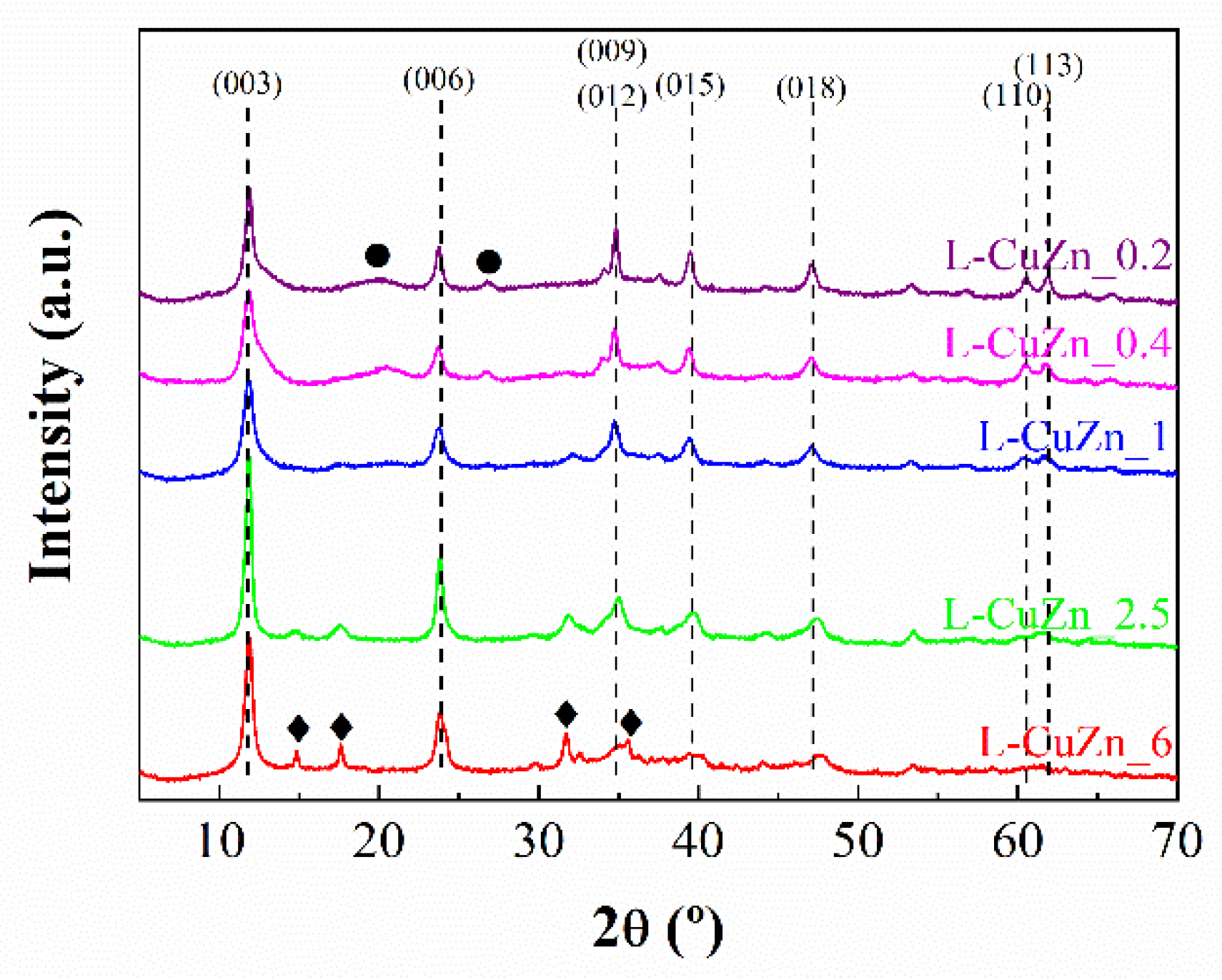
The identification of crystalline phases was carried out by powder X-ray diffraction. In all cases, the XRD patterns of LHDs synthesized (
Figure 1) are similar to those reported in the literature [
1,
45]. The most intense peaks correspond to the typical basal (00
l) planes, thereby, those located at 2θ (°) ≈ 12, 24 and 34 are attributed to the (003), (006) and (009) planes. These can be indexed in a rhombohedral symmetry, which is assigned to a (Cu,Zn)
1−xAl
x(OH)
2 (CO
3)
x/2 m H
2O layered double hydroxide (Powder Diffraction File: PDF: 00-37-0629). In addition, less intense peaks ascribed to non-basal planes of hydrotalcites are also observed, together with other low intense peaks, mainly in those LDHs whose Cu
2+ or Zn
2+ content is higher. Thus, in the case of Cu-rich LDH, diffraction peaks attributed to copper(II) hydroxide are presented, while Zn-rich LDH also exhibits broad and poorly defined diffraction peaks attributed to zinc hydroxycarbonate.
