1. Introduction
Infrared (IR) spectroscopy in its various modes—transmission, diffuse reflectance and internal reflectance—is a powerful technique among the large variety of in situ/operando methods to study materials for heterogeneous catalysis [
1]. It is traditionally exploited to determine the structure of the surface and the vibrational signature of adsorbates on solid catalysts [
2,
3]. The technique strongly relies on the interaction of probe molecule reactants and products with the material of interest to probe the fundamental vibrational modes of the resulting adsorbate. Other methods, such as X-ray absorption spectroscopy (XAS) [
4], are sensitive to coordination and oxidation state changes that are considered out of the capabilities of IR spectroscopy. Because of its nature, UV-vis spectroscopy in diffuse reflectance mode can also be used to follow the redox behavior of metal-based catalysts through changes in the baseline height in the 500–800 nm range [
5,
6] in the absence of a clear plasmonic band of metal particles.
In IR spectroscopic studies of solid catalysts, absorption level changes in IR spectra over the whole mid-IR range are often neglected [
7]. Their origin may have various explanations and may depend on several effects. In contrast to this, IR spectroscopy is used to monitor with very high time resolution (pico- to nanoseconds) electron injection into the conduction band of semiconductor materials such as those that can also be exploited as supports in heterogeneous catalysts and sensors, and the subsequent slower relaxation [
8]. Upon photo-excitation, the transmission IR spectra typically return a structureless broad absorption that increases monotonously from 4000 to below 2000 cm
−1 and decreases more sharply below 2000 cm
−1. A broad band of similar shape and position is also observed when hydrogen spillover occurs from metal nanoparticles deposited on TiO
2 to the semiconductor, which causes Ti
4+ reduction [
7,
9]. The single beam diffuse reflectance IR spectra of Au/ZnO were shown to exhibit a complete loss of reflectance in Ar at 400 °C, which re-appeared in the subsequent treatment in O
2 at the same temperature in agreement with the generation of oxygen vacancies in ZnO under reducing conditions [
10]. The changes in absorption are mirrored by changes in the baseline position (absorbance) in the corresponding background-subtracted spectra. Boccuzzi et al. [
11] showed that the spectra of the ZnO semiconductor were strongly affected by the atmosphere that the sample was exposed to, including hydrogen and CO. Bürgi et al. [
12] observed a monotonic increase in absorbance in the form of a broad-band signal centered at ca. 2500 cm
−1 when reducing in hydrogen saturated 2-propanol a 2-nm film of Pd on an Al
2O
