Abstract
A facile, sustainable method for the selective modification of aliphatic hydroxy (R–OH) groups in Kraft lignin was developed using an ionic liquid, 1-ethyl-3-methylimidazolium acetate (EmimOAc), as a solvent and catalyst. Selective R–OH modification was achieved by a one-pot, two-step homogeneous reaction: (i) acetylation of R–OH and aromatic OH (Ar–OH) groups with isopropenyl acetate (IPAc) as an acyl donor and (ii) subsequent selective deacetylation of the generated aromatic acetyl (Ar–OAc) groups. In step (i), IPAc reacts rapidly with Ar–OH but slowly with R–OH. The generated Ar–OAc is gradually deacetylated by heating in EmimOAc, whereas the aliphatic acetyl (R–OAc) groups are chemically stable. In step (ii), all R–OH is acetylated by IPAc and Ar–OAc which is a better acyl donor than IPAc, contributing to the rapid acetylation of the remaining R–OH, and selective deacetylation of the residual Ar–OAc is completed by adding a tiny amount of water as a proton source. This two-step reaction resulted in selective R–OH modification (>99%) in Kraft lignin with the remaining being almost all Ar–OH groups (93%). Selectively modified Kraft lignin was obtained with an acceptably high isolated yield (85%) and repeatability (N = 3). Furthermore, despite the lower substitution degree, it exhibited solubility in common solvents, heat-meltability, and thermal stability comparable to completely acetylated Kraft lignin.
1. Introduction
Lignin is the most abundant natural aromatic polymer and a potential renewable source of high-value chemicals and materials for significant applications [1,2]. In its native form, depending on its origin, lignin contains three types of aromatic repeating backbone units—p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) (Scheme 1a)—which have aliphatic hydroxy (R–OH) groups such as primary and secondary alcohols and aromatic hydroxy (Ar–OH) groups. During biosynthesis, these monomers are linked through ether and carbon–carbon bonds via the free-radical coupling of the resonance-stabilized structures (Scheme 1b) [3,4]. The major source of lignin is chemical pulping, 90% of which is performed via the Kraft process using sodium hydrosulfide (NaSH) and sodium hydroxide (NaOH) under high pressures and temperatures. This process affords Kraft lignin as a byproduct isolated from spent pulping liquor (i.e., black liquor), with the global annual production capacity estimated as 265 metric kilotons [5,6]. However, because of its complex heterogeneous structure, an estimated 98% of Kraft lignin is burned as a non-optimized energy resource in pulp mill boilers to recover energy and pulping chemicals [7]. Recently, demands have arisen to convert Kraft lignin into higher-utility functional polymers such as adhesives [8,9], carbon fiber [10], polymeric components in thermosets (e.g., phenolic resins [11], polyurethanes [12,13], and epoxy resins) [14], and thermally stable copolyesters [15].
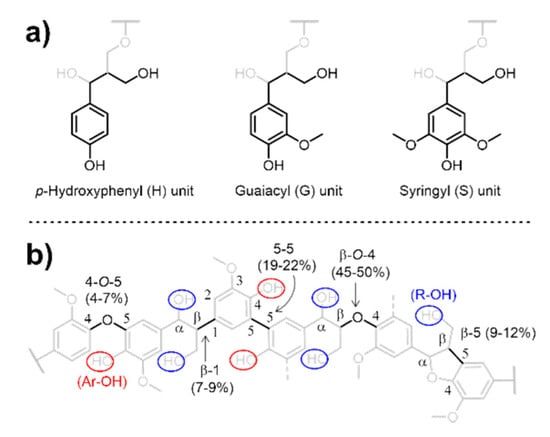
Scheme 1.
Structural representation of (a) aromatic repeating units and (b) dominant ether and carbon–carbon linkages in lignin with R–OH and Ar–OH groups.
Kraft lignin features a rigid backbone comprising aromatic units that can participate in π–π interactions and a mixture of oxygenated functional groups that can undergo hydrogen bonding, which result in a relatively high glass transition temperature (Tg) of over 100 °C [16,17]. Additionally, the molecular weight of isolated Kraft lignin is usually low since the pulping process results in significant cleavage and condensation [18]. These factors create a stiff yet fragile material that is stiff yet cannot yield without breaking [19]. Therefore, without significant modification, Kraft lignin itself has little commercial value for polymeric applications. In turn, the functionalization of Kraft lignin is hindered by its poor solubility in common organic solvents, non-heat-meltability, and heterogeneity.
