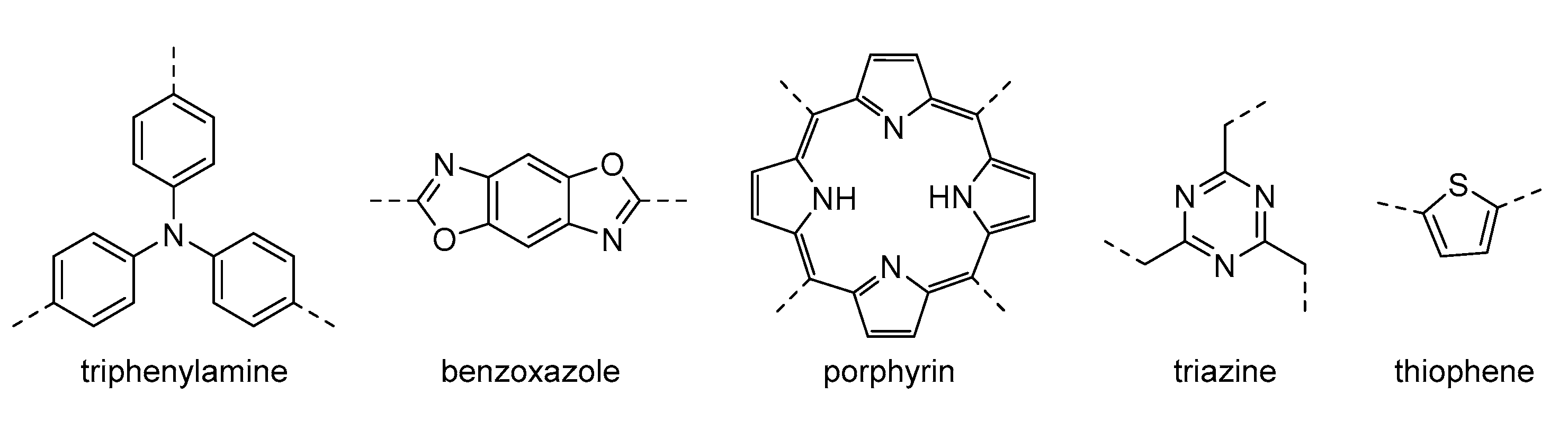
3.1.1. Selective Oxidation of Alcohols
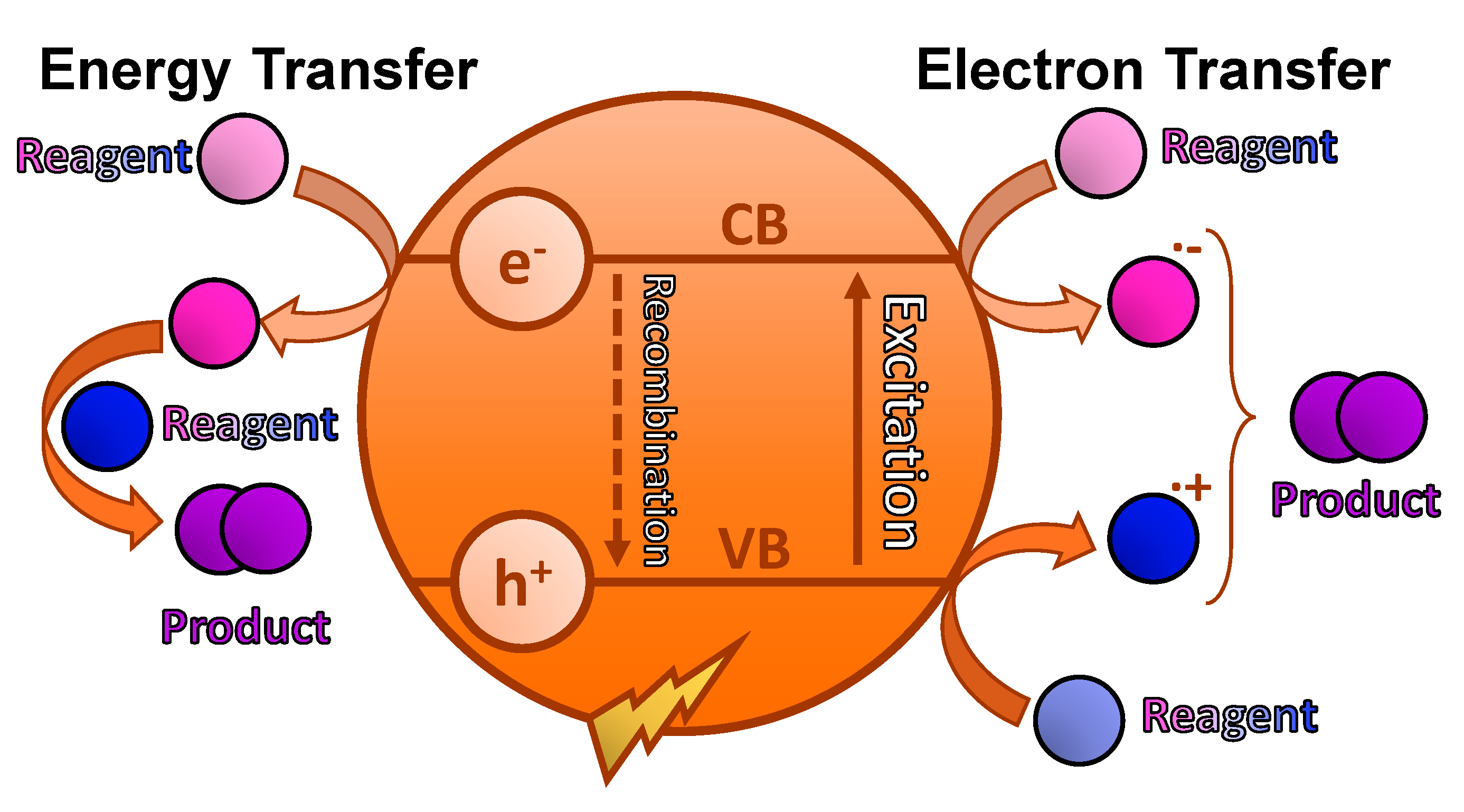
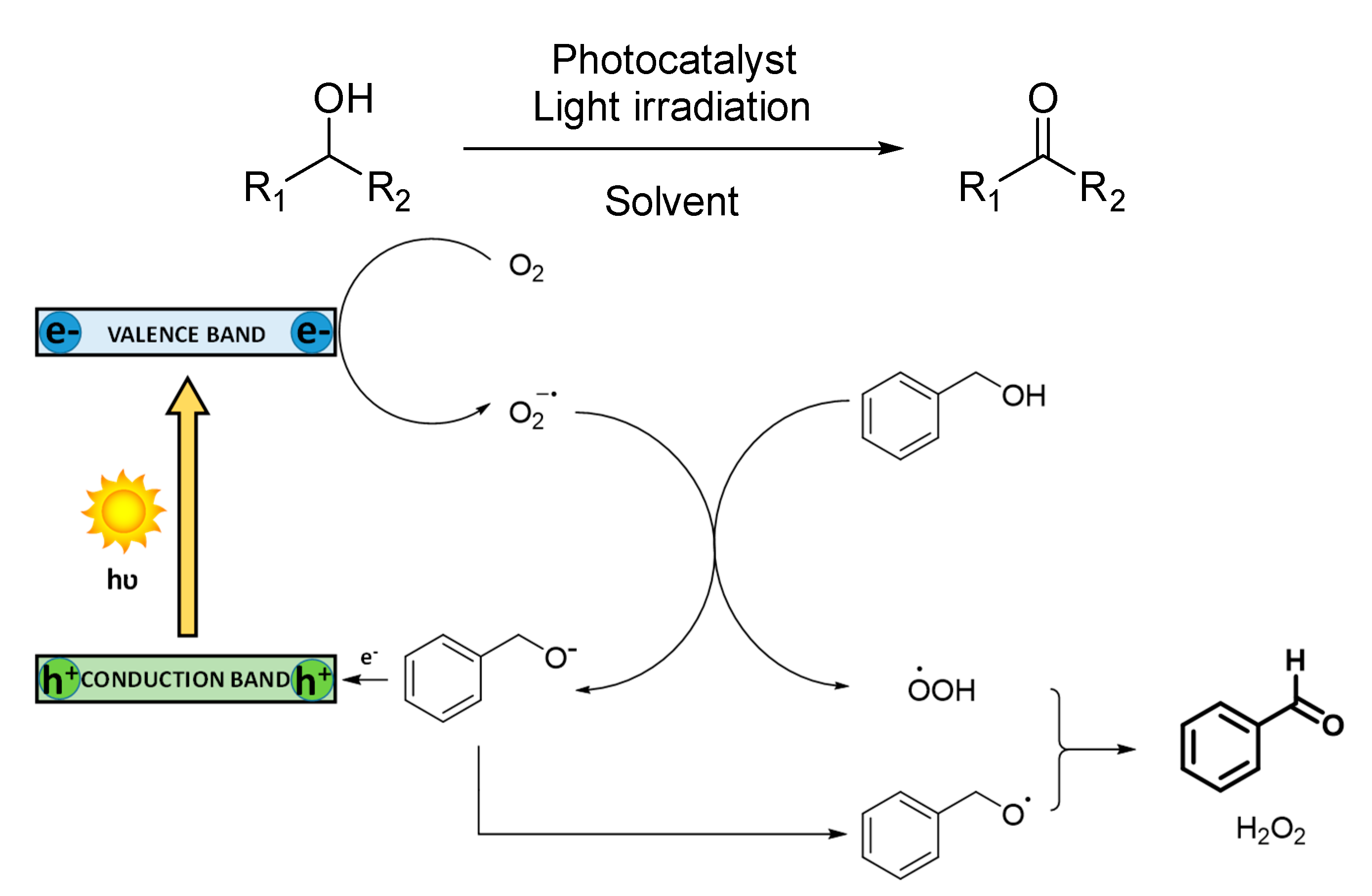
In the selective oxidation of alcohols to aldehydes or ketones, a mechanism based on the generation of superoxide radical anion is commonly reported (
Scheme 3). In this case, the photocatalyst absorbs light, and in its excited state, separation of charges is produced, promoting electrons to the conduction band and generating holes in the valance band. These electrons activate O
2 to generate superoxide radical anion (O
2−·), which deprotonates benzyl alcohol obtaining hydroperoxide radical species (·OOH). Photogenerated holes are quenched by the benzoxide anion to oxidize it to its radical form, which subsequently reacts with ·OOH, generating benzaldehyde as the final product. H
2O
2 is also usually produced. Although singlet oxygen is rarely reported, an energy transfer mechanism is observed in some photocatalytic materials, such as Bi-based MOFs. In this case, the presence of Bi suppresses the generation of O
2−· and H
2O
2, being singlet oxygen the only active oxygen species [
55].