3 layer deposited on an internal reflection element for attenuated total reflection infrared (ATR-IR) spectroscopy; this effect was partially reversed in the following oxygen containing atmosphere. Based on calculations considering a homogeneous metal layer, this behavior was associated with changes in optical properties of the metal as a result of changes in electron density. The increase of electron density upon reduction and the in situ formation of a conducting sample produced an increase in absorbance, whereas formation of the insulating PdO sample resulted in absorption decrease. Hence, changes in absorption by the sample under reducing conditions are associated with a change of electron density in the sample. The reduction and re-oxidation of TiO
2 supported Pd nanoparticles in the same ATR-IR geometry caused visible baseline changes similar to those reported later for Au/TiO
2 with transmission IR spectroscopy [
7,
9]. Polarization modulation IR reflection absorption spectroscopy measurements on polycrystalline Pt also revealed baseline changes attributable to the partial oxidation of the metal surface during CO/O
2 modulation experiments [
13]. Similar absorbance increase was observed also during reduction of Pd/Al
2O
3 under mild conditions in the liquid phase [
14]. Because in this case Al
2O
3 is an insulator, the change in density needs to be associated with the metal nanoparticles deposited on the support oxide.
In the mid-IR region, precious metals and their oxides (except Ru [
15]) do not present any specific and characteristic absorption. Radiation of higher energy, e.g., in the visible range, is required to observe the characteristic plasmonic signals of metal nanoparticles. Conversely, vibrational signatures of metal-metal and metal-ligand bonds are expected in the far-infrared region.
In this study, we show that the variations in baseline in diffuse reflectance infrared Fourier transform (DRIFT) spectra of Pd supported on insulating Al2O3 during repeated reduction-oxidation pulses can be attributed to the reversible reduction-partial oxidation of the metal component. Hence, besides probing molecular adsorbates for which IR spectroscopy is recognized to be powerful, operando DRIFT spectra could also be exploited to follow the evolution of material oxidation state.
2. Results and Discussion
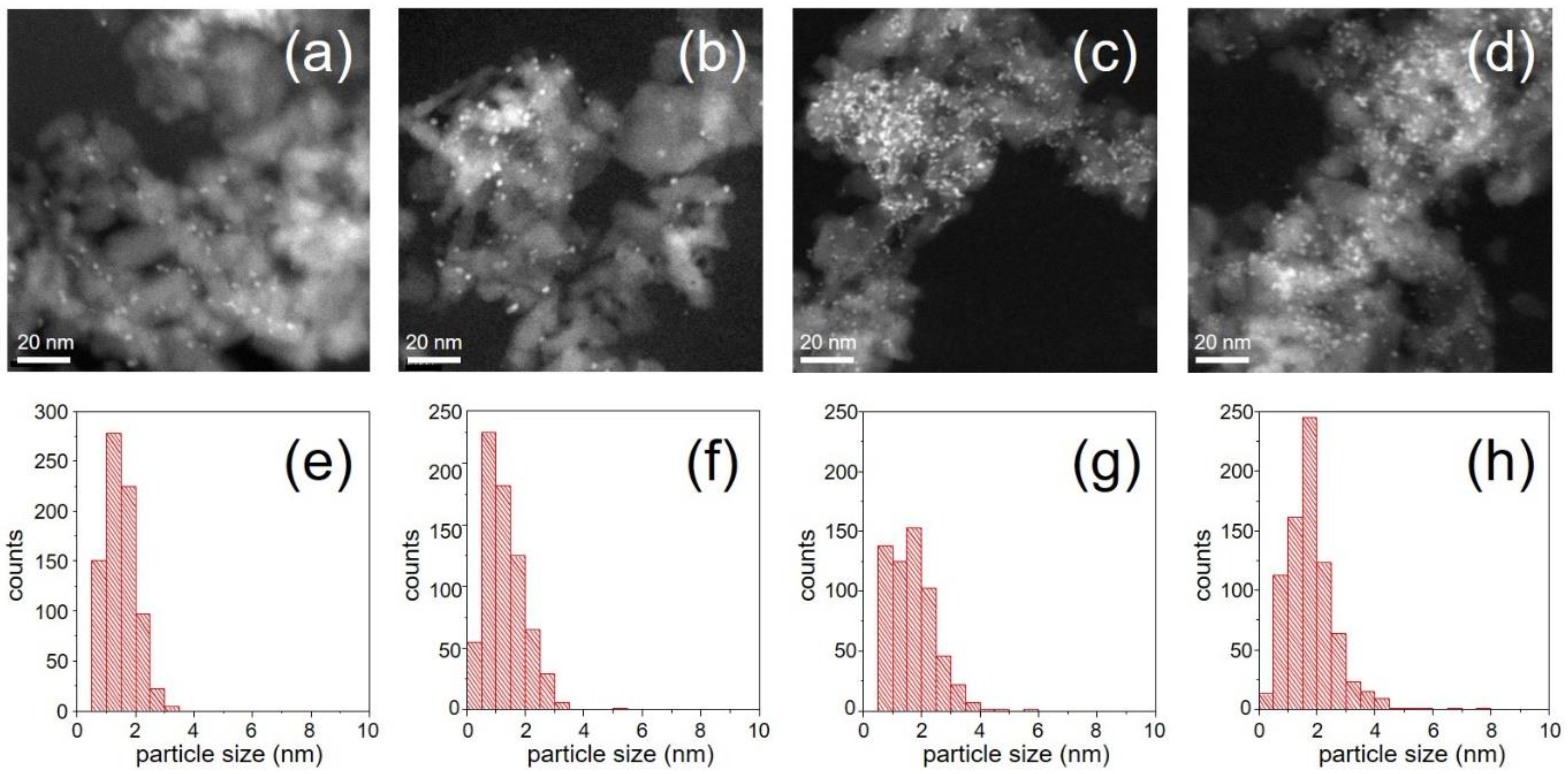
The scanning transmission electron microscopy (STEM) images collected in high-angle annular dark-field (HAADF) mode demonstrate that the Pd/Al
2O
3 samples of the variable Pd-loading series exhibit very similar Pd particle size distribution and an average Pd particle size of 1.5–2 nm irrespective of Pd loading (
Figure 1). This is useful to discard effects on the IR spectra that could be produced/complicated by particle size differences.
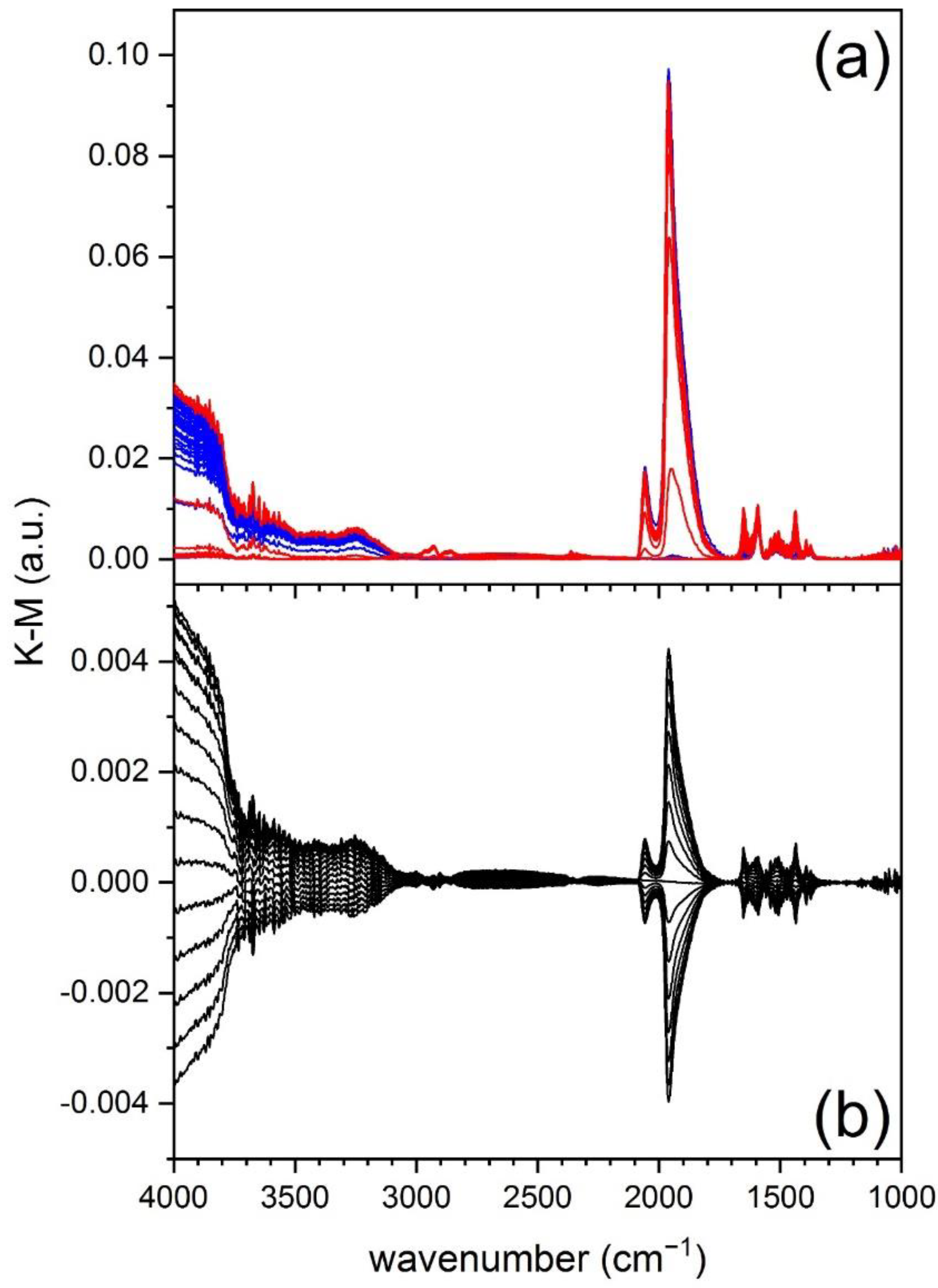
A modulated excitation DRIFTS experiment where O
2 pulses are repeatedly alternated to CO pulses on the 1 wt.% Pd/Al
2O
3 of this series after reduction at 300 °C is presented in
Figure 2, which shows the time-resolved DRIFT spectra as well as the corresponding spectra obtained after phase sensitive detection analysis (phase-resolved spectra) [
16,
17,
18].