CuZnAl-based hydrotalcites have been previously studied by FTIR and Raman spectroscopies, demonstrating that the Cu/Zn molar ratio influences their microstructure. Thus, higher Cu/Zn ratios lead to more homogeneous CuZnAl hydrotalcites, while lower values give rise to less homogeneous structures due to its composition is closer to pure ZnAl and CuAl hydrotalcites [
46]. This latter could provoke the asymmetric broadening of Bragg peak profiles of LDH for lower Cu/Zn molar ratio, also characteristic of the stacking disorder for Zn-rich compounds (
Figure 1). Moreover, this Raman study also confirmed the presence of carbonate species, not only as counterion in the interlayer region of LDH, but also in partial segregated malachite for high Cu/Zn molar ratios, which is in agreement with XRD data [
46]. Following the procedure described by Santos et al. for (Cu,Zn)
1−xAl
x(OH)
2(CO
3)
x/2·mH
2O LDHs [
46], these layered materials were thermally treated at 300 °C for 4 h. In their XRD patterns (
Figure 2), a broadening of the diffraction peak in comparison to their respective LDHs is noticeable, as a consequence of the stacking disorder of the layered structure due to decarbonation and dehydroxylation of the LDH, leading to the typical diffraction peaks of their mixed metal oxides [
1]. Thus, XRD profiles reveal an evolution from the calcined Cu-rich (P-CuZn_6) to the Zn-rich (P-CuZn_0.2) sample. In this sense, the XRD pattern of calcined P-CuZn_6 displays defined bands located at 2θ (°) = 32.5, 35.5, 38.7, 48.8, 53.5, 58.3, 61.6, 66.2 and 68.1 assigned to CuO (PDF: 00-048-1548). The progressive increase in Zn content gives rise to a much more amorphous XRD pattern, being only a broad band observed at 2θ (°) ≈ 33. These data suggest that calcined P-CuZn_2.5, P-CuZn_1 and P-CuZn_0.4 display a more amorphous structure, or are formed by smaller particles than that the calcined P-CuZn_6. For the calcined LDH with the highest Zn content (P-CuZn_0.2), broad bands at 2θ (°) = 31.8, 34.4, 36.2, 48.3, 56.5, 63.1 and 68.2, start to emerge. These peaks are assigned to hexagonal ZnO (PDF: 00-36-1451). On the other hand, no diffraction peaks could be ascribed to the presence of crystalline Al species, in such a way that these species must be amorphous, or dispersed in the other phases.