Kraft lignin possesses abundant R–OH and Ar–OH groups, which offer handles for modification to engineer its physicochemical properties. Esterification, a more than 100-year-old prototypical derivatization technique, can be employed to increase the processability and usefulness of biopolymers, as exemplified by thermoplastics and thermosets [19,20,21]. Most reported methods on lignin esterification employ acyl halides and anhydrides, typically in the presence of catalysts. Conventional acyl donors are highly activated and react with both R–OH and Ar–OH groups at comparable rates. Although a model compound study has shown that pyridine catalysts favor R–OH while triethylamine favors Ar–OH [22], conventional catalysts show no clear chemoselectivity.
Liu et al. demonstrated a chemoselective Fischer esterification of lignin R–OH groups using an excess of carboxylic acid in the absence of a catalyst [19]. However, the harsh conditions required for a satisfactory degree of conversion limit the applicability of this method, as the employed high reaction temperatures (120−160 °C) and long processing times (24−96 h) are not cost-effective. Furthermore, such severe conditions can induce the emission of environmentally hazardous organosulfur compounds due to the unavoidable pyrolysis of the sulfur links created during Kraft pulping [23]. To the best of our knowledge, apart from protection, there are no facile and sustainable methods for the chemoselective modification of R–OH or Ar–OH groups in Kraft lignin.
Ionic liquids are molten organic salts with melting points of below 100 °C [24], and can be a platform for obtaining functional materials with a wide range of applications [25,26]. In particular, 1-ethyl-3-methylimidazolium acetate (EmimOAc), is an excellent solvent for biopolymers such as cellulose and lignin [27,28], and functions as an organocatalyst for the transesterification of R–OH and Ar–OH groups with stable vinyl esters as acyl donors [9,29,30]. This reaction system is scalable and allows EmimOAc to be easily recycled [31]. Furthermore, our previous study of low-molecular-weight compounds such as 2-phenylethanol (Ph–EtOH) and phenol (PhOH) revealed that the transesterification of PhOH with isopropenyl acetate (IPAc) (Scheme 2b) is faster than that of Ph–EtOH (Scheme 2a) [29], which was attributed to the generally lower basicity and nucleophilicity of PhOH (Kb approximately 106 lower than that of methanol) [32]. Furthermore, the generated phenyl acetate (PhOAc) was found to be gradually deacetylated by heating in EmimOAc (Scheme 2c), whereas 2-phenylethyl acetate (Ph–EtOAc) remained stable. This deacetylation reactivity difference indicated that EmimOAc selectively catalyzes the deacetylation of aromatic acetyl (Ar–OAc) groups [29].
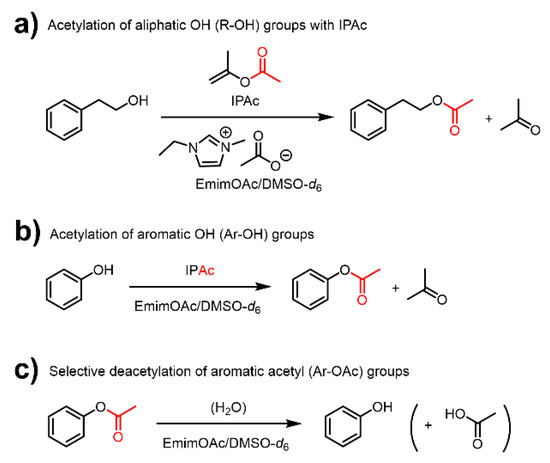
Scheme 2.
Schematic representation of the acetylation of (a) R–OH and (b) Ar–OH groups using 1-ethyl-3-methylimidazolium acetate (EmimOAc) as catalyst and isopropenyl acetate (IPAc) as an acyl donor and (c) EmimOAc-catalyzed selective deacetylation of Ar–OAc groups.
In previous studies on the transesterification of Kraft lignin using EmimOAc and excess vinyl esters, both R–OH and Ar–OH groups were esterified, although a similar selective deacylation of the generated aromatic esters was expected [9,30]. This phenomenon may be caused by the excess acyl donors continuously promoting R–OH esterification. Thus, we assumed that the selective modification of R–OH groups in Kraft lignin can be achieved if an appropriate initial concentration of the acyl donor is selected to enhance the selective deacylation of aromatic moieties. If the concentration of the acyl donor (e.g., IPAc) is properly lowered, the Ar–OH groups in Kraft lignin should rapidly react with IPAc, whereas the abundant R–OH groups should remain intact in the initial stage as their reaction with IPAc is relatively slow. However, if all the generated Ar–OAc groups are subsequently deacetylated using the remaining R–OH as a proton source, Ar–OH and R–OAc groups eventually form. Therefore, the selective R–OH modification in Kraft lignin with acyl groups can be realized in a one-pot two-step reaction (Scheme 3). If such selective modification improves the solution and thermal processabilities of Kraft lignin, it is more advantageous than the thorough modification of all OH groups, as the residual Ar–OH groups may be of value for further functionalization.