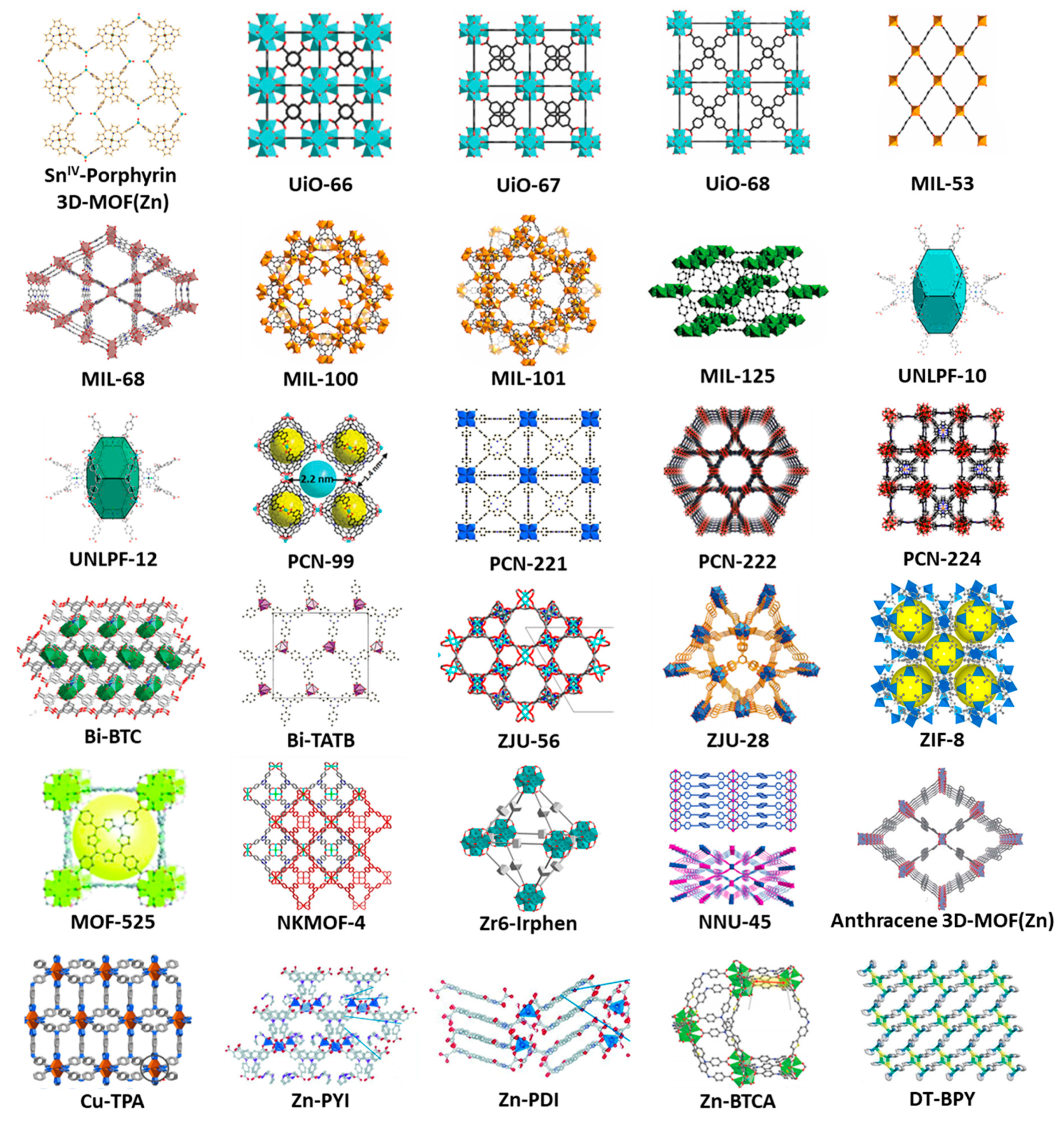
Photocatalytic oxidation of alcohols mediated by MOFs (
Table 1): the first example using the design of linkers or SBUs with photoactive fragments was reported in 2011 by Wu [
29]. A Sn
IV–porphyrin-based MOF was capable of oxidizing 1,5-dihydroxynaphthalene to the corresponding quinone with full conversion. In 2012, an outstanding versatile amine-functionalized zirconium MOF,
UiO-66-NH2 was used as photocatalyst in various oxidation reactions by Wang and coworkers. Although high catalyst loadings were needed, this report represents the first example where the introduction of amino groups into the constituting linkers of a MOF allowed its use as heterogeneous photocatalyst for several oxidation reactions [
86]. This approach was later employed on a series of
UiO-66-NH2 derivatives (consisting on mixed-linker zirconium-based MOFs synthesized through a one-pot procedure) which afforded the carbonylic products with moderate conversions [
87]. Two years later, the variation of the photocatalytic activity of
MIL-125 as a model platform was studied as a function of the presence of relative amounts of bdc-NH
2 linkers. The oxidation of different alcohols was carried out in order to demonstrate that the maximum photocatalytic activity was achieved with a 50% of bdc-NH
2, being the amination of all linkers in the MOF not required [
88]. In 2019, another three examples employing this strategy were reported. Two bismuth-based MOFs,
Bi-TATB and
Bi-BTC, afforded the product in moderate conversions after 5 h [
55]. On the other hand, a series of
Ce-UiO-66 MOFs were synthesized with a solvent-free route, achieving the oxidation of alcohols under oxygen flux and near-ultraviolet light irradiation [
89]. In addition, a further report demonstrated that was also possible the use of visible-light irradiation in these photocatalytic systems [
90].
Another strategy used to synthesize MOFs capable of oxidizing different type of alcohols consists on the design of functionalization protocols. In 2018, a series of MOFs were structurally altered via post-synthetic modification by the condensation of benzaldehyde molecules with the pending amino groups on
NH2-MIL-125(Ti). Under visible light irradiation, an enhancement of the photocatalytic activity was achieved by extending the conjugation through this post-synthetic process, carrying out the oxidation of aromatic alcohols with good results [
91]. In 2019, the encapsulation of the photoactive [Ru(bpy)
3]
2+ complex into MIL-125-NH
2 cavities improved its photocatalytic activity in the oxidation of alcohols [
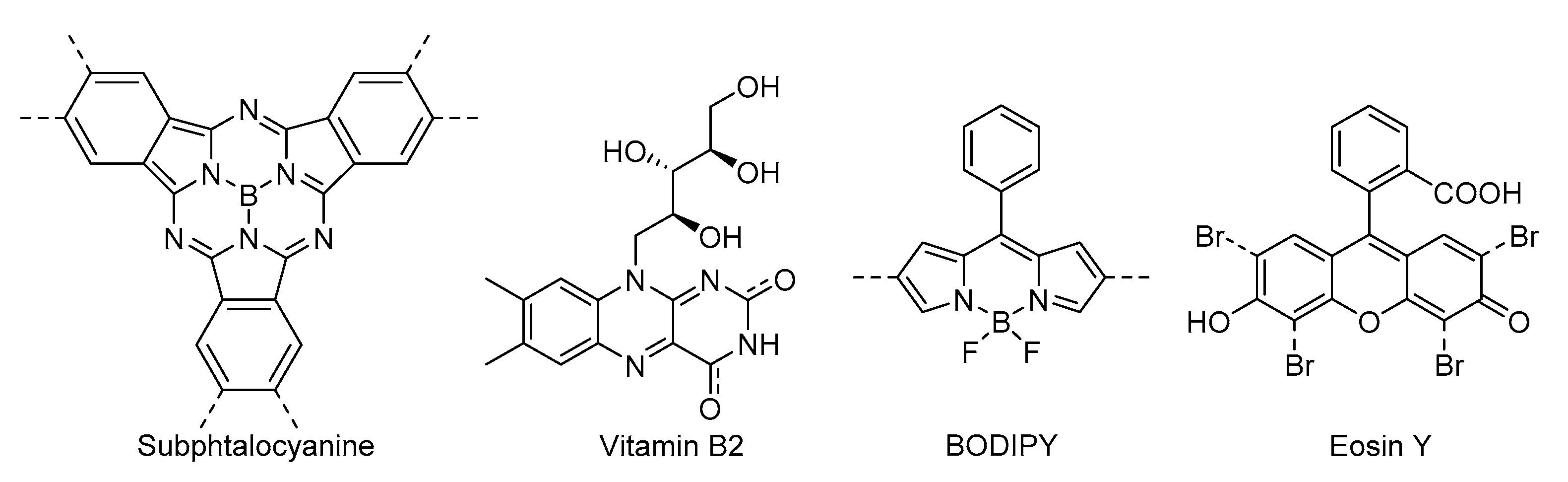
92]. Following a different approach, Huang’s group carried out the covalent anchorage and isolation of vitamin B
2 into UiO-66. Instead of generating superoxide radical anion via photoredox pathway (as observed for the homogeneous system),
VB2@UiO-66 MOF was able to uniquely give rise to singlet oxygen through an energy transfer process. This effect is due to the immobilization and isolation of VB
2, hampering triplet-triplet annihilation and increasing the stability of the photocatalyst [
68].
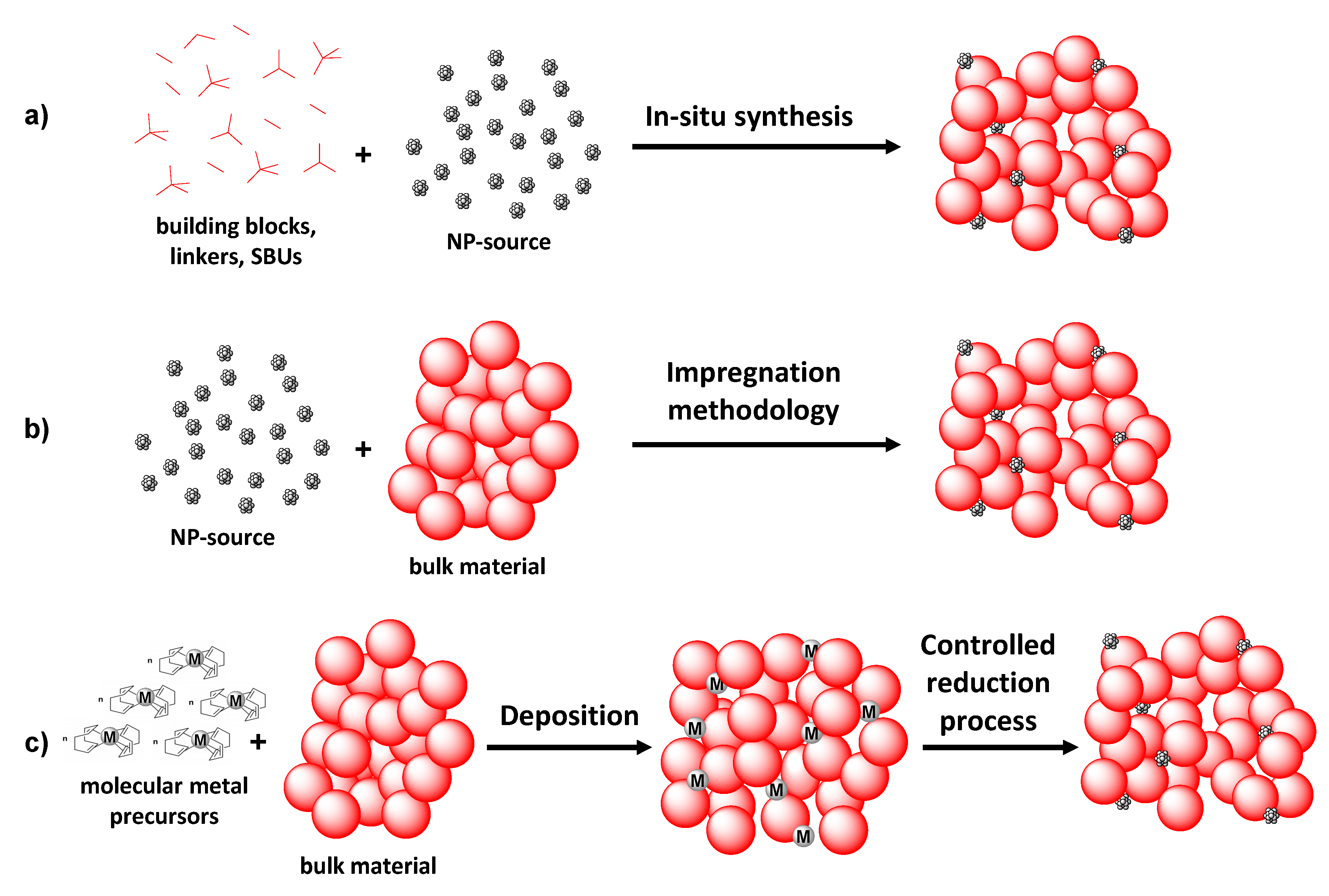
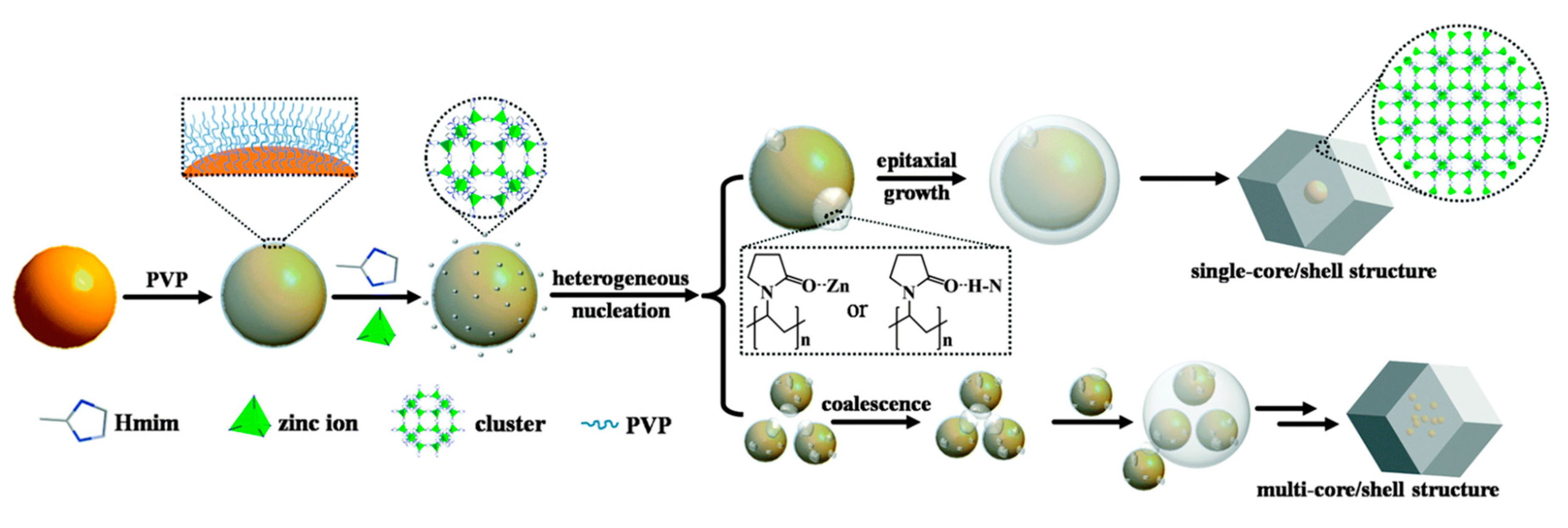
Finally, the first reported example of hybrid structures of MOFs capable of performing this oxidation reaction is from 2014. Thus, Duan and coworkers carried out the encapsulation of Au nanoparticles into the porous structure of ZIF-8. The application of epitaxial growth strategy led to single-core shell structures, while the coalescence methodology gave rise to multi-core shell structures (
Figure 11).