Once the catalyst precursors were synthesized after subsequently calcined, H
2-TPR analysis was carried out to determine the appropriate reduction temperature to obtain the Cu
0-based catalysts (
Figure 3). It has been previously reported that usually it is not possible to discern the different stages of Cu reduction (Cu
2+ → Cu
+ → Cu
0), so the different contributions observed are ascribed to Cu particles with different size and/or with different interaction with the support (ZnO/Al
2O
3) [
26,
30,
34]. From the H
2-TPR profiles, it can be inferred that the sample with the highest Cu content (CuZn_6) is more easily reducible than the rest of catalysts. Its reduction curve shows two well-defined reduction steps located at 185 and 240 °C, which can be explained by the co-existence of CuO particles with different interaction with the support, or particles with different size. As the Cu content decreases and the Zn content concomitantly increases, the H
2-TPR profile progressively changes. Thus, it seems clear that the hydrogen consumption at low temperature progressively decreased, as observed for CuZn_2.5 and CuZn_1. In the case of CuZn_0.4, this low temperature band disappears, being only noticeable the presence of a single band whose maximum is located about 245 °C. Those catalysts with a lower proportion of Zn seem to be more reducible than the richer ones, thus indicating that the presence of ZnO would modify the electronic environment around CuO particles, as previously pointed out by other authors [
47,
48]. Previous research has reported that the reduction of Cu species in CuO/ZnO takes place about 210 °C [
30], so Al species could exert an additional electronic promoter effect on the interaction between ZnO and CuO particles, in such a way that the reduction of Cu
2+ species occurs at higher temperature, as was indicated in the literature [
49]. On the other hand, as the catalyst with a lower Zn content is easily reducible, it is expected that Al species possess a less pronounced promoter effect than ZnO.
H
2 consumption data of calcined P-CuZn_X materials are always lower than theoretical values, so a fraction of Cu
2+ species could not be reduced (
Table 1). In this sense, several authors have reported that the reduction of segregated metal carbonates can take place at high temperature, about 550 °C [
46]. However, other authors have noted that Cu
2+ ions could also be embedded in the octahedral sites of Al
2O
3, ZnO, or even ZnAl
2O
4, whose reduction should require a higher temperature [
50,
51].
Taking into account the H2-TPR profiles, the precursors were reduced at 300 °C, maintaining this temperature for 1 h to ensure the complete reduction of Cu2+ species.
XRD profiles of the CuZn_X catalysts show the typical diffraction peaks of the metallic Cu
0 at 2θ (°) = 43.3 and 50.4 (PDF: 00-85-1326) (
Figure 4). The presence of Cu
2O should be discarded since, despite the main peak at 2θ (°) = 38.3 overlaps with a diffraction peak of ZnO, the secondary peaks of Cu
2O do not appear, so partially reduced copper oxide (Cu
2O) crystallites are not detected, or these are too small to be detected by XRD.
These signals are more defined than those corresponding to the hexagonal ZnO, which does not evolve to reduced Zn species. In the case of Al species, diffractograms do not reveal any characteristic peaks, so these Al species must be highly dispersed in the catalysts. The determination of the crystallite size for Cu
0 was carried out from the Williamson-Hall equation [
52], using the main diffraction peak at 2θ (°) = 43.3. The analysis of this (111) crystallographic plane reveals that the crystallinity of the Cu
0 particles increases directly with the Cu content, from 5.0 nm for the lowest content (CuZn_0.2) to 28.8 nm for the highest (CuZn_6). These value are slightly lower than those observed for Cu/ZnO or Cu/MgO synthesized by the co-precipitation method [
26,
30], so Al species, in addition of exerting an electronic promoter effect, seem to favor the dispersion of Cu nanoparticles [
49,
53].
In order to elucidate the catalyst morphology, CuZn_X catalysts were analyzed by TEM (
Figure 5). In all cases, the micrographs allow to distinguish different morphologies, including layered structures probably due to the hydrotalcite structure that did not fully collapse after the thermal treatment. These data are in agreement with the literature, where a proportion of the layered structure was reported to be maintained after the thermal treatment [
54]. In addition, it is noticeable the presence of pseudospherical and well-dispersed nanoparticles, mainly in the samples with a higher Cu content. In both cases, these particles are very small, below 15 nm.
