Eosin Y-Catalyzed Visible-Light-Mediated Aerobic Transformation of Pyrazolidine-3-One Derivatives †





Abstract
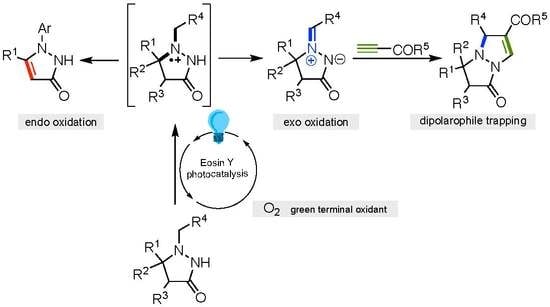
:
1. Introduction
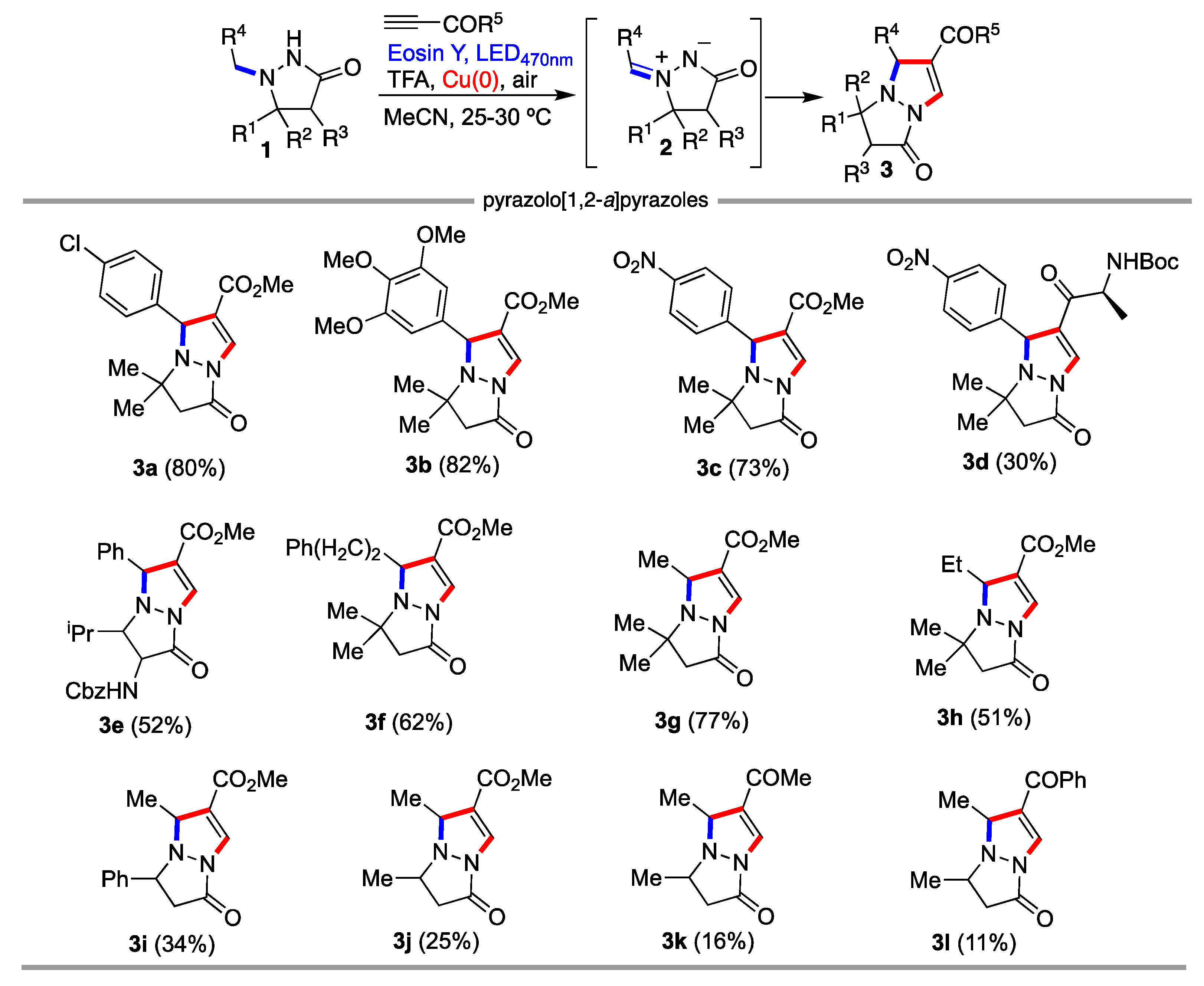
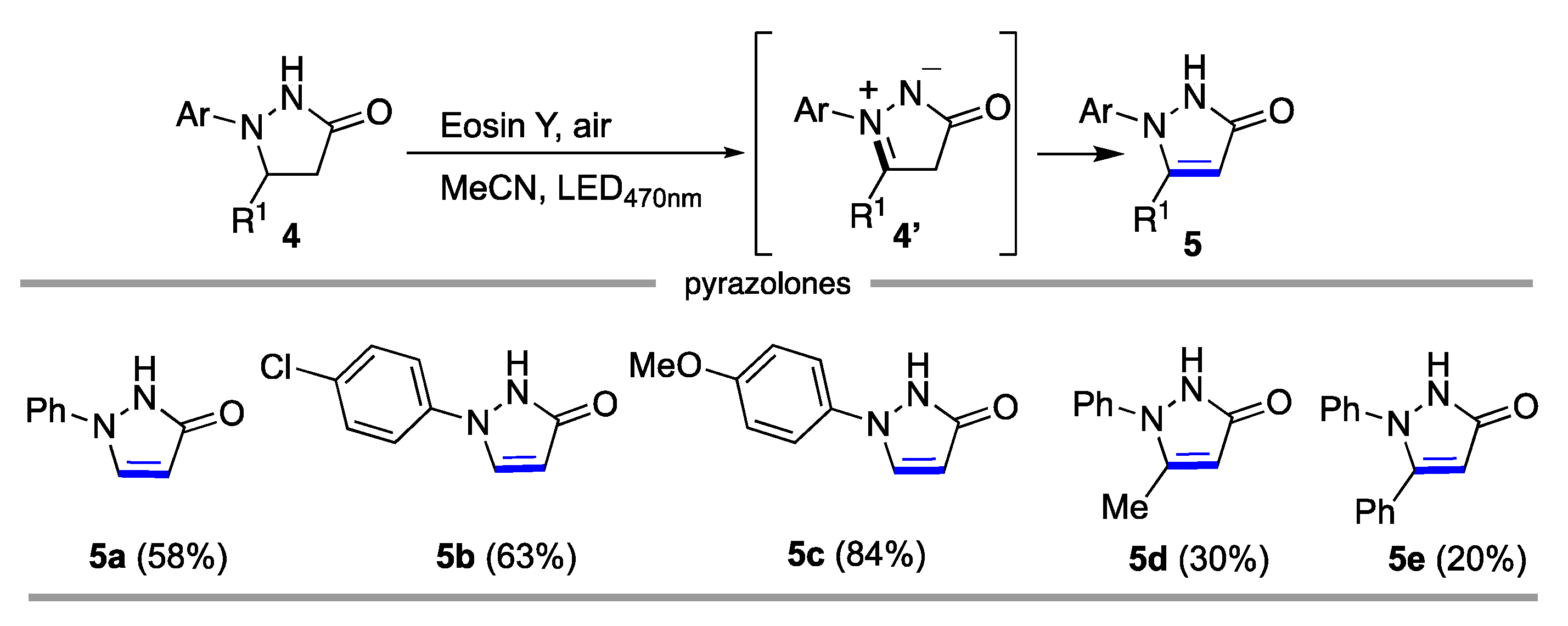
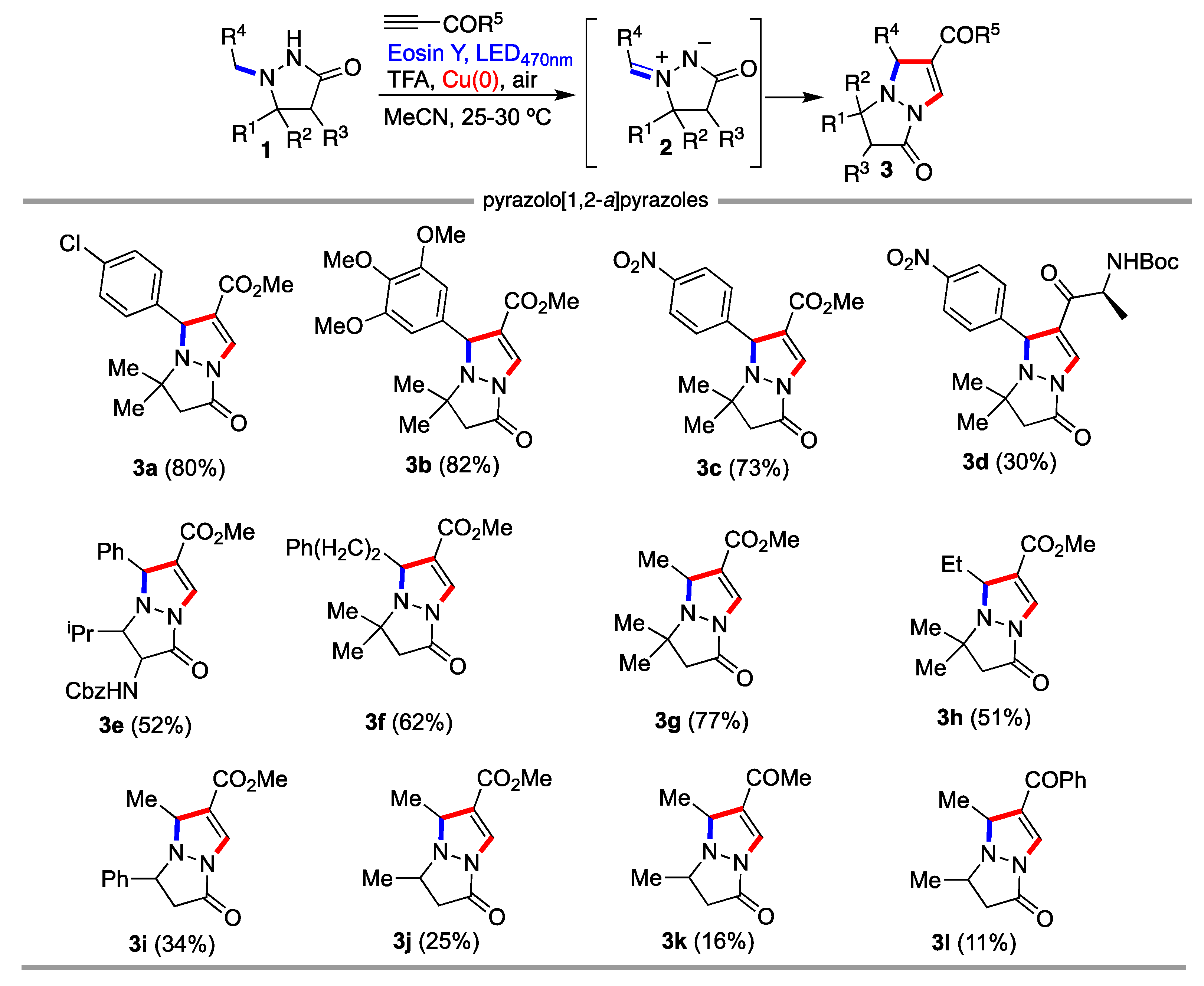
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Azomethine Imines 2
3.3. General Procedure for the Synthesis of pyrazolo[1,2-a]pyrazoles 3
3.4. General Procedure for the Synthesis of pyrazolones 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dorn, H.; Otto, A. Über die Reaktion von Pyrazolidon-(3) mit Carbonylverbindungen. Chem. Ber. 1968, 101, 3287–3301. [Google Scholar] [CrossRef]
- Dorn, H.; Otto, A. Syntheses by Means of 1-Alkylidene- and 1-(Arylalkylidene)-3-pyrazolidone N,N-Betaines, a New Type of Stable Azomethine Imine. Angew. Chem. Int. Ed. 1968, 7, 214–215. [Google Scholar] [CrossRef]
- Schantl, J.G. Advances in Heterocyclic Chemistry; Elsevier Inc.: San Diego, CA, USA, 2010; Volume 99, p. 185. [Google Scholar]
- Najera, C.; Sansano, J.M.; Yus, M. 1,3-Dipolar cycloadditions of azomethine imines. Org. Biomol. Chem. 2015, 13, 8596–8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belskaya, N.P.; Bakulev, V.A.; Fan, Z. Synthesis and (3+2) cycloaddition reactions of N,Nʹ-and C,N-cyclic azomethine imines. Chem. Heterocycl. Compd. 2016, 52, 627–636. [Google Scholar] [CrossRef]
- Ternansky, R.J.; Draheim, S.E.; Pike, A.J.; Counter, F.T.; Eudaly, J.A.; Kasher, J.S. Structure-activity relationship within a series of pyrazolidinone antibacterial agents. 2. Effect of side-chain modification on in vitro activity and pharmacokinetic parameters. J. Med. Chem. 1993, 36, 3224–3229. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, S.; Zhang, L.; Salem, M.; Greig, G.M.; Chan, C.C.; Natsumeda, Y.; Noguchi, K. Preferential inhibition of human phosphodiesterase 4 by ibudilast. Life Sci. 2006, 78, 2663–2668. [Google Scholar] [CrossRef]