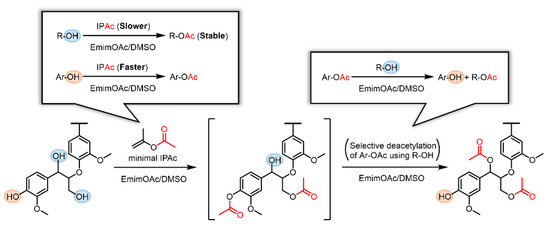
Scheme 3.
Schematic representation of the selective modification of R–OH groups in Kraft lignin via a two-step reaction in an EmimOAc/DMSO mixed solvent: transesterification of R–OH and Ar–OH groups with IPAc as an acyl donor and subsequent selective deacetylation of the generated Ar–OAc groups with the residual R–OH groups as a proton donor.
This study aimed to establish a facile and sustainable method for the selective R–OH modification in Kraft lignin with acetyl groups using EmimOAc as the solvent and catalyst. Several model reactions were conducted with low-molecular-weight compounds and analyzed by in situ 1H NMR to compare the reactivities of both types of OH groups and acetyl derivatives in EmimOAc-catalyzed transesterification reactions. The optimized reaction conditions were applied to Kraft lignin, and the physicochemical properties of selectively modified Kraft lignin derivative were compared with those of fully modified Kraft lignin derivative.
2. Results and Discussion
2.1. Selective Modification of R–OH Groups Accelerated by In Situ Generated Ar–OAc Groups as an Acyl Donor
In our previous study [29], EmimOAc was employed to catalyze the acetylation of both R–OH and Ar–OH groups using IPAc as an acyl donor for the subsequent selective deacetylation of Ar–OAc groups. However, with excess IPAc (e.g., ~10 Equation/[OH]), the deacetylation of the generated Ar–OAc was negligible, as the Ar–OH groups were continuously acetylated. Thus, the targeted selective R–OH modification was not achieved at a high IPAc concentration.
In contrast, we expected that selective modification can be realized at an appropriately low IPAc concentration. Toward this goal, the three model reactions shown in Figure 1a were performed at 80 °C for PhOH, Ph–EtOH, and an equimolar mixture of minimal amounts of IPAc and EmimOAc (1 Equation/[Ph–EtOH] or [PhOH] each). In the model reaction using the mixture of PhOH/Ph–EtOH, the concentrations of IPAc and EmimOAc were 0.5 molar equivalents of the sum of [Ph–EtOH] and [PhOH]. The changes in OH concentration were analyzed by in situ 1H NMR in deuterated dimethyl sulfoxide (DMSO-d6, see Supplementary Materials for detailed procedures and calculation methods).
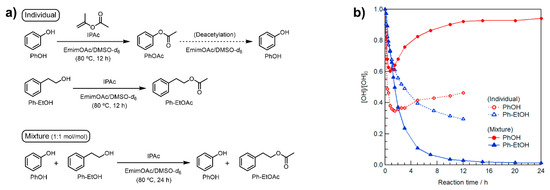
Figure 1.
(a) Schematic representation of model reactions in EmimOAc/DMSO-d6: transesterification of PhOH, Ph–EtOH, and an equimolar mixture using IPAc as an acyl donor. (b) Effect of minimal amounts of IPAc and EmimOAc (0.38 mol L−1 each) on the transesterification of individual PhOH and Ph–EtOH or their mixture (0.38 mol L−1 each) for 12 or 24 h at 80 °C determined as changes in relative contents by in situ 1H NMR in DMSO-d6.
As shown in Figure 1b, the individual acetylation of PhOH and Ph–EtOH proceeded immediately upon heating at 80 °C. The concentrations of these reactants decreased at different rates, and that of PhOH decreased faster than that of Ph–EtOH, in line with our previous study [29]. After 1h of heating, the Ph–EtOH concentration continued to decrease, whereas that of PhOH gradually increased, which suggested that the selective deacetylation of the generated PhOAc proceeded concomitantly with IPAc consumption. Interestingly, the rate of increase in PhOH concentration was faster in the presence of Ph–EtOH than that of PhOH alone. This suggests that Ph–EtOH contributed to the deacetylation of PhOAc as a proton source. Consequently, almost all the generated PhOAc was successfully reconverted to PhOH, while Ph–EtOH was completely acetylated within 24 h. Therefore, the desired selective modification of R–OH in the presence of Ar–OH groups was demonstrated in a model reaction at minimal IPAc and EmimOAc concentrations.