The analysis of these samples was also performed by Energy Dispersive X-Ray (EDX) (
Figure 6). The images show that all elements (Cu, Zn, Al and O) are well dispersed in both lamellar and pseudospherical structures. In
Figure 5 and
Figure 6, it can be seen the existence of interparticle voids between adjacent nanoparticles.
The textural properties of CuZn_X catalysts were evaluated from their N
2 adsorption-desorption isotherms at −196 °C (
Figure 7A). According to the International Union of Pure and Applied Chemistry (IUPAC) classification [
55], these isotherms can be considered as Type II, which are typical of macroporous solids, as suggests the great growing of N
2 adsorbed at high relative pressure. The shape is similar for all CuZn_X catalysts, and, consequently, the modification of the Cu/Zn molar ratio does not seem to affect the textural properties of catalysts (
Table 2). Thus, S
BET values hardly vary, being between 70 and 85 m
2 g
−1, while
t-plot data indicate that the surface ascribed to the microporosity can be considered as negligible, since values are below 10 m
2 g
−1 in all cases. In the same way, the pore volume is very similar for all catalysts, being in the range 0.474–0.679 cm
3 g
−1, while the micropore volume is very low in comparison to the total volume.
With regard to the pore size distribution, estimated by Density Functional Theory (DFT) [
56] (
Figure 7B), all CuZn_X catalysts follow the same pattern. The microporosity is barely appreciated, and only a small contribution appears at 1.2 nm. However, these samples exhibit a wide pore size distribution, extending from 3 to 150 nm, in such a way that these catalysts can be labeled as meso- and mainly macroporous. This porosity is ascribed to the voids between adjacent particles, as was suggested previously from the TEM micrographs (
Figure 5 and
Figure 6).
The quantification of surface Cu
0 species of CuZn_X catalysts was carried out by N
2O titration at 60 °C [
57] (
Table 3). The data indicate that dispersion decreases with the Cu content, going from 21% for CuZn_0.2 to 7% for CuZn_6. In the same way, the metallic surface area (m
2Cu g
Cu−1) follows a similar trend, and the catalyst with the lowest Cu content (CuZn_0.2) displays the highest value. This study also reveals that the metal particle size increases directly with the Cu loading, from 5 nm for CuZn_0.2 to 19 nm for CuZn_6, being these data slightly lower than those obtained from XRD by using the Williamson-Hall method [
52].
In order to determine the surface chemical composition of catalysts, X-ray photoelectron spectroscopy (XPS) analysis was carried out. Cu 2p core level spectra (
Figure 8A) show that Cu 2p
3/2 consists of a single contribution located at 932.0 eV, ascribed to reduced Cu species [
30,
32], since the absence of the typical shake-up satellite of divalent metals about 942–943 eV would exclude the existence of Cu(II) [
58].
However, from this contribution, it is not possible to discern between Cu
+ and Cu
0 species, so the Auger Cu
LMM signal is used to identify these oxidation states [
30,
32]. The broad Auger Cu
LMM band (
Figure 8B) can be deconvoluted in two main contributions: 918.5 eV, which is ascribed to Cu
0, and 917.0 eV due to Cu
+ [
30]. The proportion of Cu
0 increases directly with the Cu content, being about 65–70% of the total reduced Cu species. The analysis of the Zn 2p region (
Table 4) evidences a single band between 1021.6–1022.1 eV, which is assigned to ZnO. Some studies of Cu/ZnO catalysts have demonstrated that ZnO can be partially reduced, arising a new contribution located at lower binding energy. This new band is not observed in the Zn 2p core level spectra; however, the Auger Zn
LMM line (
Figure 8C) shows an asymmetric band that would confirm the existence of a small proportion of Zn partially reduced (Zn
δ+), or even a synergistic effect between Cu
0 and ZnO [
59,
60]. With regard to the Al 2p core level spectra, all CuZn_X catalysts display a contribution about 74.3 eV, which can be attributed to Al
2O
3 [
61]. In the case of O 1s, all catalysts display a main contribution located about 531.0 eV, assigned to oxide species, and another less intense at 532.6 eV, that is assigned to hydroxyl and/or carbonate [
61]. The existence of carbonates could be confirmed by the signal at a binding energy of 288–289 eV in the C 1s core level spectra [
61], which could result from a low proportion of LDH, or a possible carbonation of the catalyst surface.