- Gautam, S.; Rani, S.; Aldossary, S.A.; Saeedan, A.S.; Ansari, M.N.; Kaithwas, G. Effects of phenidone (DuCLOX-2/5 inhibitor) against N-methyl-N-nitrosourea induced mammary gland carcinoma in albino rats. Toxicol. Appl. Pharmacol. 2018, 351, 57–63. [Google Scholar] [CrossRef]
- Kosower, E.; Hershkowitz, E. 1,5-Diazabicyclo[3,3,0]octanediones and Pharmaceutical Compositions Containing Them. Isr. Patent ISXXAQ IL 94658, 7 October 1994. [Google Scholar]
- Kosower, E.M.; Radkowskyl, A.R.; Fairlambz, A.H.; Croft, S.L.; Neal, R.A. Bimane cyclic esters, possible stereologues of trypanothione as antitrypanosomal agents. Bimanes 29. Eur. J. Med. Chem. 1995, 30, 659–671. [Google Scholar] [CrossRef]
- Tong, T.M.; Soeta, T.; Suga, T.; Kawamoto, K.; Hayashi, Y.; Ukaji, Y. Formal Total Synthesis of Manzacidin C Based on Asymmetric 1,3-Dipolar Cycloaddition of Azomethine Imines. J. Org. Chem. 2017, 82, 1969–1976. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Yang, Y.; Duan, J.; Jia, Z.; Liang, J. Solvent incorporated sequential [3+2] annulation/substitution reaction of azomethine imines and propargyl sulfur ylide. J. Org. Biomol. Chem. 2018, 16, 1068–1072. [Google Scholar] [CrossRef]
- Wang, M.; Huang, Z.; Xu, J.; Chi, Y.R. N-Heterocyclic Carbene-Catalyzed [3+4] Cycloaddition and Kinetic Resolution of Azomethine Imines. J. Am. Chem. Soc. 2014, 136, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xu, X.; Li, Y.; Pan, L.; Liu, Q. [3+3]-Cycloaddition Reactions of α-Acidic Isocyanides with 1,3-Dipolar Azomethine Imines. Org. Lett. 2014, 16, 4004–4007. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Zhao, H.-T.; Nie, J.; Zheng, Y.; Fu, A.; Ma, J.-A. Allenoate-derived 1,5-, 1,7-, and 1,9-zwitterions as highly versatile coupling-partners for phosphine-triggered cycloaddition reactions. Chem. Sci. 2012, 3, 3053–3057. [Google Scholar] [CrossRef]
- Rodina, L.; Verzhba, O.A.; Korobitsyna, I.K. Photochromism in a series of azomethinimines based on 4-phenyl-1,2,4-triazoline-3,5-dione. Chem. Heterocycl. Comp. 1983, 19, 1345. [Google Scholar] [CrossRef]
- Geissler, G.; Fust, W.; Krüger, B.; Tomaschewski, G. Azomethinimine. VII. Photochemisches und thermisches Verhalten azarylsubstituierter Pyrazolidon-(3)-azomethinimine. Adv. Synth. Catal. 1983, 325, 205–210. [Google Scholar] [CrossRef]
- Shintani, R.; Soh, Y.; Hayashi, T. Rhodium-Catalyzed Asymmetric Arylation of Azomethine Imines. Org. Lett. 2010, 12, 4106–4109. [Google Scholar] [CrossRef]
- Wang, H.Y.; Zheng, C.W.; Chai, Z.; Zhang, J.X.; Zhao, G. Asymmetric cyanation of imines via dipeptide-derived organophosphine dual-reagent catalysis. Nat. Commun. 2016, 7, 12720–12729. [Google Scholar] [CrossRef] [Green Version]
- Rutjes, F.P.J.T.; Udding, J.H.; Hiemstra, H.; Speckamp, W.N. Preparation od Cyclic α-Hydrazino Acids through N-Acylhydrazonium Intermediates. Heterocycles 1992, 33, 81–85. [Google Scholar]
- Kirar, E.P.; Drev, M.; Mirnik, J.; Grošelj, U.; Golobič, A.; Dahmann, G.; Požgan, F.; Štefane, B.; Svete, J. Synthesis of 3D-Rich Heterocycles: Hexahydropyrazolo[1,5-a]pyridin-2(1H)-ones and Octahydro-2H-2a,2a1-diazacyclopenta[cd]inden-2-ones. J. Org. Chem. 2016, 81, 8920–8933. [Google Scholar] [CrossRef]
- Kawai, H.; Kusuda, A.; Nakamura, S.; Shiro, M.; Shibata, N. Catalytic Enantioselective Trifluoromethylation of Azomethine Imines with Trimethyl(trifluoromethyl)silane. Angew. Chem. Int. Ed. 2009, 48, 6324–6327. [Google Scholar] [CrossRef]