Au@ZIF-8 composites were employed as heterogeneous photocatalysts for the selective oxidation of a variety of alcohols into the corresponding aldehydes or ketones [
74]. One year later, the group of Zhu presented a series of
CdS-MIL-100(Fe) nanocomposites, formed by the superficial decoration of CdS quantum dots into the corresponding MOFs. The hybrid material presented enhanced photocatalytic activity in comparison with their separated components, because of the increased light absorption efficiency, its superior surface area and the easy charge separation and slow recombination rate. They were capable of oxidizing several alcohols with moderate conversions, reaching up to five consecutive recycling cycles [
80]. In 2016, the impregnation of
NH2-MIL-125(Ti) with nickel chloride as a metal source was performed by Zhu [
75]. After a reducing treatment with NaBH
4, Ni nanoparticles were formed. The corresponding
Ni-doped NH2-MIL-125(Ti) was employed as heterogeneous photocatalyst in the selective oxidation of several aromatic alcohols, presenting an enhanced photocatalytic activity as a result of the synergistic effect between Ti–O clusters and Ni–NPs. In 2017, a series of MIL-LIC-1(Eu) graphene oxide composites
MIL@GO were reported, yielding the final products with good conversions and short reaction times upon visible light irradiation and aqueous media. In this case, according to the authors, the molecular oxygen necessary for the transformation was generated in situ from water [
82]. In 2019, the encapsulation and the deposition of copper nanoparticles on a copper-based UiO-66,
Cu/Cu@UiO-66, was reported. This hybrid material achieved the corresponding oxidized products after 3 h in moderate conversions [
93]. Very recently, a very active photocatalytic material has been reported by Yin [
81]. The etching and regrowth process on a
3D MIL-53(Fe), that generates bismuth ferrite (BFO) nanosheets on its surface, was used as a strategy in order to avoid fast electron–hole recombination.
Photocatalytic oxidation of alcohols mediated by COFs (
Table 2): The use of COFs as photocatalysts in organic oxidation reactions is still underexplored and most of the works are very recent. The first report is dated to 2017, where an undecorated pristine thiophene-containing CTF (
CTF-Th@SBA-15) was constructed using mesoporous silica as template. In this work, the performance of this heterogeneous photocatalyst was comparable to the most used non-metal-based catalysts reported up to that date, reaching a similar turnover frequency [
64]. The only two examples of hybrid COFs as heterogeneous photocatalysts for synthetic organic transformations using as model reaction the oxidation of alcohols were reported by Wang and coworkers [
85]. The first of them was devoted to the coating of TiO
2 nanobelts, with an imine-based COF, obtaining the hybrid material
TiO2@COF-3. This material presents an enhanced photocatalytic activity due to the improvement on photon absorption related to the possibility of charge transfer from COF to TiO
2. Such effect makes possible to perform benzylic alcohol oxidation under visible light irradiation. On the other hand, in 2020, a hybrid MOF–COF,
NH2-MIL-125@TAPB-PDA-3, shown similar photocatalytic properties. In this case, amino groups of the previously synthesized MOF served as linking points to the growth of the imine-based COF [
84].
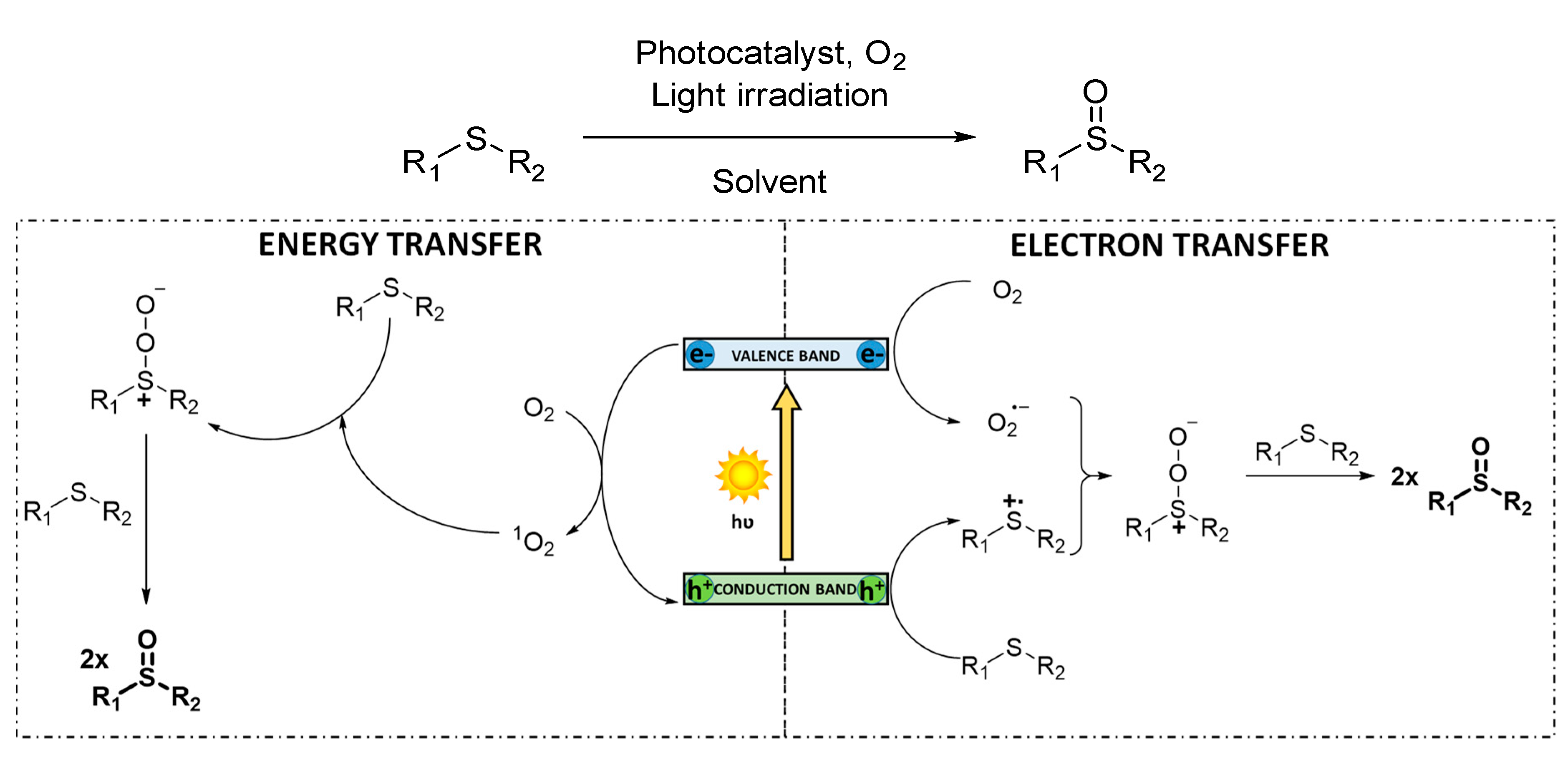
3.1.2. Selective Sulfoxidation Reactions
Sulfoxides are very interesting compounds since they are prevalent structures in agriculture and pharmaceutical industries [
94]. Thus, the selective oxidation of sulfides to sulfoxides is a recurrent model reaction in organic synthesis, since it allows to simply evaluate the photocatalytic activity of the new materials synthesized. Sulfoxidation also allows easy determination of chemoselectivity and to differentiate between the two possible mechanisms involved: an electron transfer or an energy transfer [
62] (
Scheme 4). On the one hand, in an electron transfer mediated mechanism, charge separation is produced in the photocatalyst under light irradiation. The generated holes oxidize the sulfide to its corresponding cationic radical and the photogenerated electrons reduce O
2 to O
2−·. Finally, this active oxygen species reacts with the cationic radical intermediate, generating the persulfoxide species which reacts with a second sulfide to form two molecules of the final sulfoxide. On the other hand, energy transfer process can also take place under visible light irradiation, converting
3O
2 into
1O
2 through triplet–triplet annihilation between the triplet excited state of the catalytic fragment and
3O
2. Then, singlet oxygen reacts with the organic sulfide generating the same persulfoxide intermediate, which account for the formation of the corresponding sulfoxide product. The occurrence of one or another pathway can be precluded using selective scavengers (e.g., DABCO for singlet oxygen [
95], 1,4-dimethoxybenzene for superoxide radical anion [
96]). Such mechanistic probes have also been essayed for other oxidation processes. Interestingly, in many cases, there is not an exclusive pathway, and both mechanisms can be simultaneously operative. In some reports, the question of which mechanism produces the sulfoxide product is not addressed.