The atomic concentration data show that the surface Cu content progressive increases with the Cu loading, as expected (
Table 4), although surface molar ratios seem to be lower than the theoretical values. This fact could be ascribed to both ZnO and Al
2O
3 exhibit a smaller crystallite size than Cu
0, in such a way that both metal oxides should be better dispersed in catalysts. This fact can lead to lower than expected surface Cu concentrations.
As both acid and basic sites could also influence on the gas-phase FUR hydrogenation reaction, NH
3-TPD and CO
2-TPD studies were performed (
Table 2). The NH
3-TPD data show a relatively low amount of acid sites, decreasing from 78 μmol g
−1 for CuZn_0.2 to 60 μmol g
−1 for CuZn_6. The presence of these acid sites is ascribed to Al
2O
3 and Cu
+, which provide Lewis acid sites, and even to ZnO due to its amphoteric character, similar to alumina. Several authors have reported that the modulation of the acidity plays a key role in FUR conversion, since a high number of acid sites can cause the polymerization of FUR, mainly in gas-phase, leading to the formation of a high proportion of carbonaceous deposits [
29,
37,
62]. These polymerized FUR species interact strongly with the active sites, in such a way that catalysts tend to be deactivated relatively fast. However, the presence of weak acid sites could exert a beneficial effect on the catalytic behavior, since the interaction between FUR, or reaction products, and the catalyst is weakened in comparison with catalyst with higher acidity, thus favoring the desorption of products adsorbed on the catalyst surface. In this sense, several supports, such as SiO
2 or clay minerals, with similar amount of acid sites [
29,
37], have provided good catalytic activity in FUR hydrogenation, under similar experimental conditions.
Similarly, a basic support (MgO, CaO, ZnO or CeO
2) [
25,
26,
30,
63,
64] also seems to have a positive effect in this catalytic process. CuZn_X catalysts display a small concentration of basic sites, between 15 and 39 μmol g
−1, raising as the Cu content decreases. The presence of these basic sites is ascribed to the amphoteric character of both ZnO and Al
2O
3, as previously mentioned [
65,
66].
4. Materials and Methods
4.1. Preparation of Catalysts
A series of Cu/ZnO/Al
2O
3 catalysts has been synthesized by co-precipitation method, according to the methodology described by Santos et al. [
46]. The LDH precursors were synthesized from aqueous solutions of Cu(NO
3)
2·2H
2O (99%, Aldrich, Saint Louis, MI, USA), Zn(NO
3)
2·6H
2O (99%, Aldrich) and Al(NO
3)
3·9H
2O (99%, Aldrich) with a total metal concentration of 0.3 M. In all cases, (Cu
2++Zn
2+)/Al
3+ molar ratio was 3, whereas the Cu
2+/Zn
2+ molar ratio was varied between 0.2 and 6. In the next step, an aqueous solution of Na
2CO
3 (1.0 M) was slowly added to precipitate metal hydroxides and form the LDH structure. Later, the obtained gel was aged at room temperature for 48 h (in two different synthesis, the gel was aged 1 and 168 h). The obtained solid was filtered and washed with distilled water until reach a neutral pH to ensure the total removal of Na
+ ions. Finally, the solid was dried overnight at 90 °C and calcined at 300 °C for 4 h, using a ramp of 10 °C min
−1.