Note that the transesterification of the above mixture was performed at an IPAc loading of only 1 molar equivalent with respect to Ph–EtOH. Although the PhOH concentration at one point significantly decreased because of transesterification with IPAc, all Ph–EtOH was ultimately acetylated. This suggests that the generated PhOAc was sequentially consumed as an acyl donor for Ph–EtOH acetylation. The Ph–EtOH conversion rate in the presence of PhOH in the middle stage after ~1 h was notably faster than that of Ph–EtOH alone at the same IPAc concentration, which was attributed to the fact that the generated PhOAc is a more active acyl donor for Ph–EtOH than IPAc (see Figure S1).
2.2. Key Factors in the Selective Modification of R–OH Groups
The above model study using a mixture of Ph–EtOH and PhOH in the presence of 1 Equation/[Ph–EtOH] of IPAc and EmimOAc realized the facile selective modification of R–OH groups through the transesterification of PhOH and Ph–EtOH with IPAc followed by the selective deacetylation of the generated PhOAc using Ph–EtOH. In this reaction system, the concentration of IPAc is key. When the IPAc concentration was increased to 3 Equations/[Ph–EtOH], all Ph–EtOH was rapidly acetylated, as the abundant IPAc and the in situ generated PhOAc functioned as acyl donors. However, as excess IPAc continued to react with PhOH and finally reached equilibrium, the reconversion of PhOAc to PhOH was largely prevented (Figure S2).
To apply the above reaction to actual Kraft lignin, it is also necessary to determine the proper amount of EmimOAc as it functions not only a catalyst but also as a lignin solvent. Another model reaction with a ten-fold molar concentration of EmimOAc was performed on a mixture of Ph–EtOH and PhOH in the presence of 3 Equation/[Ph–EtOH] of IPAc. As shown in Figure 2, regardless of the presence of PhOH, all Ph–EtOH was immediately acetylated because of the significant increase in EmimOAc, and the generated Ph–EtOAc remained intact for up to 10 h. Although a certain amount of PhOH was converted to PhOAc immediately upon heating, all generated PhOAc was gradually deacetylated as this deacetylation was promoted by the increased amount of EmimOAc [33]. Therefore, the appropriate combined amount of IPAc and EmimOAc was demonstrated to be essential for selective R–OH modification.
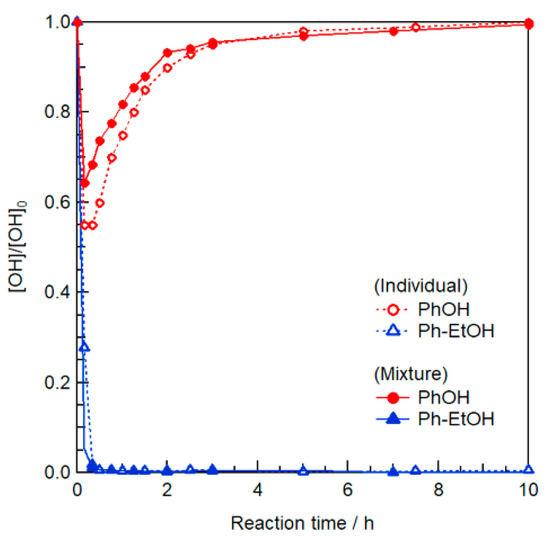
Figure 2.
Effect of the amount of EmimOAc (3.8 mol L−1) on the transesterification of individual PhOH and Ph–EtOH or their mixture (0.38 mol L−1 each) under excess IPAc (1.14 mol L−1) for 10 h at 80 °C determined as changes in relative contents by in situ 1H NMR in DMSO-d6.
Prior to the application of the above reaction to Kraft lignin, the PhOAc deacetylation mechanism should be discussed. As shown in Figure 2, after Ph–EtOH as an effective proton source for PhOAc deacetylation was completely acetylated in the early stage, PhOAc deacetylation proceeded until 10 h. This implies that the existence of another proton source (e.g., traces of water) for PhOAc deacetylation. However, when the EmimOAc concentration was low (0.38 mol L−1), the deacetylation of PhOAc was not significantly promoted (Figure S2). Thus, the significant deacetylation of PhOAc illustrated in Figure 2 could not be mainly ascribed to water. Considering all other chemicals present in the reaction system, the weakly acidic proton at the C2 position of the Emim cation [33] is most likely proton source.