Photocatalytic selective oxidation of sulfides mediated by MOFs (
Table 3): Focusing on the field of MOFs, there are some examples in which the design of linkers or SBUs with photoactive fragments is the chosen strategy. In 2011, the versatile Sn
IV-porphyrin-based MOF previously presented in the alcohol oxidation part was also able to oxidize sulfides to sulfoxides with good results. In addition, the process became more selective by the immobilization of porphyrinic fragment (that acts as photosensitizer) into the porous framework, without overoxidation into sulfone product. In contrast, sulfone is found when the analogous molecular porphyrin was employed [
29]. In the same year, Lin and coworkers reached the obtaining of bimetallated MOFs, by doping of
UiO-67 with Ir, Re and Ru centers through the combination of both pre-synthesized metal-coordinated and naked linkers with Zr nodes. These doped MOFs were used as photocatalyst in the selective photosulfoxidation reaction, among other model test reactions, proposing an energy transfer mechanism [
59]. Three years later, an anionic indium porphyrin framework,
UNLPF-10, was capable of selectively oxidizing sulfides under low power visible light irradiation [
31]. In 2019, three bimetallic Ir
III-Zr
IV MOFs were synthesized (
Zr6-Irbpy,
Zr6-IrbpyOMe, and
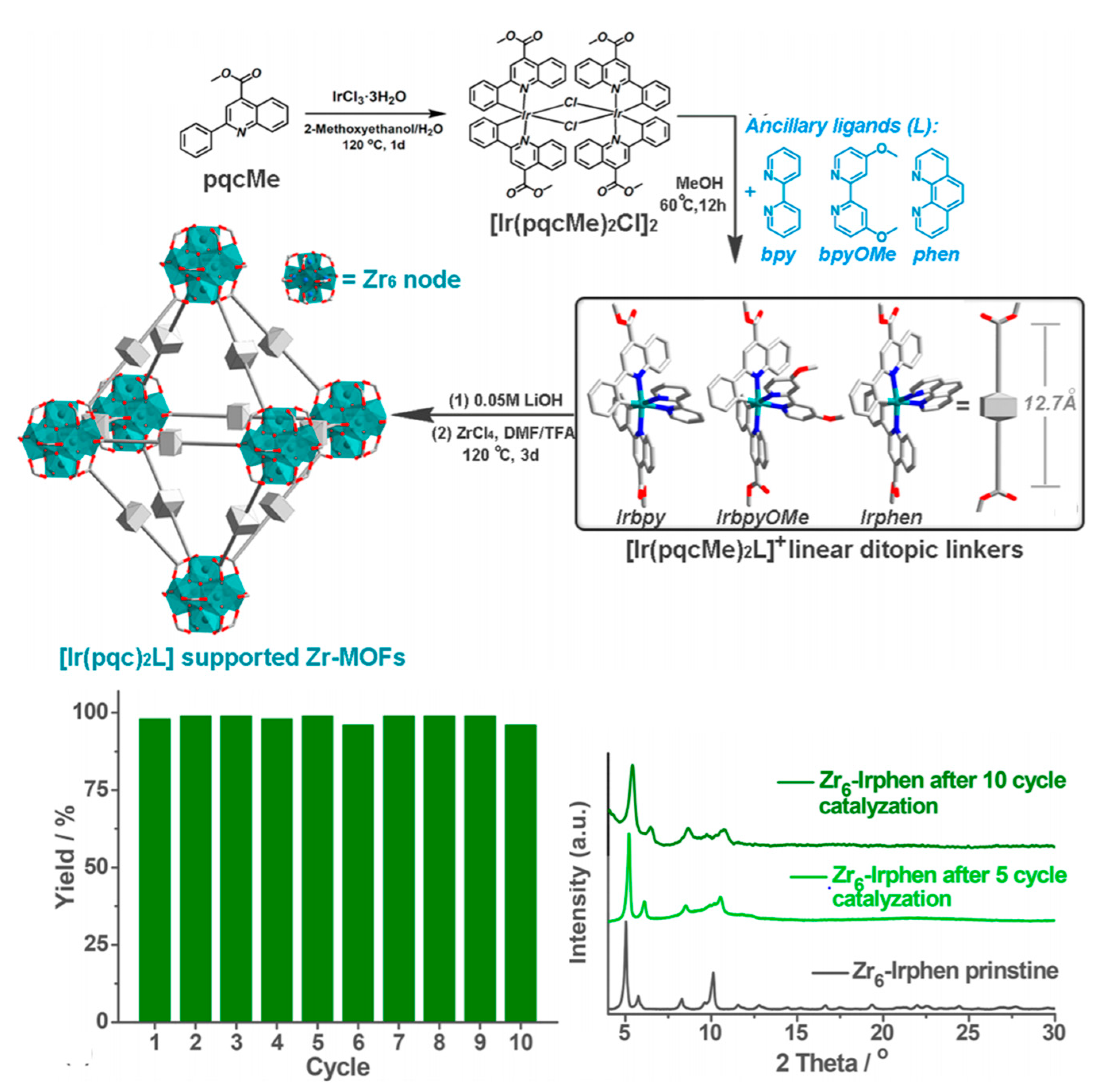
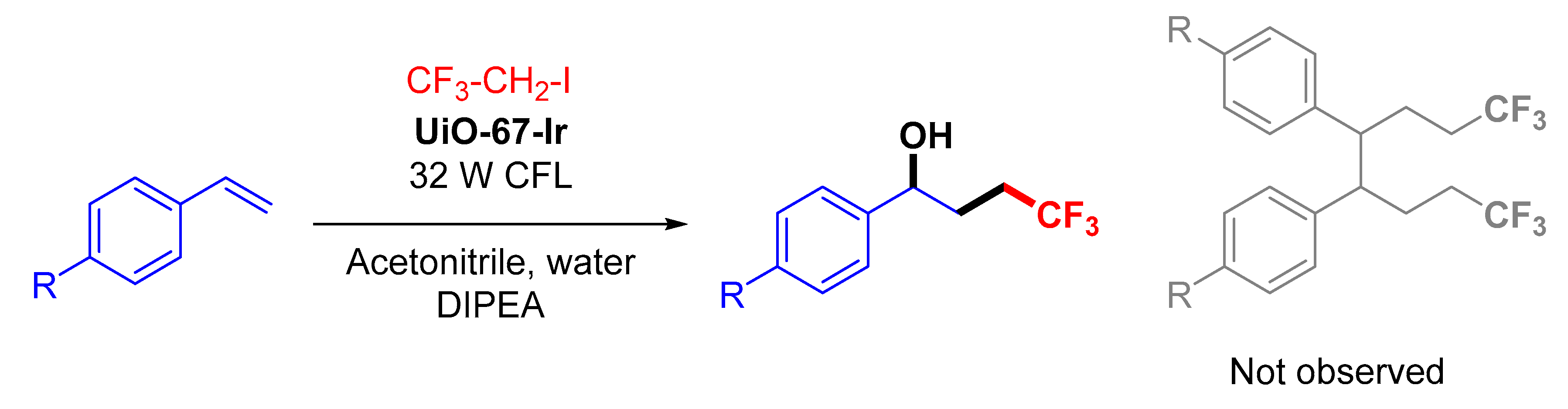
Zr6-Irphen) by the combination of predesigned Ir-phenylpyridine complexes as ancillary linkers and Zr as metal nodes. Their extreme stability allowed the use of water as solvent in the sulfoxidation tests, observing a photoredox pathway as the preferred reaction mechanism. In addition, the
Zr6-Irphen catalyst was recycled up to 10 times without losing its effectivity, neither its structural integrity (
Figure 12). [
35]. The same exclusive photoredox activity was observed for an anthracene-based MOF,
NNU-45, that was lately reported [
36].
The last example consists on a hybrid MOF obtained by the integration of guest C
60 molecules and a porous porphyrin-based MOF,
C60@PCN-222, which selectively achieved sulfoxides [
83]. This composite takes advantage of the special properties of fullerene C
60, due to the fact of its highly delocalized conjugated structure that accounts for the generation of reactive oxygen species. In particular, the high electron affinity of C
60 is a key factor to enhance photoinduced charge separation with slow recombination. Thus, the combination of fullerenes with semiconductor MOFs resulted in an increased photocatalytic activity.
Photocatalytic selective oxidation of sulfides mediated by COFs (
Table 4): Our research group published in 2019 three different undecorated imine-based pristine materials: a layered-COF, a spherical-COF, and a 3D-COF that demonstrated photocatalytic activity in oxidation reactions. In particular, the oxidation of sulfides to sulfoxides efficiently proceeded using an ecofriendly mixture of solvents (ethanol and water) [
62]. Remarkably, despite the use of water as a solvent, imine-based COFs were stable enough to perform several consecutive catalytic runs without losing activity. In the same year, two N,N’-bicarbazole-based CTFs,
BC-CTF and
Ph-BC-CTF, carried out the same oxidation reactions [
97]. Very recently, a 2D and a 3D Pd-containing porphyrinic COFs,
2D-PdPor-COF and
3D-PdPor-COF, were capable of oxidizing the sulfides in very short reaction times (25 min), using low power visible light irradiation [
98]. Also, a nanostructured COF,
h-LZU1, which was obtained by using nanocrystals of ZIF-8 as template to yield capsule structures was obtained. This material was reported as photocatalytically active towards sulfoxidation under high power light UV-Visible irradiation with low recyclability and not complete selectivity [
99]. Finally, our group reported the covalent in situ anchorage of photoactive Pt
II-hydroxyquinoline complexes as defects into imine-based COFs, presenting TON up to 8000 and good stability and recyclability in comparison with molecular complex and pristine material [
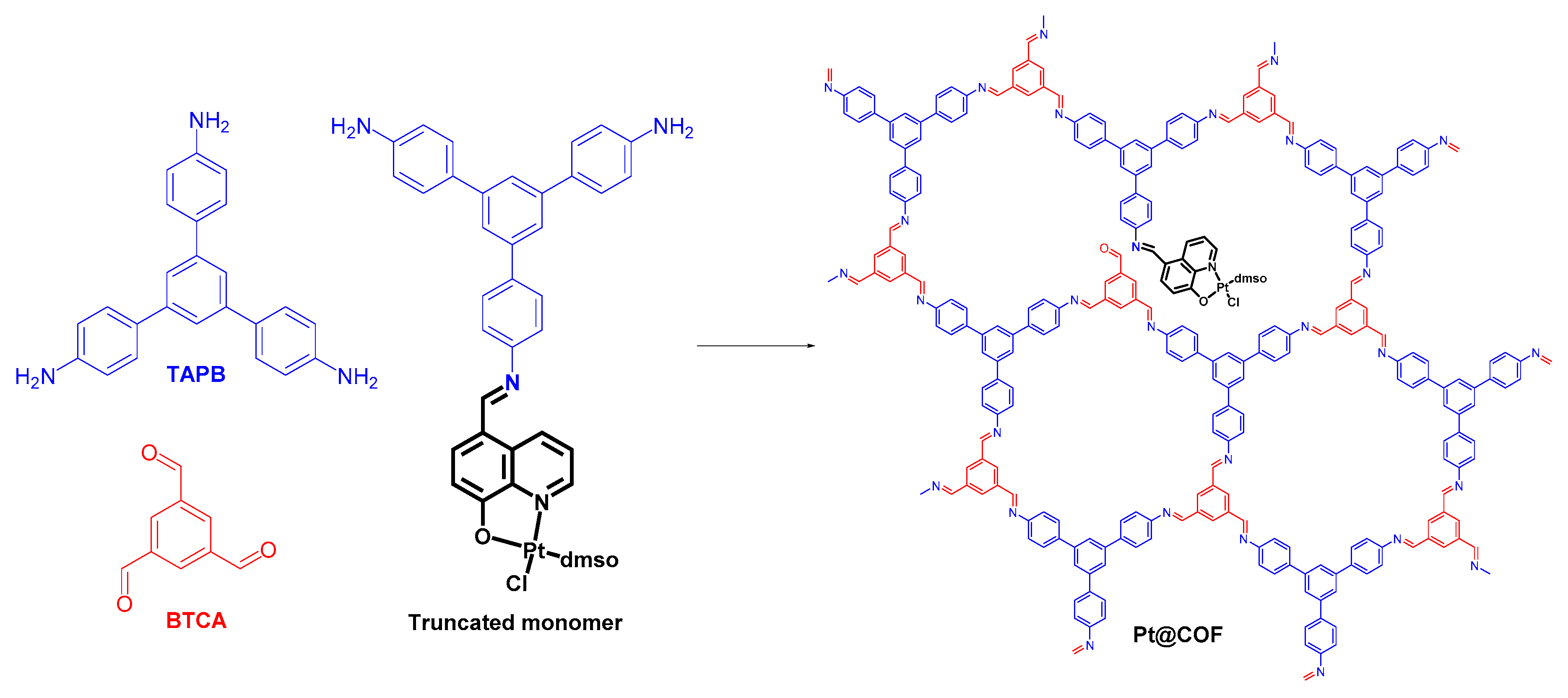
72].