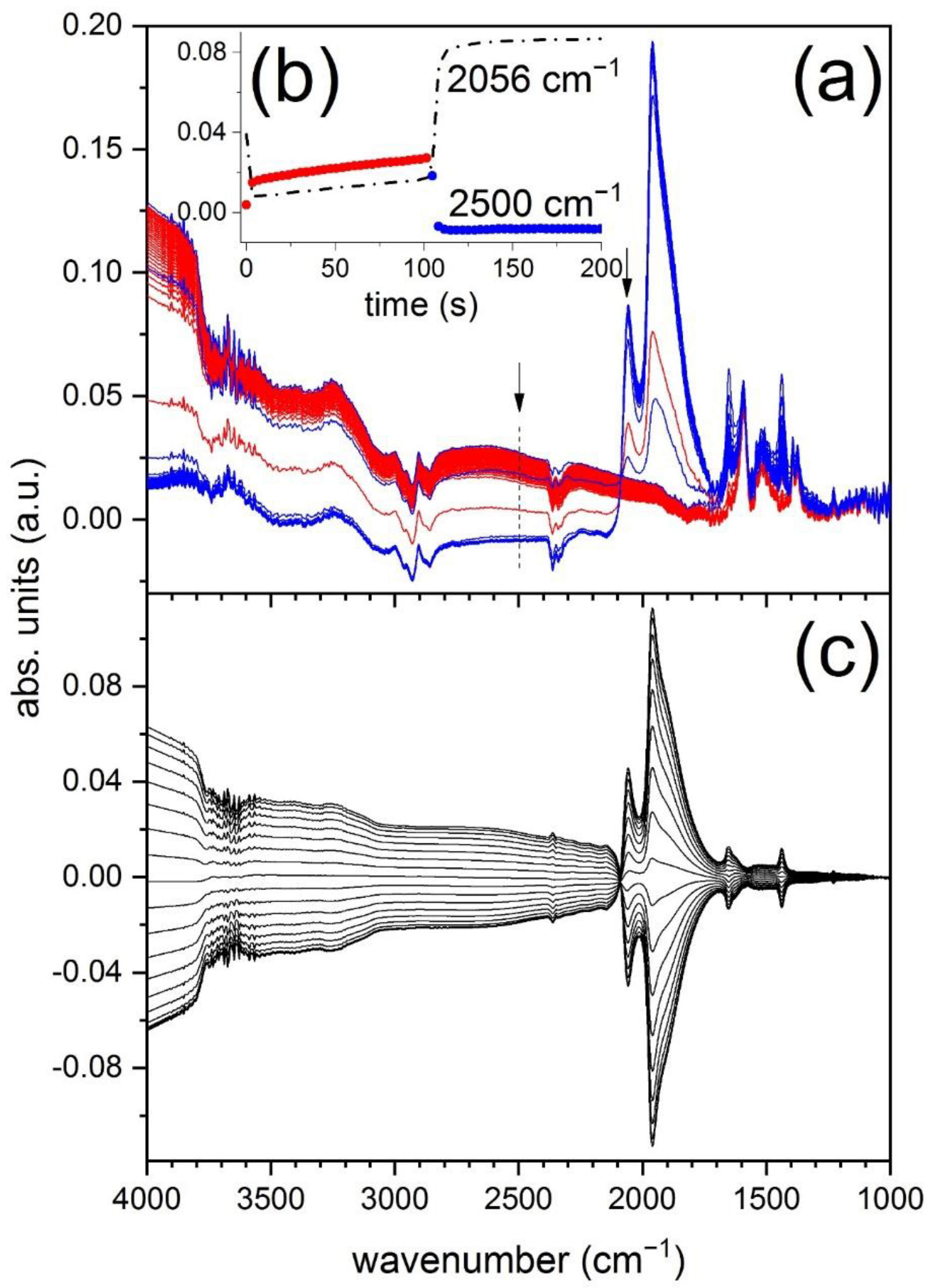
The time-resolved DRIFT spectra (
Figure 2a) are clearly dominated by the signals of adsorbed CO on reduced Pd nanoparticles [
19]. The signal at 2056 cm
−1 corresponds to adsorption of CO in a-top geometry (CO
L), while those at 1958 and (shoulder) 1900 cm
−1 to CO coordinated in two-fold (CO
B) and three-fold mode, respectively. The signals grow in the CO pulse and disappear in the following O
2 pulse. Both adsorption and consumption of CO are very quick at 300 °C as it is indicated by the temporal profile of the freestanding CO
L signal (
Figure 2b). In the reduction pulse CO coordinates to metallic Pd, in the oxidation pulse O
2 consumes CO to produce CO
2 that is detected in the online MS (not shown). Additional IR signals to those of adsorbed CO appear in the region below 1800 cm
−1 that are readily assigned to bicarbonates (1652, 1437, 1229 cm
−1), carbonates (1520 cm
−1) and formates (1592, 1393, 1374 cm
−1) likely adsorbed on Al
2O
3 [
20]. Only the signals of 1652 and 1437 cm
−1 change evidently in response to the O
2/CO pulses, indicating that these species are repeatedly produced and consumed during the modulation experiment. On the contrary, the other species appear as static species not stimulated by the O
2/CO pulses. The change of the species in response to CO and O
2 can be taken as evidence that at this temperature CO oxidation involves such species.
It is clear from
Figure 2a that the baseline of the spectra also changes while CO adsorption/reaction/desorption proceeds. Taking the signal at 2500 cm
−1 as reference for the discussion, a point where no contribution from vibrational signals is expected, a two-step intensity increase in O
2 (absorbance gain) is observed. The initial fast increase slows down after 5 s and is then followed by a fast decrease during the CO pulse (absorbance loss).
While changes in these time-resolved spectra are already visible, the phase-resolved spectra (
Figure 2c) improve the spectral quality and show only that part of the spectra that responds to the external stimulus, in this case the modulation of the CO and O
2 concentrations. Please note that the intensity of the phase-resolved spectra is scaled by a factor ca. 1.6 compared to the time-resolved spectra. The phase-resolved spectra are dominated not only by the signals of adsorbed CO but also by the increasing intensity of the baseline (increase of absorbance) above 2000 cm
−1 that corresponds to the baseline changes reflected by the behavior of the signal at 2500 cm
−1 (
Figure 2b). Additionally, the signals at 1652 and 1437 cm
−1 also appear clearly and are accompanied by that at 1229 cm
−1 indicating they belong to bicarbonates.
The use of (pseudo-)absorbance is not common in DRIFTS, but it is justified [
21]. Because of the different interaction between matter and IR radiation compared to the transmission mode, DRIFT spectra are typically reported in reflectance units using the Kubelka-Munk (K-M) function. This is of utmost importance when quantification is carried out.