Multiple reaction pathways are expected for PhOAc deacetylation. In the presence of a sufficient amount of a proton source such as Ph–EtOH (Figure 1b), PhOAc deacetylation should proceed through a concerted mechanism involving both the cation and the anion of EmimOAc, as depicted in Scheme 4 (i). This concerted mechanism is based on the insights into EmimOAc-catalyzed transesterification reported by Kakuchi et al. obtained by their experimental and computational investigations [34].
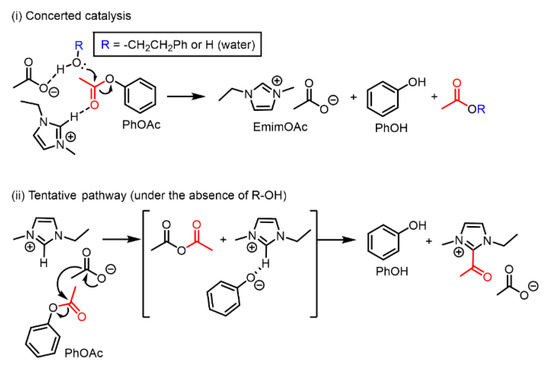
Scheme 4.
Schematic representation of the proposed mechanisms of PhOAc deacetylation in the presence of EmimOAc: (i) concerted catalysis with the coexistence of R–OH groups and water as proton sources and (ii) another tentative pathway utilizing a weakly acidic proton at the C2 position of the Emim cation as another proton source.
For the EmimOAc-catalyzed transesterification of OH groups with vinyl acetate as an acyl donor, Chen et al. proposed that the acetate anion of EmimOAc can deprive the Emim cation of a proton at the C2 position, forming an N-heterocyclic carbene and reacting with vinyl acetate to generate an acylated Emim cation (eventually, the OH group is acetylated by the acylated Emim cation) [35]. Furthermore, Hirose et al. proposed another minor pathway via a mixed acid anhydride, which can be generated by a direct nucleophilic attack from the acetate anion of EmimOAc on the vinyl ester [36]. Considering these two EmimOAc-catalyzed transesterification pathways, we proposed a tentative pathway for PhOAc deacetylation without Ph–EtOH as a proton source, as shown in Figure 2. As depicted in Scheme 4 (ii), PhOAc deacetylation could be promoted by a weakly acidic proton from EmimOAc as a proton source, and the C2 position of the Emim cation could be substituted with the acyl group derived from PhOAc.
In tentative deacetylation pathway (ii), first, the acetate anion nucleophilically attacks PhOAc, forming acetic anhydride and a phenoxide anion as intermediates. Subsequently, the generated phenoxide anion takes the weakly acidic proton from the C2 position of the Emim cation and becomes PhOH. Although the generation of an N-heterocyclic carbene cannot be clarified, the deprotonated Emim could react with acetic anhydride, generating the acylated Emim cation and acetate anion. In other words, EmimOAc could function as a kind of substrate in the deacetylation of PhOAc. However, there is insufficient evidence to prove reaction pathway (ii) because the in situ 1H NMR performed in this study could not detect clear peaks corresponding to the acylated Emim cation or the other associated intermediates. It is an important future task to verify proposed mechanism (ii) using analytical techniques with a higher time resolution.
More importantly, if EmimOAc functions as a proton donor for PhOAc deacetylation, the consumed EmimOAc could not be reused without subsequent regeneration. To improve the efficiency and sustainability of our proposed method for selective R–OH modification, it would be better to promote the main reaction pathway (i) or another step for the subsequent hydrolyzation of the acylated Emim cation by adding other proton sources such as water.
2.3. Effects of Methoxy Groups at o-Positions of Ar–OH Groups
A further concern in applying the polymer reaction is the effects of the abundant methoxy (OMe) groups at the o-positions of the Ar–OH groups in Kraft lignin. The Kraft lignin used in this study contains a few H-type Ar–OH groups whose o-positions are not substituted like PhOH, but it is rich in G- and S-type Ar–OH groups with mono- or di-OMe groups at the o-positions. To investigate the effect of the OMe groups on the selective R–OH modification, two more model reactions (Figure 3a) were performed using molar equivalent mixtures of Ph–EtOH and guaiacol (GuOH) or 2,6-dimethoxyphenol (syringol, SyOH) in a similar manner to the previous model reactions using 3 Equations/[Ph–EtOH] of IPAc and 10 Equations/[Ph–EtOH] of EmimOAc.