4.2. Characterization of Catalysts
An X´Pert Pro automated diffractometer (PANanalytical, Bruker, Rheinstetten, Germany) was used to obtain powder X-ray diffraction patterns, this equipment are composed of a Ge (111) primary monochromator (Cu Kα1) and a X´Celerator detector with a step size of 0.017° (2θ), between 2θ = 10° and 70° with an equivalent counting time of 712 s per step. Williamson-Hall Equation (1) [
52] was applied to calculate the crystallite size (D):
where B is the full width at half maximum (FWHM) of the XRD peaks, θ the Bragg angle, B is, K is the Scherrer constant, λ is the wavelength of the X ray and ε is the lattice strain.
A FEI Talos F200X (Thermo Fisher Scientific, Waltham, MA, USA) system was used to study the catalyst morphology by transmission electron microscopy (TEM). The images obtained with this technique show a high resolution; moreover, this equipment allows the combination of STEM and TEM imaging and, therefore, a 3D characterization with chemical composition mapping. The samples were dispersed in isopropyl alcohol and each one of them was put on a carbon grid.
The temperature of catalysts reduction was evaluated by H2-TPR (hydrogen temperature-programmed reduction). To carry out the experiments, 0.080 g of sample has been used. First, the catalyst precursor is treated under a He flow (35 mL min−1) at 100 °C for 30 min, after this, it is cooling to room temperature to start the analysis, where the H2 consumption is monitored between 50 and 800 °C, by using an Ar/H2 flow (48 mL min−1, 10 vol.% H2) with a heating rate of 10 °C min−1. An on-line thermal conductivity detector (TCD) (Shimadzu, Kioto, Japan) was used to carry out the H2 quantification. It is necessary to trap the water formed in the process to avoid equipment contamination, so the outcoming flow is passed through a cold finger immersed in a liquid N2/isopropanol bath (−80 °C).
In previous research [
30,
37], N
2O titration has been employed to determine the metallic surface area and the dispersion of Cu
0 species, where the superficial oxidation of Cu
0 under a N
2O flow takes place according to the Equation (2):
Prior to the analysis, the catalyst precursor was treated under a He flow (35 mL min−1) at 100 °C for 30 min, followed by a reduction step under a 10 vol.% H2/Ar-flow (48 mL min−1) at 300 °C for 1 h, with a heating rate of 5 °C min−1, being monitored the H2 consumption by TCD. When the catalyst was reduced, it was cooled until 60 °C under a He flow to carry out the Cu0 oxidation to Cu+ performed by titration N2O (5 vol.% N2O/He), at 60 °C for 1 h. At last, a second sample reduction was carried out by heating from room temperature to 300 °C with a heating rate of 5 °C min−1, being also monitored its H2 consumption by TCD.
The textural parameters were determined from the N
2 adsorption-desorption isotherms at −196 °C by using an automatic ASAP 2020 Micromeritics apparatus (Micrometrics, Norcross, GA, USA). The sample was previously outgassed at 200 °C and 10
−4 mbar for 12 h. The Brunauer-Emmet-Teller (BET) equation [
72] was utilized to determine surface area taking a N
2 cross section of 16.2 Å
2. Micropore volume was determined from alpha-s- method. Pore size distribution was determined using the density functional theory (DFT) [
56].
X-ray photoelectron spectra were obtained using a Physical Electronic PHI 5700 spectrometer (Physical Electronics, Eden Prairie, MN, USA), equipped with an Electronics 80-365B multichannel hemispherical electron analyzer and an Mg Kα X-ray excitation source (300 W, 15 kV, hv = 1253.6 eV). High-resolution spectra were recorded by a concentric hemispherical analyzer in a 29.35 eV constant energy mode, using a 720 µm diameter analysis area, and the pressure in the analysis chamber was kept below 5 × 10−6 Pa. Binding energies (BE) were determined to an accuracy of ± 0.1 eV, using the adventitious carbon C 1s signal at 284.8 eV as reference. PHI ACCESS ESCA-F V6 software (Eden Prairie, Minnesota, USA) was used for data acquisition and analysis. A Shirley-type background was subtracted from the signals. The recorded spectra were always analyzed with Gauss-Lorentz curves, in order to determine more precisely the binding energy of the atomic levels of the different elements. To avoid sample oxidation after reduction, they were stored in sealed vials with an inert solvent; moreover, samples were prepared in a dry box under a N2 flow, where the solvent was evaporated prior to its introduction into the analysis chamber, and directly analyzed without previous treatment.