Figure 3 shows the result of PSD analysis on the same set of spectra of
Figure 2a after K-M conversion. While the presence of adsorbates is confirmed in the K-M spectra, the K-M function stretches strong signals at the expenses of weaker signals. The ratio between the signals of adsorbed CO and of the species in the region below 1700 cm
−1 is clearly changed compared to the absorbance spectra. As a major result, the baseline changes that are dominating the pseudo-absorbance data are almost inexistent in the phase-resolved K-M spectra and become visible only in the high wavenumber region (>3000 cm
−1).
To address the nature of the baseline changes, modulation experiments were performed by alternating H
2 and O
2 pulses at 300 °C. O
2-H
2 modulation makes it possible to stimulate reduction-oxidation without interference of adsorbates such as CO on the IR spectra.
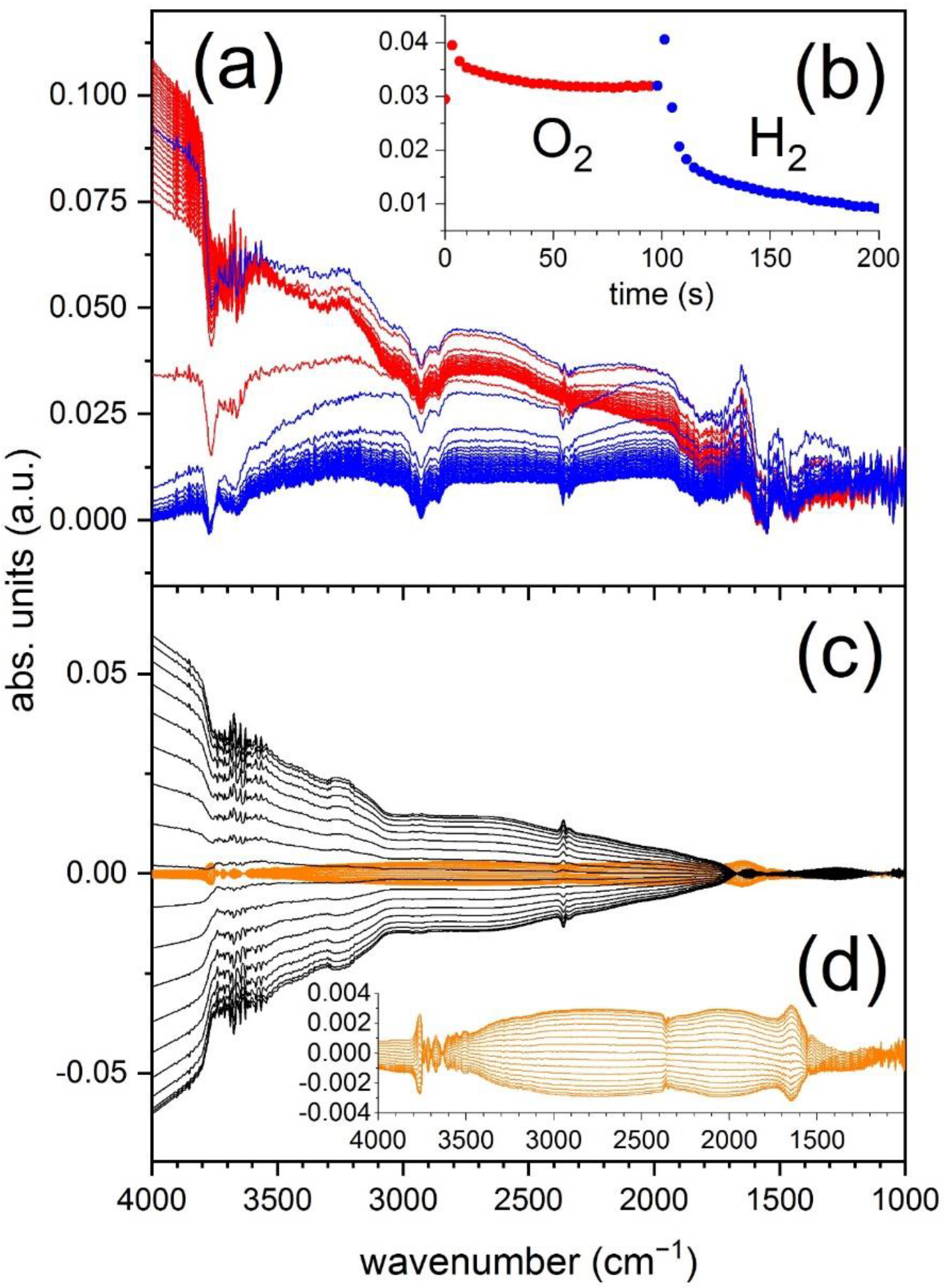
Figure 4 shows the result of a modulated excitation experiment on the same 1 wt.% Pd/Al
2O
3. The time-resolved spectra (
Figure 4a) clearly confirm that at this temperature large baseline changes occur. The absorbance at 2500 cm
−1 increases in O
2 to reach a pseudo steady state (
Figure 4b), after which the intensity returns slowly to the initial value when the gas feed is switched to H
2. The two transitions (H
2→O
2 and O
2→H
2) are accompanied by spikes and the spectra are not featureless. The H
2-O
2 combination produces water, which is visible by the signal at ca. 1640 cm
−1 (δ
O-H). In the region at ca. 3000 cm
−1, negative signals of C–H groups are observed, which most likely originate from differences with respect to the background spectrum (obtained before starting the modulation experiment, after reduction at 300 °C). The signals of atmospheric CO
2 at ca. 2350 cm
−1 are also artefacts due to bad atmospheric compensation.
Negative signals are also visible at 3764 and 3662 cm
−1, which are assigned to Al–OH groups consumed by the interaction of Al
2O
3 with water [
22]. In this case, the phase-resolved spectra clean the time-resolved spectra greatly (
Figure 4c). For example, the signal of physisorbed water at 1640 cm
−1 is only poorly visible. This is expected based on the fact that water removal within the short time of the pulses is not efficient and thus the signal is not strongly modulated. Only the portion that can be formed and then effectively removed appears in the phase-resolved spectra. Additionally, the signals at 3000 cm
−1 completely vanish, confirming that they are artefacts of background subtraction rather than signals of adsorbed species.