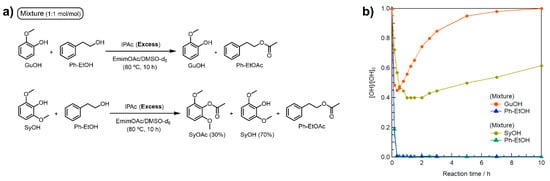
Figure 3.
(a) Schematic representation of model reactions in EmimOAc/DMSO-d6: transesterification of equimolar mixtures of GuOH or SyOH with Ph–EtOH using IPAc as an acyl donor. (b) Effect of mono- or di-OMe groups at the o-positions of Ar–OH groups on the transesterification of mixtures of GuOH or SyOH with Ph–EtOH (0.38 mol L−1 each), excess IPAc (1.14 mol L−1), and EmimOAc (3.8 mol L−1) for 10 h at 80 °C determined as changes in relative contents by in situ 1H NMR in DMSO-d6.
As shown in Figure 3b, all Ph–EtOH in the presence of GuOH was immediately acetylated, like that with PhOH (Figure 2). Although the rate of decrease in GuOH concentration in the early stage was faster than that of PhOH, all the acetylated GuOH (GuOAc) returned to GuOH by 10 h. On the other hand, Ph–EtOH in the presence of SyOH was completely acetylated, and the initial reduction rate of SyOH was slightly slower but the maximum conversion rate was the highest. The SyOH concentration gradually increased after 2 h of heating, but approximately 30% of the initial SyOH concentration remained in the acylated state (SyOAc) after heating for 10 h. These results imply that an increase in the number of electron-donating OMe groups improves the chemical stability of the generated Ar–OAc groups against subsequent deacetylation. Thus, complete SyOAc deacetylation was prevented. To realize facile and selective R–OH modification in Kraft lignin, the initial concentration of IPAc should be minimized to prevent excess acetylation of S-type Ar–OH groups in the early reaction stage.
2.4. Selective R–OH Modification in Kraft Lignin by One-Pot Two-Step Reaction
To improve the solution and thermal processabilities of Kraft lignin without substituting the valuable Ar–OH groups, a facile and sustainable method for selective R–OH modification using EmimOAc was developed (Scheme 5). In this system, (i) fast Ar–OH acetylation and relatively slow R–OH acetylation are first performed at 80 °C for 1 h using a determined amount of IPAc in an EmimOAc/DMSO mixed solvent. Subsequently, (ii) selective deacetylation of the generated Ar–OAc groups is conducted at 80 °C for more than 1 h, which is promoted by adding a tiny amount of water as a proton donor.
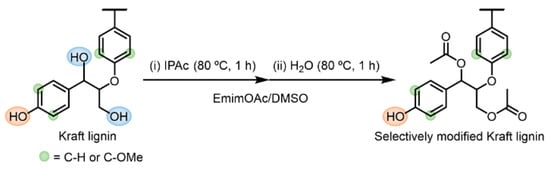
Scheme 5.
One-pot two-step reaction for selective R–OH modification in Kraft lignin: (i) acetylation of Ar–OH and R–OH groups using IPAc and (ii) subsequent selective deacetylation of generated Ar–OAc groups promoted by adding 2 Equations/[Ar–OH] of water as a proton source in an EmimOAc/DMSO mixed system.
Importantly, a certain amount of Ar–OH groups is expected to be acetylated during step (i). Thus, subsequent deacetylation of the produced Ar–OAc groups must be completed to realize selective R–OH modification in Kraft lignin. However, the content of R–OH groups (2.7 mmol g−1) in the employed Kraft lignin is less than that of Ar–OH groups (3.4 mmol g−1) [30]. This unbalance can cause a lack of proton sources for the complete deacetylation of Ar–OAc groups if excess Ar–OH groups are acetylated in the first step.
The model experiment (Figure 2) demonstrated that the use of excess EmimOAc enabled the complete deacetylation of Ar–OAc groups, even after all R–OH groups were acetylated. However, to increase the reusability of EmimOAc, another proton source should be used instead. Water can act as an optimal proton source in terms of greenness and cost-effectiveness, but excess water would significantly hydrate EmimOAc and interfere with its excellent catalytic activity for R–OH acetylation. Therefore, this series of reactions—acetylation of R–/Ar–OH groups with IPAc and subsequent selective deacetylation of Ar–OAc groups with water—was divided into two steps, and the added amount of water in the second step was adjusted to 2 Equations/[Ar–OH] to appropriately promote Ar–OAc deacetylation.