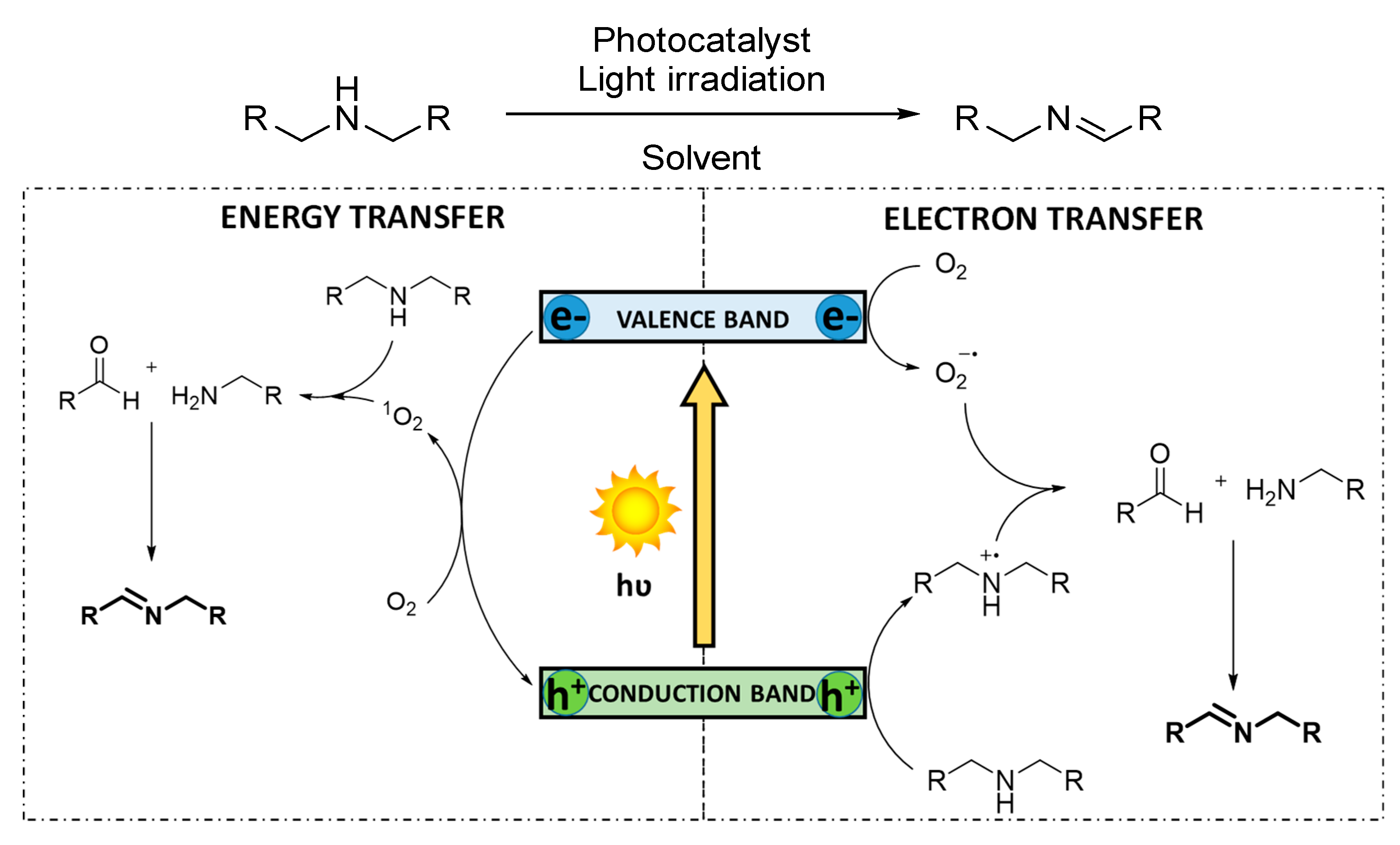
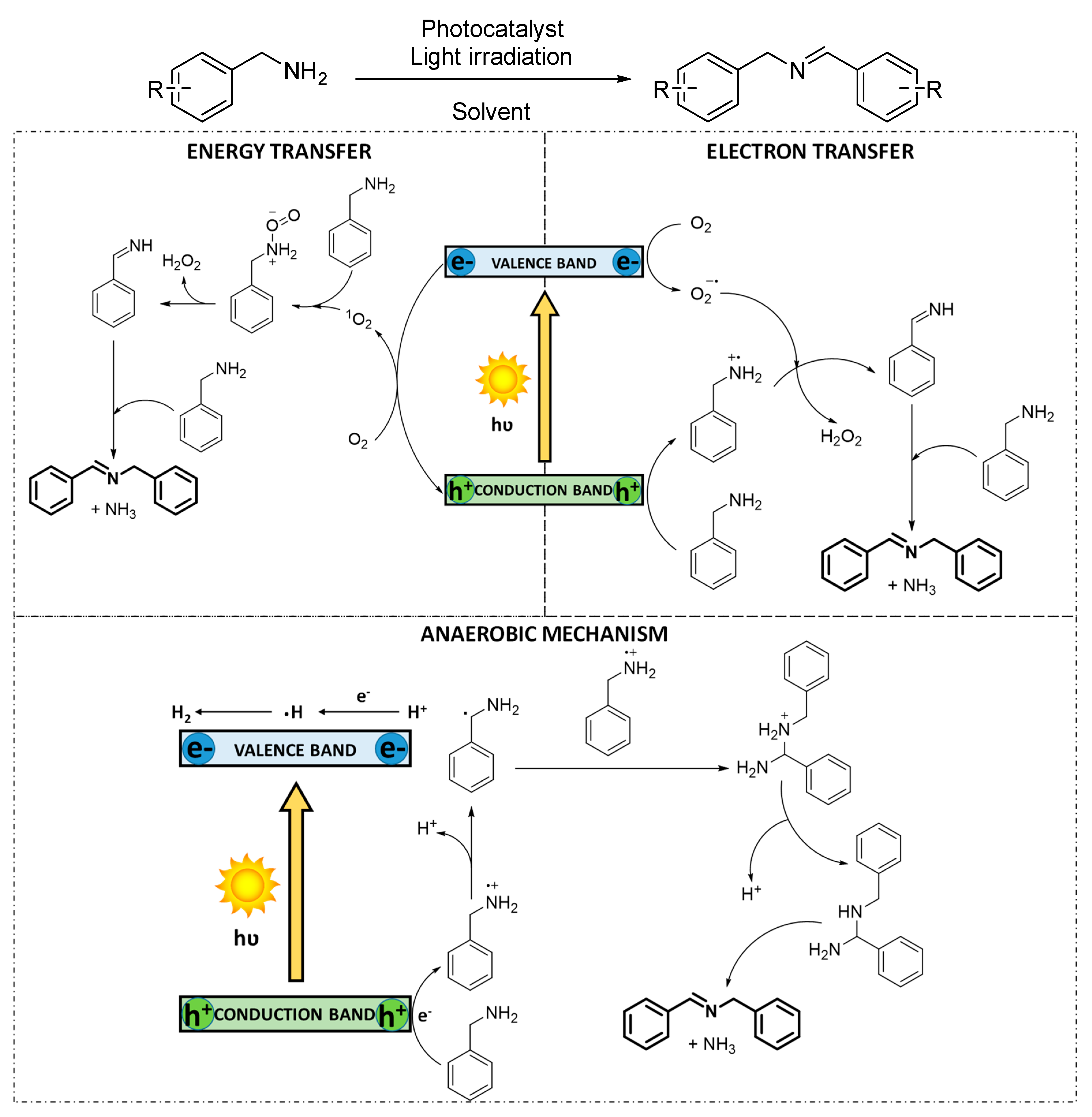
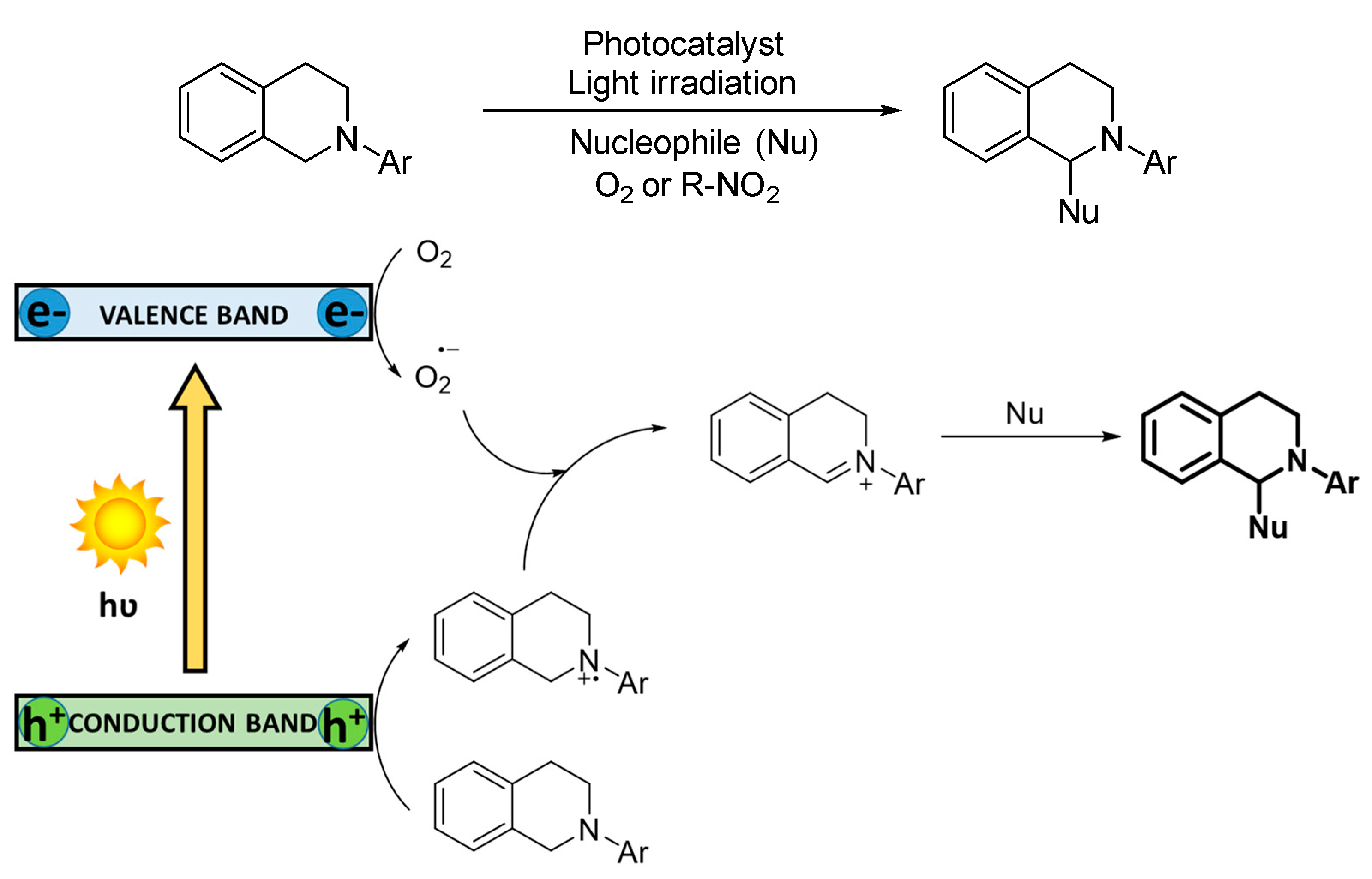
3.1.3. Photocatalytic Dehydrogenation of Secondary Amines
The selective oxidative dehydrogenation of secondary amines to imines is feasible, due to the stability of the products and the impossibility of the substrates to generate nitriles. However, the substrate conversion is influenced by the steric hindrance around the N−H bond. In addition, the chemoselectivity for the oxidation of asymmetric dibenzylamines is often hampered because two different α-CH bonds can lead to distinct products. Therefore, examples of oxidation of symmetric dibenzylamines are common, while the selective oxidation of asymmetric dibenzylamines still remains challenging. The mechanism of the reaction has been reported for both reactive oxygen species, O
2−· and
1O
2 [
46,
73] (
Scheme 5). In the first case, superoxide radical anion is concomitantly formed to the amine radical cation species. Such activated intermediates furtherly react triggering heterolytic cleavage, generating a mixture of benzaldehyde and benzylamine, which ultimately condensate to the corresponding imine. For the energy transfer mechanism, the singlet oxygen species directly reacts with the amine, provoking the subsequent heterolytic cleavage.