To calculate the total acid sites present in the samples, thermo-programmed desorption of ammonia (NH3-TPD) was carried out. In a typical procedure, 0.08 g of catalyst is placed in a U-shape quartz reactor and cleaned flowing He (40 mL·min−1) up to 400 °C with a heating rate of 10 °C·min−1. Then, the sample is cooled until 100 °C under the same He flow, and once the temperature is stabilized at 100 °C, the sample is saturated with ammonia for 5 minutes and then physisorbed NH3 was removed under He. Ammonia desorption is performed by heating the sample from 100 to 400 °C, with a rate of 10 °C·min−1, registering the signal using a GC-14B instrument (Shimadzu, Kioto, Japan) equipped with a thermal conductivity detector (TCD), previously calibrated with Ni(NH3)6Cl2 (Aldrich) in order to quantify total acid sites.
Thermo-programmed desorption of CO2 (CO2-TPD) was employed for the quantification of basic sites. In a typical procedure, 0.03 g of catalyst is pretreated under a He flow (40 mL min−1) at 400 °C for 15 min (10 °C·min−1). Later, the sample was cooled to 100 °C and a pure CO2 stream (60 mL min−1) was subsequently introduced into the reactor for 30 min. Finally, the amount of CO2 evolved was analyzed using a TCD detector between 100 and 600 °C and a helium flow (10 °C min−1).
4.3. Catalytic Tests
The furfural hydrogenation was carried out at atmospheric pressure in a tubular quartz reactor with an internal diameter of 6.35 mm. For this, 150 mg of pelletized catalyst (325–400 μm) were placed inside the reactor between two layers of quartz wool, which is placed inside a programmable temperature tubular furnace, controlled with a thermocouple. Before starting the reaction, catalysts were reduced in-situ with a hydrogen flow (99.99%, Airgas, Paris, France) of 60 mL min−1 for 1 h at the reduction temperature deduced from their corresponding H2-TPR profiles (350 °C). Subsequently, the desired reaction temperature is set and once the system is stable, the reaction is carried out under a flow of H2 that ranges between 10 and 60 ml min−1 and a feed flow of 3.87 mL h−1 of a furfural solution in cyclopentyl methyl ether (CPME, 5 vol.%), which is introduced with the help of an HPLC piston pump, model 307 SC-10 (Gilson, Middleton, WI, USA).
To avoid problems, such as blockage of the lines in the equipment, furfural was dissolved in cyclopentyl methyl ether (CPME). This solvent is environmentally friendly and has been used in different organic reactions [
30]. Reaction samples were collected every hour, dissolved with a chloroform and o-xylene solution (internal standard), stored in sealed vials to be subsequently analyzed by gas chromatography, using a Shimazu GB-14A chromatograph equipped with a flame ionization detector and a CP-Wax 52 CB capillary column.
In a preliminary test, the CuZn_1 catalyst was chosen to evaluate the stability of this solvent at 190 °C, after 5 h of time-on-stream (TOS), but in the absence of furfural. The analysis of the collected samples confirmed the full recovery of CPME; without any products different from this ether, thus demonstrating its stability. The conversion, selectivity and yield values were determined using the following expressions:
The turnover frequency was calculated as follows (6):
where F is the molar rate of furfural (mol h
−1), W is the catalyst weight (g), X is the conversion and M is the mole of sites loaded (mol g
−1). This equation, in which –ln (1 − X) substitutes for X assumes a pseudo first-order reaction which may be justified by the excess of hydrogen [
63].


 and
and 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}