As suggested by the model experiments, the key to selective R–OH modification should be the IPAc concentration. Our preliminary examination using excess IPAc (12 Equations/[R–OH]) achieved 100% R–OH acetylation, while 90% of the Ar–OH groups remained by extending the deacetylation time in the second step to 24 h (see Figure S3). However, excess IPAc and long reaction times are not time- or cost-effective. Thus, the reaction time for each step was decided as 1h, and the minimum amount of IPAc required for selective R–OH modification in Kraft lignin was investigated from 1 to 12 Equations/[R–OH].
As shown in Figure 4, when 10–12 Equations/[R–OH] of IPAc were added, 20–30% of Ar–OH groups were lost to acetylation because the degree of deacetylation of the generated Ar–OAc groups was insufficient in the limited reaction time. On the other hand, almost all Ar–OH groups were retained when the IPAc amount was 1–7 Equations/[R–OH]. However, R–OH acetylation was incomplete using 1–2 Equations/[R–OH] of IPAc. Therefore, 3 Equations/[R–OH] was determined as the minimum amount of IPAc for the efficient selective modification of R–OH groups in Kraft lignin.
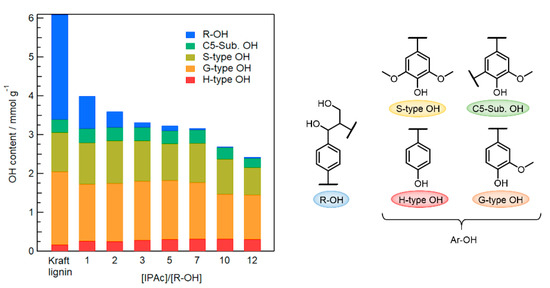
Figure 4.
Changes in contents of R–OH and Ar–OH (H/G/S-type and C5-Sub. OH) groups in Kraft lignin before and after acetylation using different amounts of IPAc (1–12 Equations/[R–OH]) at 80 °C for 1 h and subsequent selective deacetylation with added water (2 Equations/[Ar–OH]) at 80 °C for 1 h in an EmimOAc/DMSO mixed system.
2.5. Isolated Yield and Repeatability of the Reaction with Kraft Lignin
Under the optimized reaction conditions, the synthesis of selectively modified Kraft lignin, Lig-Ac (Selective), succeeded with a good isolated yield (~85%) and reproducibility (N = 3). During the one-pot two-steps reaction, Kraft lignin was completely dissolved in the EmimOAc/DMSO mixed system and sufficiently reacted. Thus, the obtained product should not have contained the unreacted material. In step (ii) for promoting selective deacetylation of the generated ArOAc, water (Scheme 5) was added into the reaction system. However, since the amount of water was very tiny, nothing was precipitated, which allowed that the selective deacetylation proceeded in totally homogeneous reaction. Therefore, the finally obtained water-precipitate is expected not to have contained fully acetylated Kraft lignin, either. The reason why the isolated yield of the Lig-Ac (Selective) was not reached to 100% might be because a part of the products dissolved in water and/or were uncollectable.
To clarify the advantage of the selective modification of R–OH groups with Ac groups in terms of physicochemical properties, fully modified Kraft lignin, Lig-Ac (Full), in which all OH groups were acetylated using excess IPAc, was also prepared as a reference. Table 1 shows the isolated yields, average OH contents, and molecular weight distributions of the Kraft lignin and selectively and fully modified Kraft lignin derivatives. The OH contents were estimated by quantitative 31P NMR analysis (see Supplementary Materials for the assignments in Table S1, and spectra in Figure S4) according to previous studies [37,38]. The Ar–OH content in Lig-Ac (Selective) was 3.2 mmol g−1, whereas the R–OH content was less than 0.1 mmol g−1. On the other hand, Lig-Ac (Full) had less than 0.1 mmol g−1 of both types of OH groups. These results strongly support that the selective modification of R–OH groups and full acetylation of both R–/Ar–OH groups succeeded, respectively.

Table 1.
Average yields, OH contents, and molecular weight distributions of Kraft lignin and acetyl derivatives.