- Pirnot, M.T.; Rankic, D.A.; Martin, D.B.C.; MacMillan, D.W.C. Photoredox Activation for the Direct β-Arylation of Ketones and Aldehydes. Science 2013, 339, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Nicewicz, D.A.; MacMillan, D.W.C. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Ischay, M.A.; Anzovino, M.E.; Du, J.; Yoon, T.P. Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yoon, T.P. Crossed Intermolecular [2+2] Cycloadditions of Acyclic Enones via Visible Light Photocatalysis. J. Am. Chem. Soc. 2009, 131, 14604–14605. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Narayanam, J.M.R.; Stephenson, C.R.J. Visible-light-mediated conversion of alcohols to halides. Nat. Chem. 2011, 3, 140–145. [Google Scholar] [CrossRef]
- Narayanam, J.M.R.; Tucker, J.W.; Stephenson, C.R.J. Electron-Transfer Photoredox Catalysis: Development of a Tin-Free Reductive Dehalogenation Reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. [Google Scholar] [CrossRef]
- Zou, Y.-Q.; Lu, L.-Q.; Fu, L.; Chang, N.-J.; Rong, J.; Chen, J.-R.; Xiao, W.-J. Visible-Light-Induced Oxidation/[3+2] Cycloaddition/Oxidative Aromatization Sequence: A Photocatalytic Strategy To Construct Pyrrolo[2,1-a]isoquinolines. Angew. Chem. Int. Ed. 2011, 50, 7171–7175. [Google Scholar] [CrossRef]
- Zou, Y.-Q.; Chen, J.-R.; Liu, X.-P.; Lu, L.-Q.; Davis, R.-L.; Jørgensen, K.A.; Xiao, W.-J. Highly Efficient Aerobic Oxidative Hydroxylation of Arylboronic Acids: Photoredox Catalysis Using Visible Light. Angew. Chem. Int. Ed. 2012, 51, 784–788. [Google Scholar] [CrossRef]
- Fan, W.; Li, P. Visible-Light-Mediated 1,2-Acyl Migration: The Reaction of Secondary Enamino Ketones with Singlet Oxygen. Angew. Chem. Int. Ed. 2014, 53, 12201–12204. [Google Scholar] [CrossRef]