Figure 4d also shows the phase-resolved spectra obtained for an identical O
2/H
2 experiment on Al
2O
3. The baseline changes are modest (ca. 25%) compared to those observed for 1 wt.% Pd/Al
2O
3. This confirms that Pd contributes significantly to the baseline variation in the DRIFT spectra.
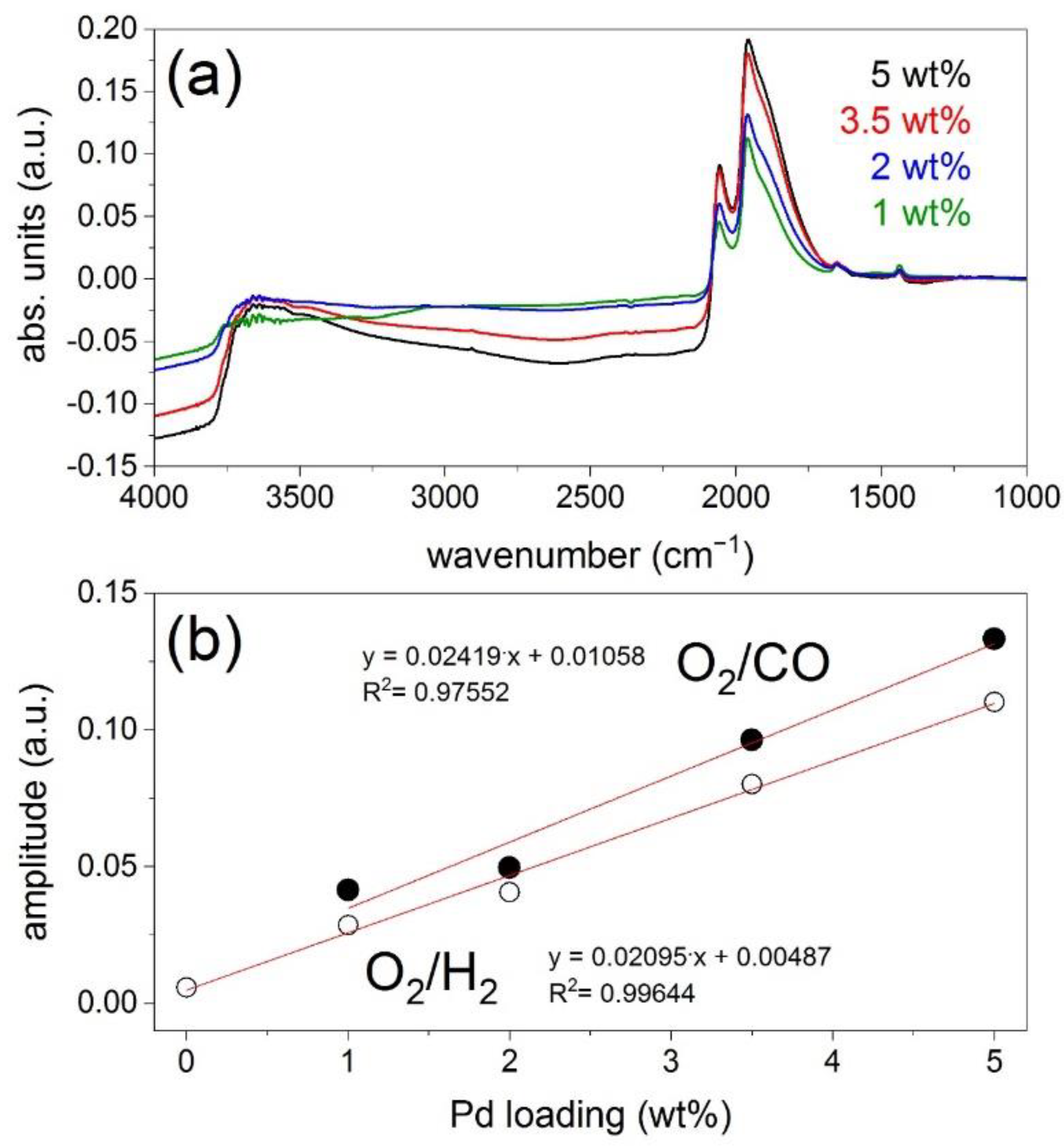
These DRIFTS modulation experiments were repeated for all catalysts with various Pd loadings (
Figure 1) as shown in
Figure 5a. The phase-resolved spectra clearly demonstrate that the extent of baseline changes increases with increasing Pd loading, in parallel to an increased adsorption of CO as the amount of Pd increases (at comparable particle size, hence dispersion). If we consider the signal at 2500 cm
−1, take the difference of absorbance in the phase-resolved spectra of maximum divergence (φ
PSD 0 and 180°) and plot this difference against the Pd loading, a satisfactory linear correlation is obtained in both sets of O
2/CO and O
2/H
2 modulations (
Figure 5b). Therefore, irrespective of reducing agent and of the nature of adsorbates, the phase-resolved DRIFT spectra show that baseline changes are proportional to the amount of Pd in the sample.
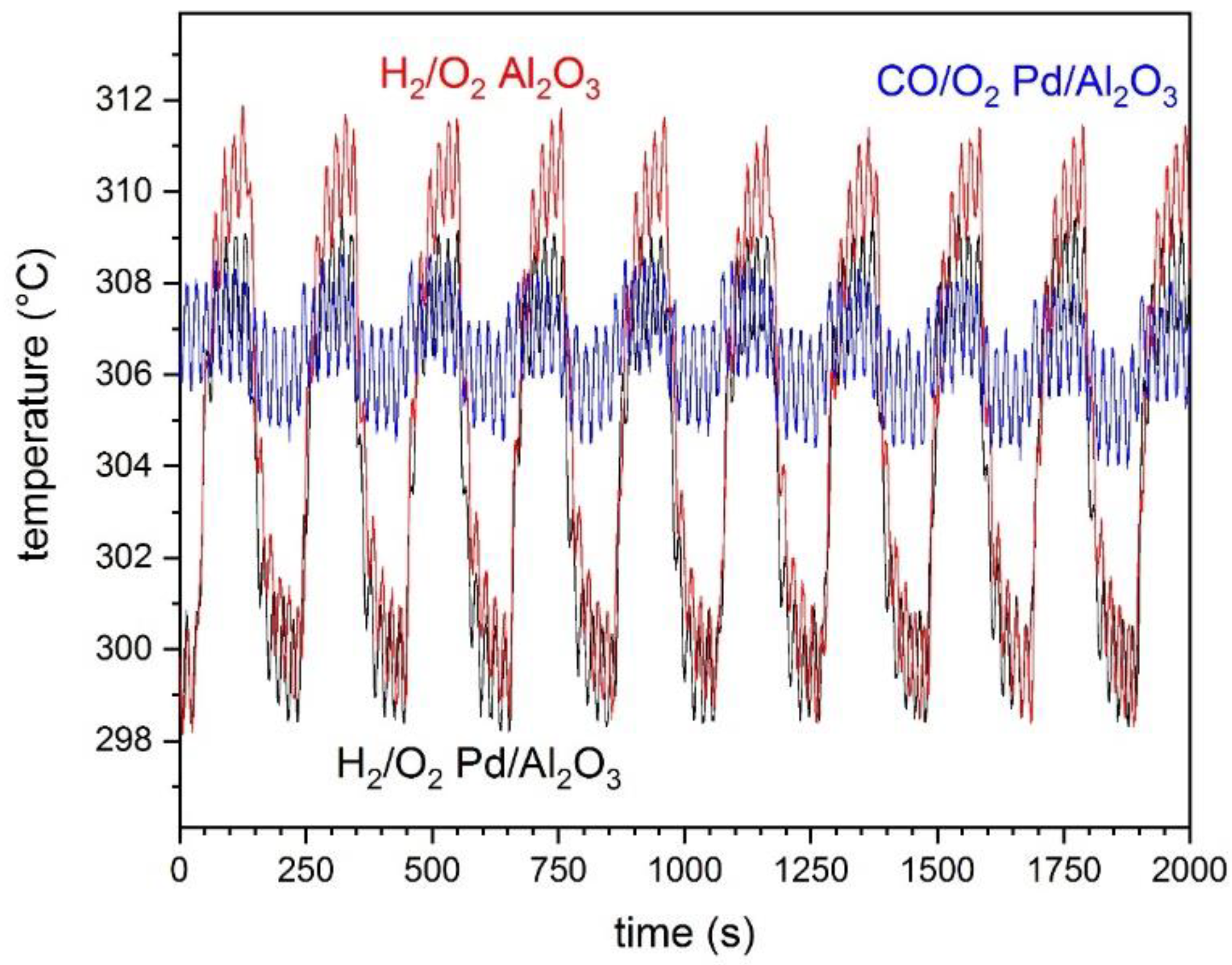
The origin of these changes can be manifold. On the one hand, the aspect of IR spectra strongly depends on the sample temperature. To account for such effect, the sample temperature was monitored using a thermocouple installed below the sample cup near the control thermocouple [
23]. The temperature changes detected during the modulation experiments are shown in
Figure 6 for Al
2O
3 and 1 wt.% Pd/Al
2O
3. In the case of O
2/H
2 modulations, the temperature changed as much as by ca. 11–14 °C irrespective of the presence of Pd, but more in the case of Al
2O
3. Far smaller changes (ca. 3 °C) where recorded for O
2/CO modulations as shown in the case of 1 wt.% Pd/Al
2O
3. While we do not know the response of these IR spectra to a difference in temperature of ca. 11 °C (14 °C–3 °C), we exclude the effect of exothermicity on the DRIFT spectra. The fact that the extent of spectral changes was significantly reduced in the case of Al
2O
3 compared to those produced by the Pd/Al
2O
3 samples (
Figure 4d) supports our conclusion. Moreover, it is clear that the average increase and decrease in temperature follows the same behavior in the reduction and in the oxidation pulses, which is not what we observed for the baseline changes.