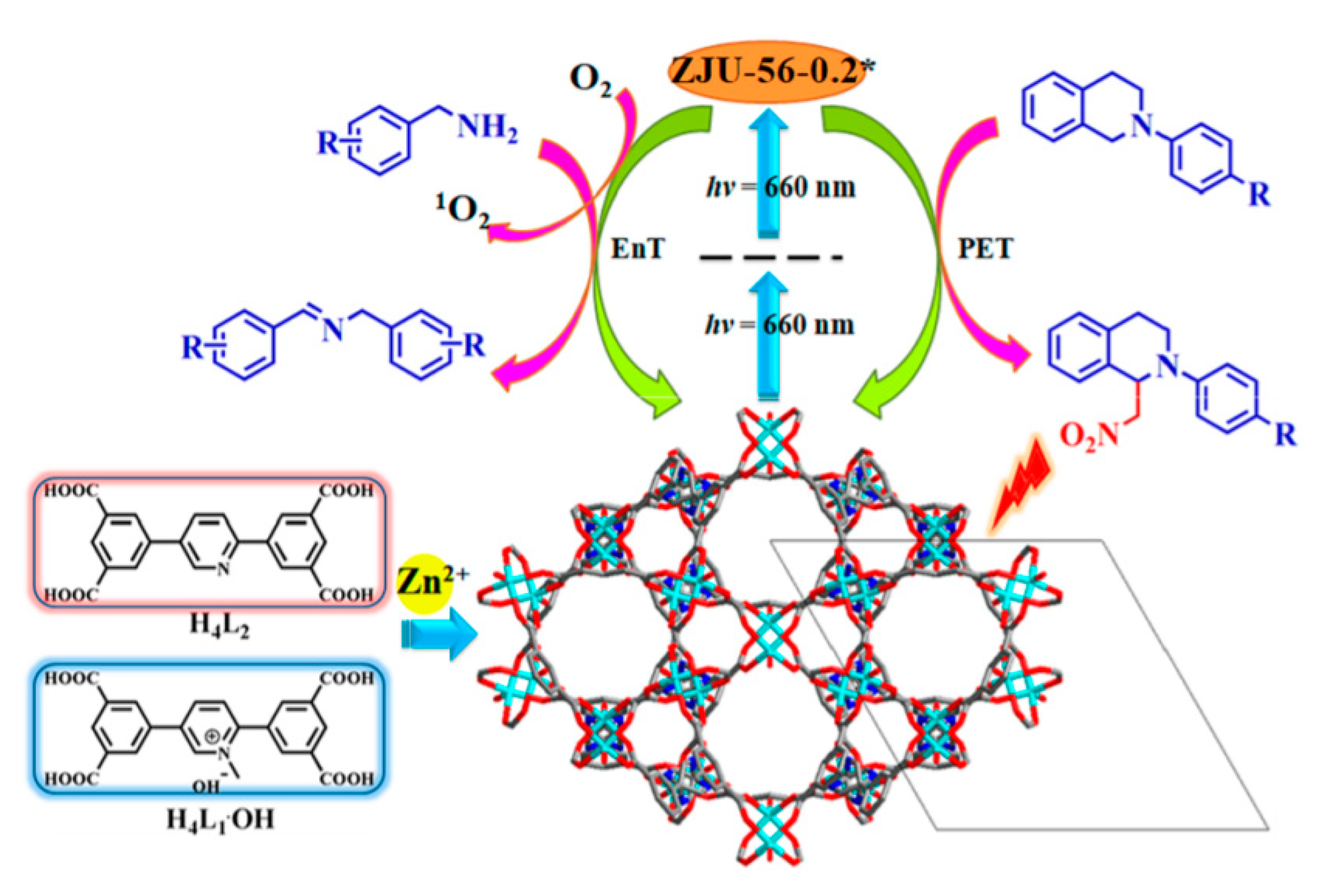
Photocatalytic oxidation of amines into imines mediated by MOFs (
Table 5): A pristine zinc-based MOF containing anthracene linkers as photosensitizers was reported in 2019, carrying out the photooxidation of secondary amines to the corresponding imines in short reaction times and great yields [
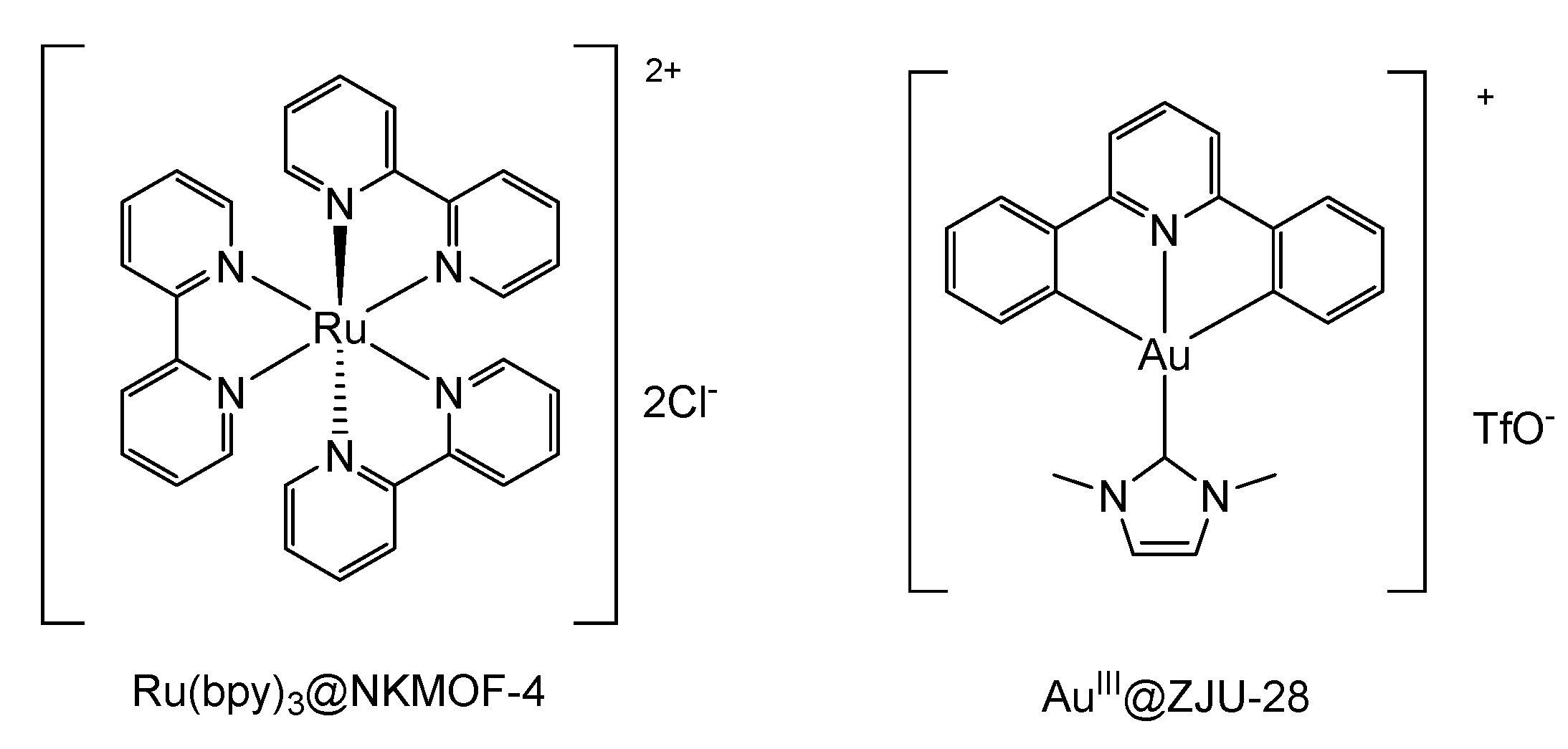
46]. A different strategy, which is related to the encapsulation of photoactive Au complexes into the porous structure of two MOFs, was achieved by Su and collaborators. The isolation and immobilization of emissive Au complexes into
AuIII@MOF1 and
AuIII@ZJU-28 resulted on the improvement of emission intensity, quantum yield, life-time of the excited state, and selective photocatalytic activity with respect to the complex in solution [
73].
Photocatalytic oxidation of amines into imines mediated by COFs (
Table 6): A very stable 2D porphyrin-containing COF,
Por-sp2c-COF, was synthesized by Wang and collaborators [
63] through the Knoevenagel condensation, giving rise to a cyanovinylene-based COF. This material was firstly used to generate the imine products in only 30 min under low power visible light irradiation. In 2020, they also reported that the same
Por-sp2c-COF was also capable of oxidizing secondary amines in combination with TEMPO as co-catalyst in 18 min using low-power red light source through a two-photon absorption mechanism [
28].