- Zhu, S.; Das, A.; Bui, L.; Zhou, H.; Curran, D.P.; Rueping, M. Oxygen Switch in Visible-Light Photoredox Catalysis: Radical Additions and Cyclizations and Unexpected C–C-Bond Cleavage Reactions. J. Am. Chem. Soc. 2013, 135, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yang, C.; Gao, F.; Li, Z.; Xia, W. Oxidative C–C Bond Cleavage of Aldehydes via Visible-Light Photoredox Catalysis. Org. Lett. 2013, 15, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Riser, O. Shining Light on Copper: Unique Opportunities for Visible-Light-Catalyzed Atom Transfer Radical Addition Reactions and Related Processes. Acc. Chem. Res. 2016, 49, 1990–1996. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Bhattacharyya, A.; Reiser, O. Copper’s rapid ascent in visible-light photoredox catalysis. Science 2019, 364, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Nicewicz, D.A.; Nguyen, M.T. Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis. ACS Catal. 2014, 4, 355–360. [Google Scholar] [CrossRef]
- Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Metal-Free, Cooperative Asymmetric Organophotoredox Catalysis with Visible Light. Angew. Chem. Int. Ed. 2011, 50, 951–954. [Google Scholar] [CrossRef]
- Romero, N.A.; Margrey, K.A.; Tay, N.E.; Nicewicz, D.A. Site-selective arene C-H amination via photoredox catalysis. Science 2015, 349, 1326–1330. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, I.; Ghosh, T.; Bardagi, L.J.; König, B. Reduction of aryl halides by consecutive visible light-induced electron transfer processes. Science 2014, 346, 725–728. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, C.; Tang, C.; Jiao, N. Recent advances in transition-metal catalyzed reactions using molecular oxygen as the oxidant. Chem. Soc. Rev. 2012, 41, 3381–3430. [Google Scholar] [CrossRef]