Alternately to temperature, a change in colour could also be at the origin of the baseline changes. Before reduction the samples were of very similar light brown colour, while after reduction they were all dark grey. It is known that in Pd-based catalysts visual inspection of the sample during this type of experiments assists to observe appreciable colour changes during the pulses [
6,
24], which we attribute to Pd reduction-oxidation.
To verify this, we performed a similar O
2/CO modulation experiment using Pd K-edge XAS under equivalent experimental conditions on a 2 wt.% Pd/Al
2O
3. Additionally, this sample was reduced in situ at 300 °C. This sample was of different origin and exhibited larger average particle size (ca 3.5 nm); nevertheless, the results obtained serve to illustrate the changes in oxidation state, coordination number and bond distances upon pulsing CO and O
2. The DRIFTS experiment on this sample (not shown) also presents the baseline changes described in
Figure 1, but does not fit in the linear correlation of
Figure 5, probably because it does not belong to the series of Pd/Al
2O
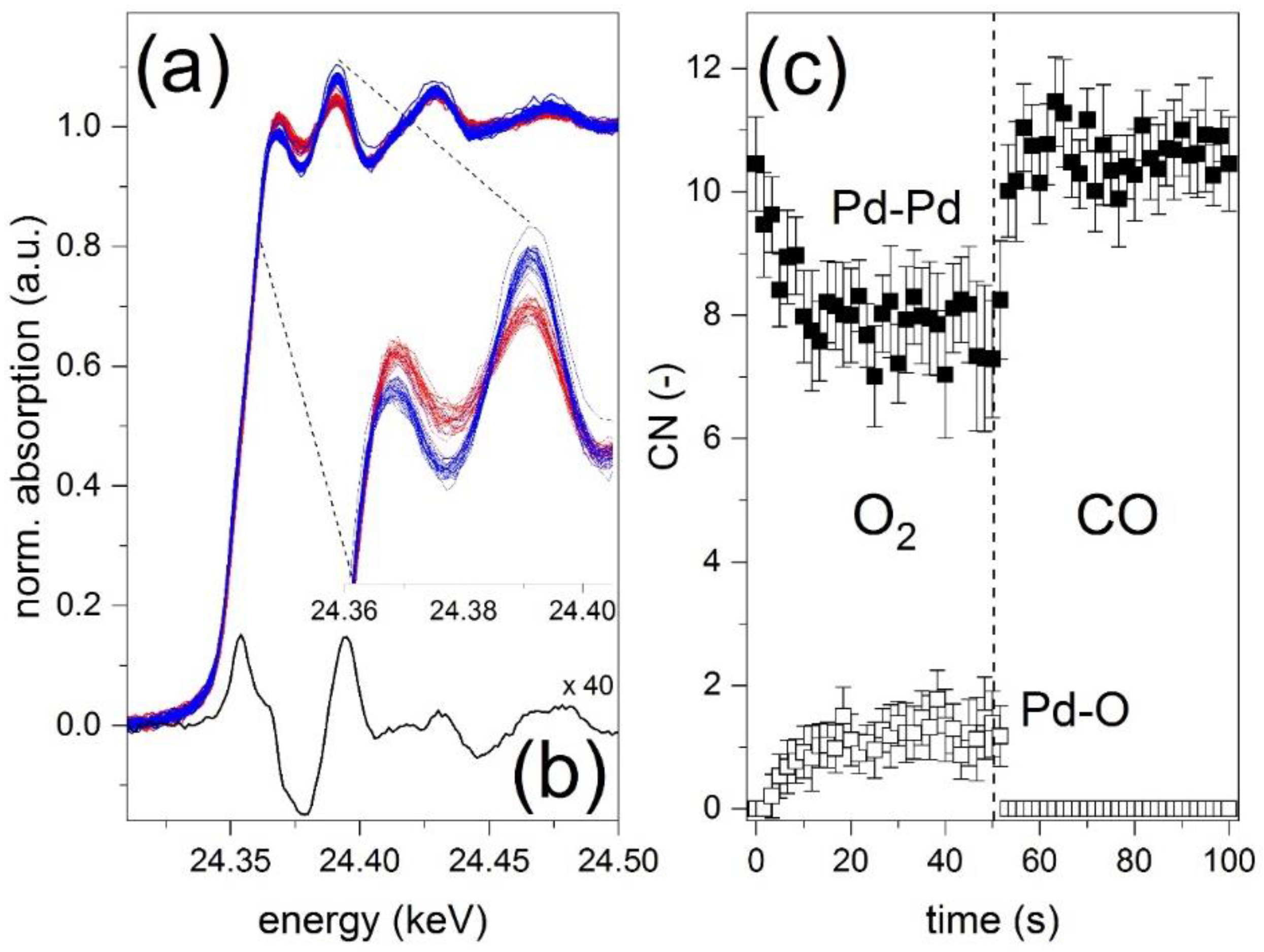
3 samples; the experiment was performed much later and on a different instrument. The Pd K-edge absorption near edge structure (XANES) of the spectra averaged to one modulation period demonstrates that Pd is partially oxidized in the O
2 pulse and reduced back in the CO pulse (
Figure 7a). As observed in past studies [
25], after reduction at 300 °C the changes around the whiteline, which in the case of Pd are ideal for identifying PdO and Pd
0, are only faint. The phase-resolved spectra of the normalized XANES region provide more information because the edge jump is removed by PSD and changes such as in difference spectra can be observed more easily. A selected PSD spectrum in
Figure 7b exhibits lobes of opposite sign in the white line region that are identical to those obtained by subtracting the spectrum of a PdO reference from that of a Pd foil [
25]. The overall lower intensity is due to the nano size of the Pd particles. The profile of
Figure 7b also indicates the absence of carbide like species that we observed previously on 2 wt.% Pd/Al
2O
3 during CO/NO modulation experiments at 300 °C [
25]. The redox behavior observed in XANES is quantified by determination of the first nearest neighbor coordination numbers (CN) by fitting of the extended X-ray absorption fine structure (EXAFS).
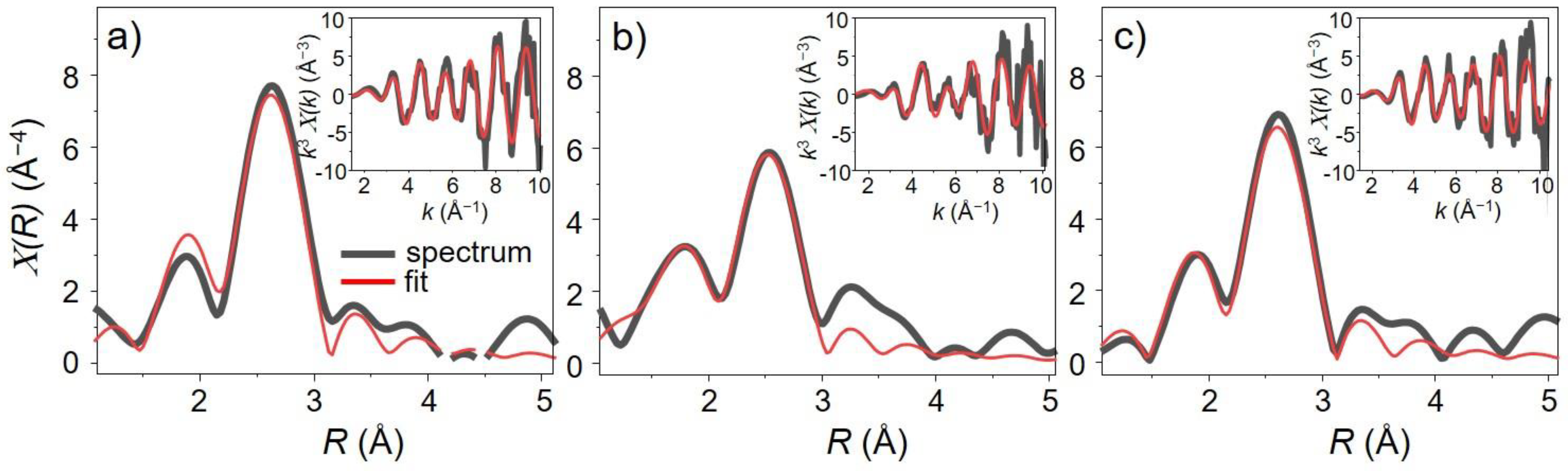
Table 1 presents all the fit parameters for three selected spectra corresponding to the first and last spectrum in the O
2 pulse and the last spectrum in the CO pulse. The first shell Pd–Pd and Pd–O bond distances obtained are consistent with previous reports on supported Pd nanoparticles [