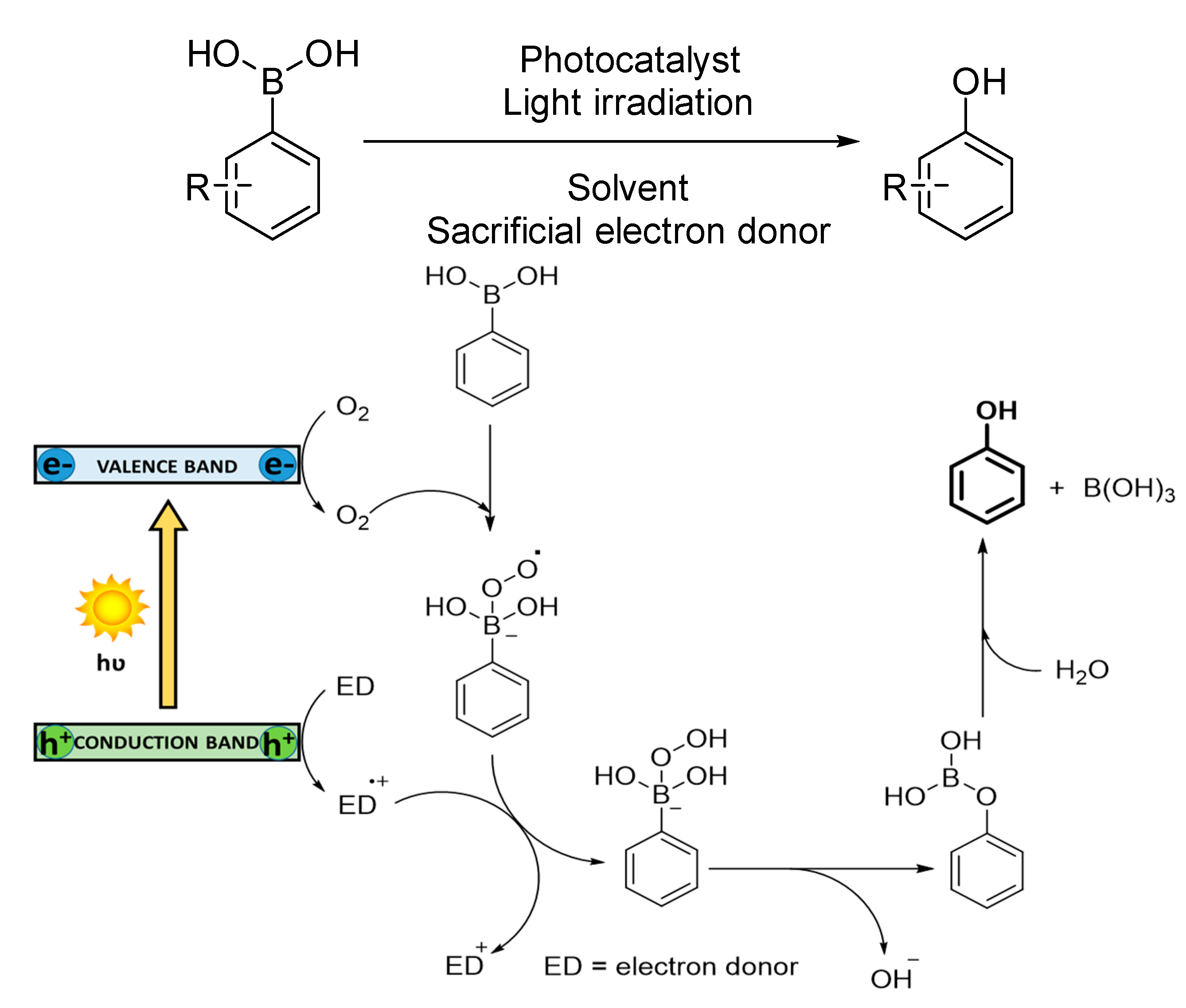
3.1.4. Oxidation of Arylboronic Acids
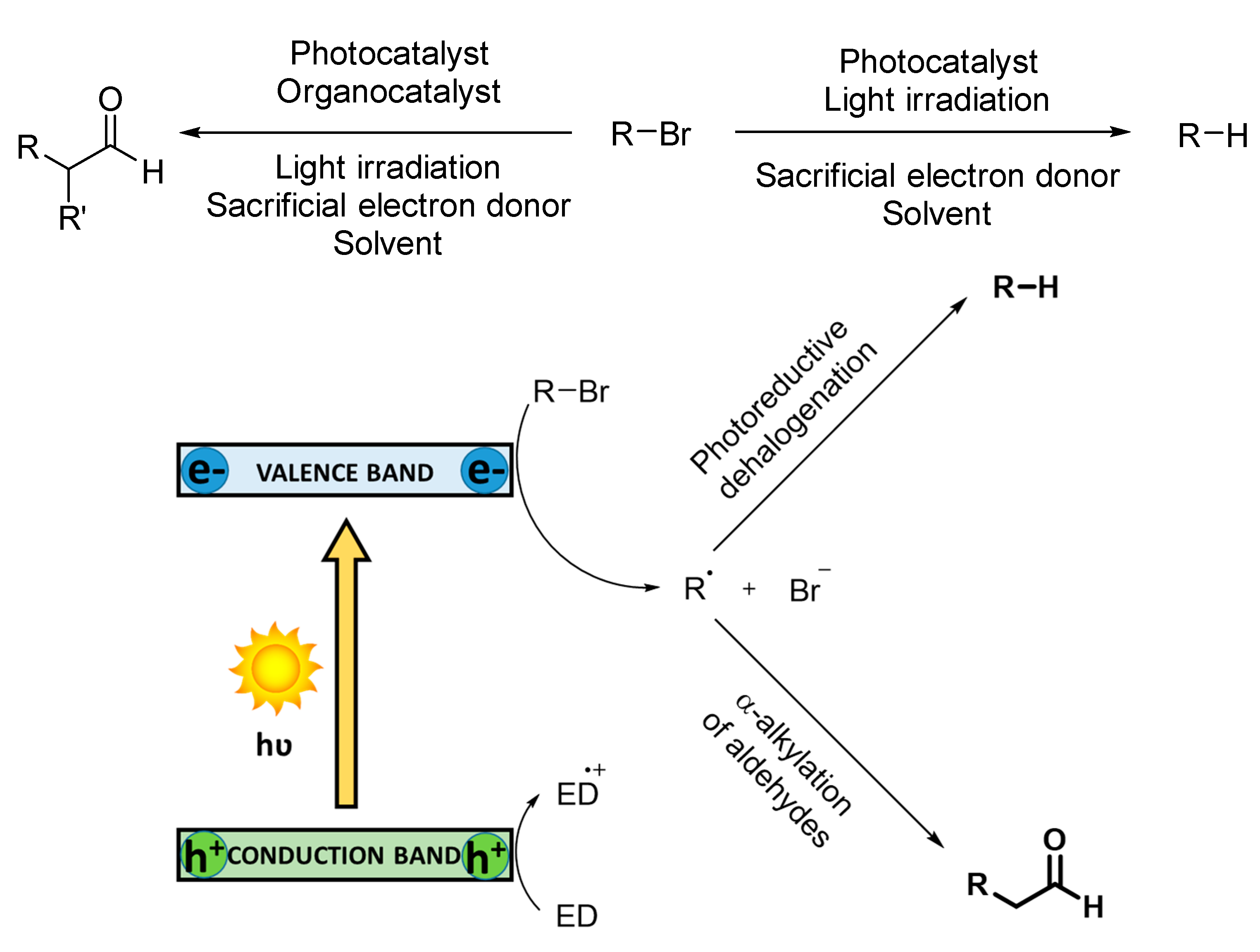
The oxidation of arylboronic acids to phenols is known to proceed exclusively via superoxide radical anion [
100]. Charge separation occurs in the photocatalyst under light irradiation, generating holes that oxidize a tertiary amine as sacrificial electron donor to its corresponding cationic radical and the photogenerated electrons reduce O
2 to O
2−·. The transient reactive oxygen species directly reacts with arylboronic acid, forming an anionic radical intermediate. This radical species abstracts one hydrogen atom from the cationic oxidized amine. Finally, an irreversible rearrangement ion migration (OH
−) is subsequently followed by a hydrolysis affording the final phenol (see
Scheme 6).
Photocatalytic oxidation of arylboronic acids mediated by MOFs (
Table 7): In 2015, two research groups, Matsuoka and Zhang, reported two different porphyrin-containing MOFs able to oxidize arylboronic acids to phenols.
Zr-MOF-TCPP (also known as
MOF-525) achieved the final phenol product under green LEDs irradiation using triethylamine as sacrificial electron donor [
101]. In the other case, a family of UNLPF-MOFs were synthesized by variating the amount and the nature of the coordinated metal (Sn
II, Sn
IV or In
III). The photophysical properties of the materials synthesized were evaluated, and further translated into their application as photocatalyst in different organic transformations. As a result of this analysis, it was found that
UNLPF-12 (which contains Sn
IV coordinated to the porphyrin and In
III as metal node) was the optimal photocatalyst for the oxidative hydroxylation of different substituted arylboronic acids [
32].
Functionalization processes related to the different incorporation of [Ru(bpy)
3]
2+ complexes are also described in this reaction. In 2015, Cohen reported a UiO-67-based MOF with coordinated Ru centers (
UiO-67-Ru(bpy)3). This material was capable of oxidizing arylboronic acids under near-UV and visible light irradiation with DIPEA as sacrificial electron donor [
102]. In this case, the complex is covalently bonded to the material, since the linker contain bipyridine ligands. One year later, a different strategy was applied by Zhou [
42], consisting on the occlusion of molecular [Ru(bpy)
3]
2+ that stablished coulombic interactions with the anionic indium-based MOF,
PCN-99. This functionalized framework was able to catalyze the oxidation of arylboronic acids, using also diisopropylethylamine (DIPEA) as sacrificial electron donor.
Photocatalytic oxidation of arylboronic acids mediated by COFs (
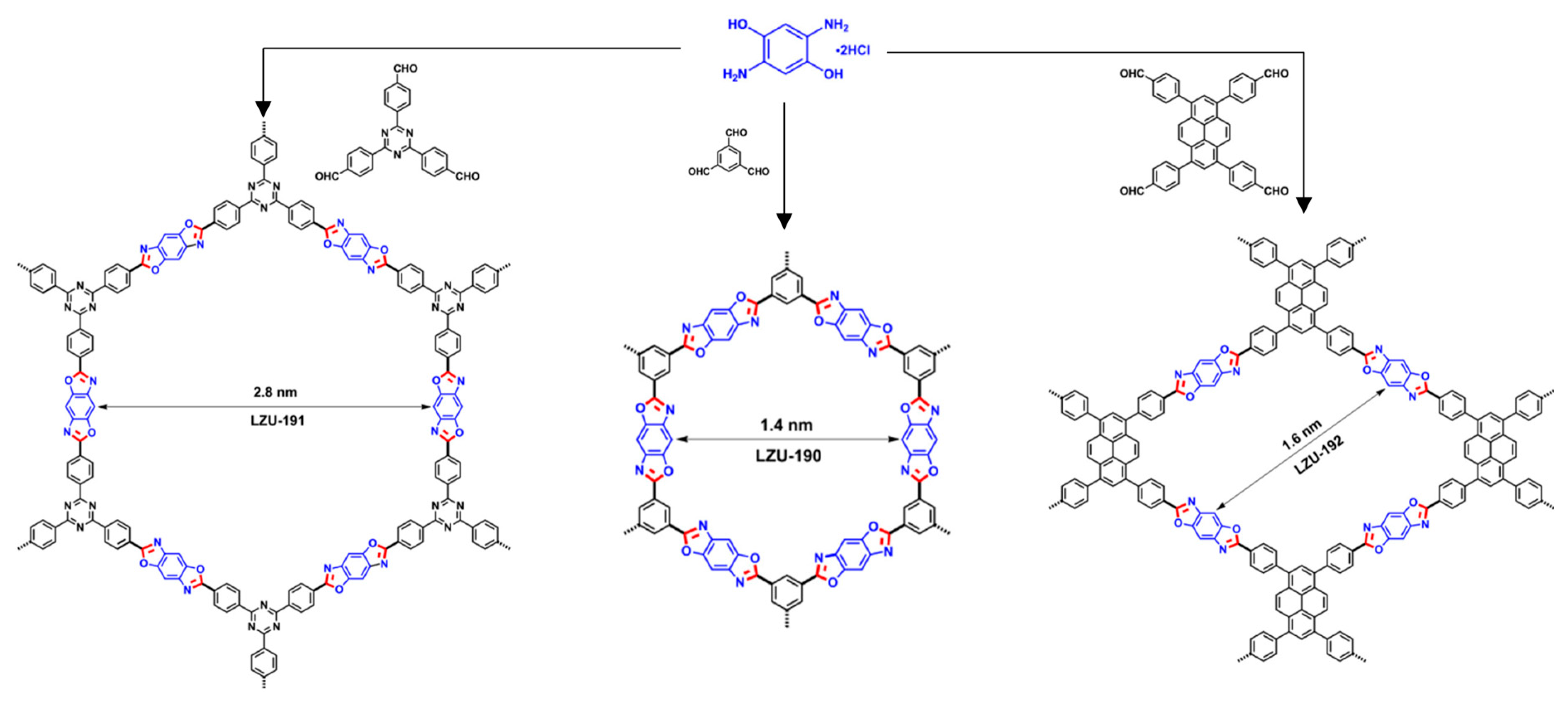
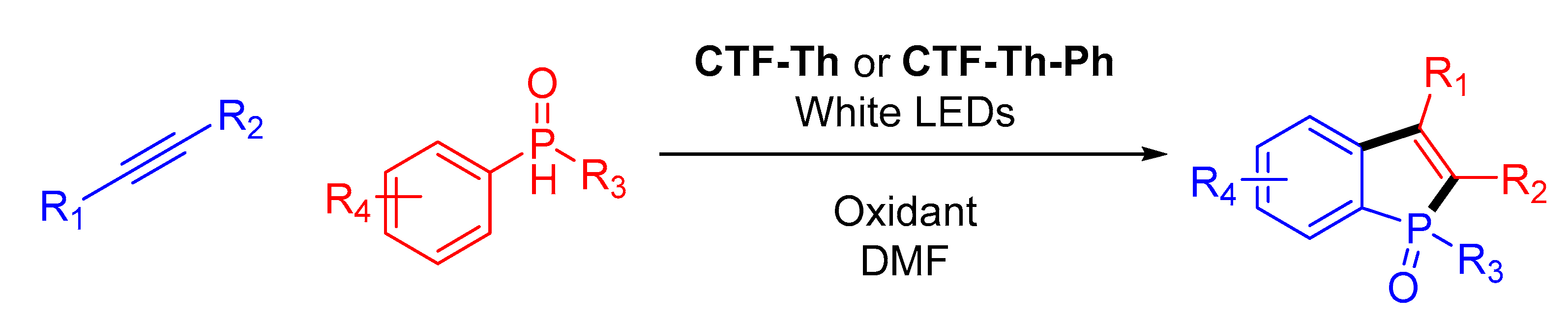
Table 8): in 2018, three new benzoxazole-linked COFs, (
LZU-190 to
192), were obtained by Wang and coworkers [
66] through cascade-type reactions (see
Figure 13). The initially imine-based formed COF evolved to the corresponding benzoxazole linkage thanks to the hydroxyl groups present in the amine-containing building block, conferring an outstanding stability to the material. This stability was confirmed by running until 20 photocatalytic cycles in the oxidation of phenylboronic acids to phenols with full conversion in each cycle. A different approach was employed in 2019, where an imine-linked COF was obtained through the self-condensation of a tetratopic building block, which contains both aldehydes and amino groups into its structure. The
BBO–COF achieved the corresponding phenolic product with quite long reaction times [
97]. Finally, three imine-based undecorated COFs with different architectures and morphologies carried out the oxidation of phenylboronic acid to phenol using a household white bulb as irradiating system with moderate yields, confirming the photoredox activity of these materials [
62].