- Wang, D.; Weinstein, A.B.; White, P.B.; Stahl, S.S. Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions. Chem. Rev. 2018, 118, 2636–2679. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, J.; Nguyen, T.H.; Zheng, N. The chemistry of amine radical cations produced by visible light photoredox catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. [Google Scholar] [CrossRef] [PubMed]
- Majek, M.; Filace, F.; Jacobi von Wangelin, A. On the mechanism of photocatalytic reactions with eosin Y. Beilstein J. Org. Chem. 2014, 10, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Lazarides, T.; McCormick, T.; Du, P.; Luo, G.; Lindley, B.; Eisenberg, R. Making Hydrogen from Water Using a Homogeneous System Without Noble Metals. J. Am. Chem. Soc. 2009, 131, 9192–9194. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Jin, H.; Xu, P.; Zhu, C. When C–H bond functionalization meets visible-light photoredox catalysis. Tetrahedron Lett. 2014, 55, 36–48. [Google Scholar] [CrossRef]
- Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. Copper-Catalyzed Oxidative Direct Cyclization of N-Methylanilines with Electron-Deficient Alkenes Using Molecular Oxygen. J. Org. Chem. 2011, 76, 6447–6451. [Google Scholar] [CrossRef]
- Ju, X.; Li, D.; Li, W.; Yu, W.; Bian, F. The Reaction of Tertiary Anilines with Maleimides under Visible Light Redox Catalysis. Adv. Synth. Catal. 2012, 354, 3561–3567. [Google Scholar] [CrossRef]
- Shimidzu, T.; Iyoda, T.; Koide, Y. An advanced visible-light-induced water reduction with dye-sensitized semiconductor powder catalyst. J. Am. Chem. Soc. 1985, 107, 35–41. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Deviation from “Standard Conditions” | NMR Yield (%) a |