26,
27]; as illustration of the fit quality selected experimental and simulated spectra are plotted in
Figure 8.
Figure 7c shows the evolution of Pd–Pd and Pd–O CN during the O
2/CO modulation. At the beginning of the O
2 pulse the Pd appears in a fully reduced state; this is evidenced by the Pd–Pd CN of ca. 11 and by the fact that the best fit was obtained when no O neighbors were included in the fit (i.e., CN
Pd–
O = 0). During the O
2 pulse, the Pd–Pd CN slowly decreases from ca. 10.5 to ca. 7, which indicates gradual partial oxidation of Pd, consistently the Pd–O CN increases up to 1.5. After switching to CO the initial CNs values (10.5 and 0 for Pd–Pd and Pd–O, respectively) are rapidly restored revealing a fast reduction and confirming the reversible nature of the redox process. No changes in the Pd–Pd distance are observed in the CO pulse (not shown) in agreement with the interpretation of the phase-resolved XANES data that carbide like species did not form under these conditions.
The gradual oxidation followed by a fast reduction observed by EXAFS analysis is compared qualitatively with the slow increase in baseline in the DRIFT spectra of
Figure 2, while the rapid reduction is mirrored by the faster decrease in baseline. We use this XAS experiment to support our interpretation that the non-negligible baseline changes observed in the DRIFT spectra of
Figure 2 and
Figure 4 need to be associated with the redox behavior of the catalyst, in the current case essentially with the presence of the redox active Pd component. Because this is a recurrent behavior that we and others observe irrespective of Pd/Al
2O
3 sample, the XAS experiment supports the results of the DRIFTS measurements. The overall slower oxidation process in O
2 and the two steps with which it is accomplished are also reminiscent of similar behavior of Pd/Al
2O
3 catalysts in modulation experiments using XAS [
26,
28] and X-ray diffraction [
28,
29], further confirming our assignment of the baseline changes in DRIFT spectra. Therefore, as demonstrated previously for ATR-IR spectroscopy [
12] and PM-IRRAS [
13], we can use DRIFTS to follow changes in the oxidation state of metal active phases especially on a material composed of an insulating support such as Al
2O
3. The presence of a semiconducting metal oxide support could contribute significantly to the spectral variations because it takes part in the redox behavior of the whole catalytic material. This contribution may even predominate over the one we report here.