Other photocatalytic oxidation reactions mediated by MOFs (
Table 9): In addition, there are some examples of oxidation reactions that have only been carried out using MOFs, remaining unexplored in the field of COFs. The first example dates from 2012, where large catalytic loadings of
UiO-66-NH2 were employed to achieve the oxidation of cyclohexane to cyclohexanone and epoxidation of cyclooctene and styrene derivatives. They described the mechanism of these transformations as an electron transfer process through the oxidation of the hydrocarbon concomitantly with superoxide radical anion formation. Such mixture of transient species eventually evolved to the corresponding products [
86].
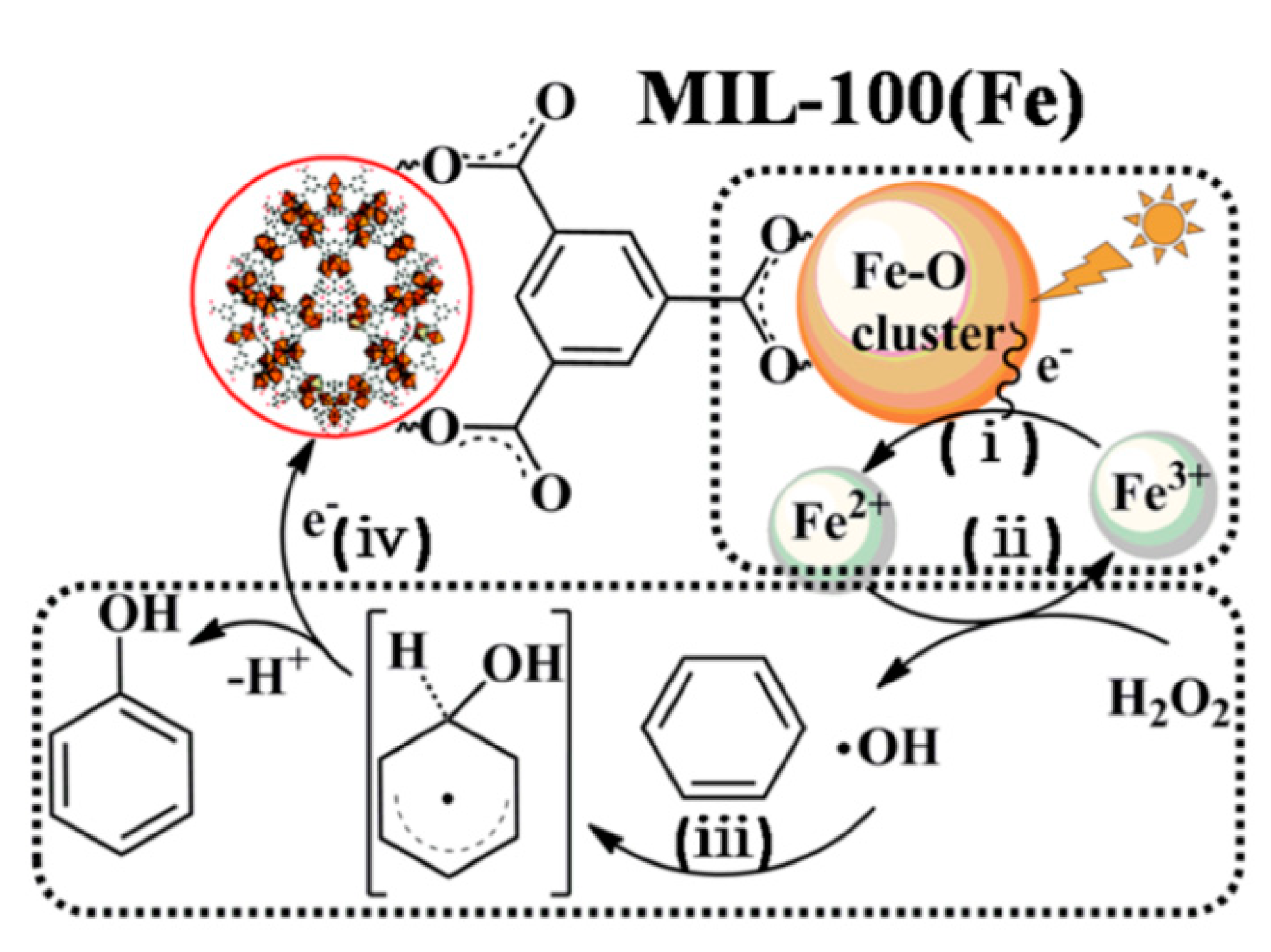
In 2015, two pristine Fe-based MOFs,
MIL-100(Fe) and
MIL-68(Fe), were capable of oxidizing benzene to phenol using a mixture of acetonitrile and water as solvent and H
2O
2 as reagent [
54]. These MOFs produce the homolytic cleavage of hydrogen peroxide to generate active hydroxyl radicals (·OH) and the oxidation of Fe
II centers, forming Fe
III–OH species via photo-Fenton-like reaction. The hydroxyl radical attacks the benzene ring to form the hydroxycyclohexadienyl radical, that finally evolves to the phenol product through a hydrogen atom abstraction. This process also leads to the reduction of Fe
III–OH into Fe
II and the release of a water molecule, regenerating the original catalytic species (see
Figure 14).
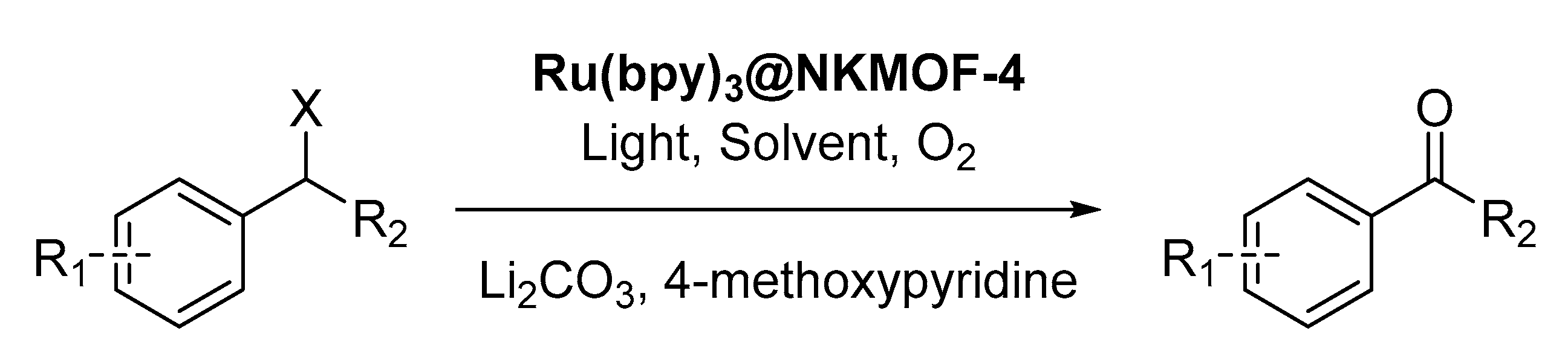
In 2019, the encapsulation of [Ru(bpy)
3]
2+ into the porous structure of a ZIF was performed by Zhang [
34]. It was achieved by the
in-situ construction of the framework in presence of the mentioned complex, allowing the isolation of
Ru(bpy)3@NKMOF-4. The photocatalytic activity of this functionalized material was evaluated using the oxidation of benzyl halides to the corresponding acetophenones as a model reaction (
Scheme 7). As co-catalyst, 4-methoxypyridine was used, and Li
2CO
3 was added as a base under white light irradiation, resulting in the full conversion of various benzyl halides. They proposed a complex mechanistic landscape, in which the superoxide radical anion is involved, and the 4-methoxypyridine acts as electron transfer mediator.
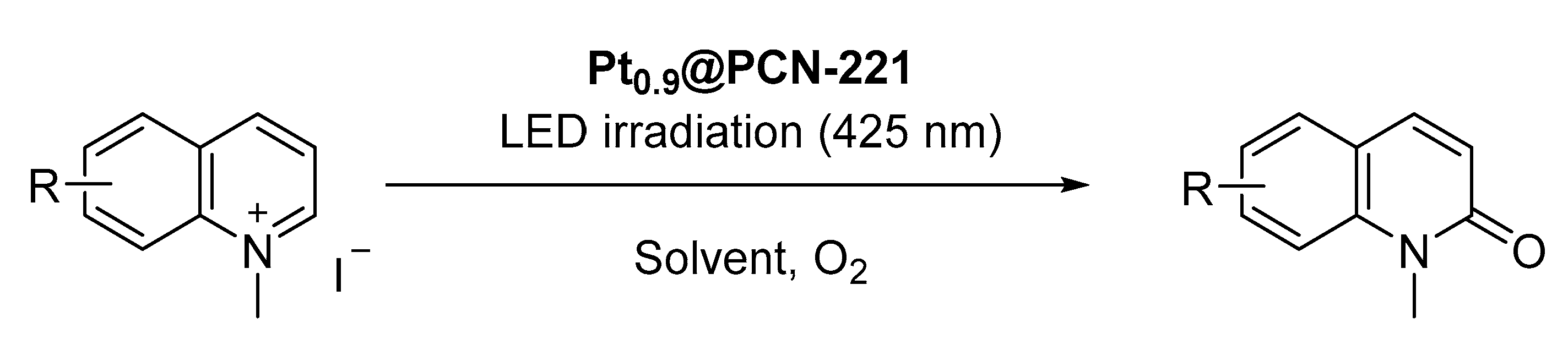
Very recently, the generation of Pt nanoclusters supported on the surface of
PCN-221 through photoreduction was achieved. This hybrid material was employed as photocatalyst for the oxidation of different
N-alkyl(iso)quinolinium halides into the corresponding
N-alkyl(iso)quinolones via singlet oxygen generation (
Scheme 8) [
77].








































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}