| 1 | no deviation | 62 b |
| 2 | 0.5 h | 36 |
| 3 | DMSO-d6, 0.5 h | trace |
| 4 | MeOH, 0.5 h | 20 |
| 5 | acetone, 0.5 h | trace |
| 6 | THF, 0.5 h | trace |
| 7 | DCM, 0.5 h | 20 |
| 8 | toluene, 0.5 h | 35 |
| 9 | 2 mol% eosin Y | 50 |
| 10 | 2 mol% MeMesAcrClO4 | 38 |
| 11 | 2 mol% Ru(bpy)3Cl2·6H2O | 30 |
| 12 | 2 mol% Ir(ppy)3 | 20 |
| 13 | no acid added | 0 c |
| 14 | 4 equiv. TFA | 63 |
| 15 | 2 equiv. TCA | 52 |
| 16 | 2 equiv. Oxalic acid | 33 |
| 17 | 2 equiv. Citric acid | trace |
| 18 | 2.0 mL of MeCN | 63 b |
| 19 | reaction carried out in the dark | trace |
| 20 | reaction mixture purged with nitrogen | trace |
| 21 | no catalyst added | trace |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petek, N.; Grošelj, U.; Svete, J.; Požgan, F.; Kočar, D.; Štefane, B. Eosin Y-Catalyzed Visible-Light-Mediated Aerobic Transformation of Pyrazolidine-3-One Derivatives. Catalysts 2020, 10, 981. https://doi.org/10.3390/catal10090981
Petek N, Grošelj U, Svete J, Požgan F, Kočar D, Štefane B. Eosin Y-Catalyzed Visible-Light-Mediated Aerobic Transformation of Pyrazolidine-3-One Derivatives. Catalysts. 2020; 10(9):981. https://doi.org/10.3390/catal10090981
Chicago/Turabian StylePetek, Nejc, Uroš Grošelj, Jurij Svete, Franc Požgan, Drago Kočar, and Bogdan Štefane. 2020. "Eosin Y-Catalyzed Visible-Light-Mediated Aerobic Transformation of Pyrazolidine-3-One Derivatives" Catalysts 10, no. 9: 981. https://doi.org/10.3390/catal10090981
APA StylePetek, N., Grošelj, U., Svete, J., Požgan, F., Kočar, D., & Štefane, B. (2020). Eosin Y-Catalyzed Visible-Light-Mediated Aerobic Transformation of Pyrazolidine-3-One Derivatives. Catalysts, 10(9), 981. https://doi.org/10.3390/catal10090981