Finally, it should be noted that the shape of the baseline variations over the whole energy range of, for example, the spectra in
Figure 1 is different compared to transmission [
11] and ATR-IR [
12] experiments, where the changes show a rising absorption from 4000 to ca. 2000 cm
−1 and a decreasing absorption below 2000 cm
−1. This difference could be indicative of the different source of the phenomenon. In diffuse reflectance, spectra are the result of both absorbance and scattering of IR radiation. The two processes overlap, and each can prevail under certain experimental conditions. The rising baseline from 2000 to 4000 cm
−1 in the case of the DRIFT spectra in the modulation experiments, the absence of an absorption centered below 2000 cm
−1 that is typical of semiconductors and the opposite sign of absorption gain–loss compared to transmission/ATR-IR spectra may reveal that the observed changes derive predominantly from variations of scattering properties of the sample upon reversible reduction and partial oxidation.
3. Materials and Methods
Catalysts were kindly provided by Chimet S.p.A. (Viciomaggio, Italy) and were prepared on a transition Al
2O
3 support (specific surface area, 116 m
2g
−1; pore volume, 0.41 cm
3g
−1) through a deposition-precipitation method [
30] with an amount of Na
2PdCl
4 as the metal precursor to obtain 1, 2, 3.5 and 5 wt.% Pd/Al
2O
3. An additional 2 wt.% Pd/Al
2O
3 sample (specific surface area, 135 m
2g
−1; Pd particle size of ca. 3.5 nm) was kindly provided by Umicore AG & Co. KG (Hanau, Germany).
Electron microscopy images of the Pd/Al
2O
3 series were obtained with a Jeol 2200FS transmission electron microscope (Jeol, Tokyo, Japan) equipped with a 200 kV field emission gun and a high-angle annular dark field detector in STEM mode providing images with atomic number (Z
1.5–1.8) contrast [
31]. The samples were prepared by evaporation of an alcohol suspension on a Cu grid coated with a holey carbon film. The palladium particle size distribution was obtained by measuring up to 200 particles using the software ImageJ [
32].
Diffuse reflectance infrared Fourier transform spectra were obtained with a Vertex 70 (Bruker Optics, Fällanden, Switzerland) spectrometer equipped with a liquid N
2 cooled HgCdTe detector and a diffuse reflectance unit (Harrick Inc.) at 4 cm
−1 spectral resolution and 80 kHz scanner velocity. The sample was loaded on the cup of a custom made diffuse reflectance cell [
23] equipped with a CaF
2 window and two thermocouples inserted below the cup to control heating and to read out the temperature. Three solenoid valves (Parker, Series 9) installed in front of the cell and controlled by the software OPUS of the spectrometer enabled the repeated change of gas feed to the sample. If not otherwise specified, spectra are reported in (pseudo-)absorbance [
21]. The DRIFT spectra were also resampled in the form of Kubelka-Munk (K-M) function defined as F(R) = (1 − R)
2/(2R), where R is the ratio between sample and background scans. A mass spectrometer (MS, Pfeiffer Omnistar) installed after the cell was used to determine the nature of the gas species (
m/
z = 2–H
2, 4–He, 18–H
2O, 28–CO, 32–O
2, 44–CO
2). After reduction at 300 °C in 5 vol% H
2/He for 30 min, modulation experiments were started at 300 °C by introducing a reducing environment (either 5 vol% H
2 or 1 vol% CO) alternated to an oxidizing environment (5 or 1 vol% O
2), all gas mixtures being balanced with He. Ten modulation periods consisting of 101 s under reducing and 101 s under oxidizing conditions were carried out for each modulation experiment. During one period, 60 DRIFT spectra were collected by accumulating 25 interferograms. All spectra were then averaged to one modulation period and subjected to phase sensitive detection (PSD, Equation (1)) [
16] using a Matlab script.
where
I(φPSD) is the absorbance in the phase-resolved spectra,
T the modulation period,
I(
t) the absorbance in the time-resolved spectra,
k the demodulation index,
ω the stimulation frequency and
φPSD the phase angle. In this work, we report PSD data for
k = 1 (fundamental harmonic).
X-ray absorption spectra were acquired at the Pd K-edge (24.35 KeV) in quick transmission mode (quickXAS) using a Si(311) monochromator at beamline SuperXAS of the Swiss Light Source (SLS, Villigen, Switzerland). A Pd foil inserted between the second and the third ionization chamber was used for energy calibration throughout the measurement. The 2 wt.% Pd/Al
2O
3 sample (ca. 60 mg; sieved to 50–100 µm) was firmly fixed within two plugs of quartz wool in a homemade cell equipped with two graphite windows to allow transmission of X-rays [
33]. After heating the sample in 5 vol% H
2/He to 300 °C, the modulation experiment was started by repeated 1 vol% O
2/He pulses alternated to 1 vol% CO/He pulses (total flow, 100 mL/min) using solenoid valves (Parker, Series 9). During one modulation period (ca. 100 s), 120 quickEXAFS spectra were collected at a sampling rate of 5 kHz and at a time resolution of ca. 1.2 s while moving the monochromator continuously forward and backward. For the data analysis, we consider only the spectra collected in forward mode for simplicity. Thirty modulation periods were performed. After alignment and normalization, all spectra were averaged to one modulation period (60 spectra). The coordination number (CN) and the Pd-Pd distance (d
Pd) were obtained from these averaged spectra.
E0 was chosen as the first maximum in the first derivative. Spectrum number 60 (corresponding to metallic Pd after full reduction under CO) was used for calibration and E
0 of this spectrum was calibrated to 24.35 keV. The resulting shift (−1.4 eV) was applied to all spectra. Normalization and background subtraction were performed in the Athena software package [
34]. To follow the reduction/oxidation during CO and O
2 pulses EXAFS fitting was performed using the Artemis software package (S
02 = 0.8; fit ranges = 3.0 < k/Å
−2 < 9.2, 1.3 < R/Å < 3.5) [
34]. As reference for bond distance (R) and coordination numbers (CN) of scatter atoms, crystallographic data obtained from ICSD database was used for Pd and PdO.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}