There was no significant change in the weight average molecular weight (Mw) or polydispersity (Ð) between Lig-Ac (Selective) and the original Kraft lignin, suggesting that side reactions such as degradation and crosslinking did not occur during the mild one-pot two-step reaction. On the other hand, Mw of Lig-Ac (Full) decreased slightly (see details in Figure S5). The use of excess IPAc for the full acetylation of Kraft lignin could cause unexpected side reactions, and/or the apparent Mw could decrease due to a change in cohesiveness in the SEC eluent (0.01 mol L−1 LiBr/DMF solution).
2.6. Chemical Identification
To provide clear chemical structural information, Lig-Ac (Selective) and Lig-Ac (Full) were analyzed by ATR-mode FT-IR, 1H NMR, and 13C NMR and compared with Kraft lignin, as shown in Figure 5. The FT-IR spectrum of Lig-Ac (Selective) contains bands at 1589, 1513, 1426, 1144, and 851 cm−1 corresponding to the aromatic ring vibrations of the phenylpropane skeleton; [39] the bands for Lig-Ac (Full) were observed at 1593, 1508, 1420, 1124, and 852 cm−1, and those of Kraft lignin were observed at 1593, 1511, 1420, 1124, and 854 cm−1. The bands at 2939–2938 and 2839–2838 cm−1, which were observed in all samples, are related to the C–H vibration of CH2/CH3 groups and OMe groups, respectively [39]. The sharp band at 1032–1029 cm−1 arising mainly from C–O(C) stretching, which was also observed in all samples, is due to the C–O–C linkages in lignin molecular [40]. The large, broad band at approximately 3401 cm−1 due to O–H stretching in the FT-IR spectrum of Kraft lignin disappeared in the spectrum of Lig-Ac (Full), indicating that both R–OH and Ar–OH groups were acetylated. On the other hand, a broad band at approximately 3502 cm−1 was observed in the spectrum of Lig-Ac (Selective) corresponding to the retained Ar–OH groups. Notably, the strong bands at approximately 1221 and 1216 cm−1 in the spectra of Lig-Ac (Selective) and Lig-Ac (Full) correspond to the C–O(C) symmetric stretching of the C–O–C ester linkage in Ac groups. Furthermore, the bands at 1367 and 1368 cm−1 in both spectra arise from CH3 in Ac groups. Interestingly, Lig-Ac (Full) has C=O stretching bands at 1735 and 1761 cm−1, while Lig-Ac (Selective) shows only one peak at 1736 cm−1, suggesting that the band at 1736–1735 cm−1 arises from Ar–OAc groups. The distinct differences between the bands of Lig-Ac (Selective) and Lig-Ac (Full) indicate that the R–OH groups in Lig-Ac (Selective) were acetylated in the two-step reaction, while the Ar–OH groups remained mostly intact.
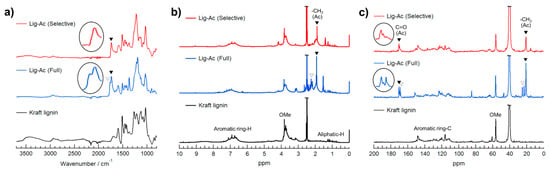
Figure 5.
(a) ATR-mode FT-IR, (b) 1H NMR, and (c) 13C NMR spectra of Lig-Ac (Selective), Lig-Ac (Full), and Kraft lignin.
The 1H NMR spectra (Figure 5b) contained broad peaks at 6.2–7.5 ppm from the aromatic skeleton of lignin and strong peaks at approximately 3.7 ppm from OMe groups. There were also sharp peaks at 1.7–2.1 ppm from CH3 in R–OAc groups and another peak at 2.1–2.4 ppm from CH3 in Ar–OAc groups in the spectrum of Lig-Ac (Full). The strong peak at 1.7–2.1 ppm from R–OAc groups was also observed in the spectrum of Lig-Ac (Selective), but the Ar–OAc peak was hardly present. These observations proved that the selective modification of R–OH groups in Kraft lignin succeeded.
For additional chemical investigation of Lig-Ac (Selective) and Lig-Ac (Full), 13C NMR analysis was performed. All the 13C NMR spectra (Figure 5c) contained several peaks in the aromatic carbon region from 100 to 150 ppm, strong peaks at approximately 56 ppm corresponding to OMe groups on G/S units, and characteristic aliphatic carbon peaks of β-O-4 linkages from 60 to 85 ppm. Notably, Lig-Ac (Full) showed two carbonyl carbon (C=O) peaks from Ac groups at 169.6 and 168.9 ppm, as well as a sharp peak at 20.6 ppm and two small peaks at 20.1 and 21.0 ppm, which correspond to the methyl carbons (CH3–) of Ac groups. On the other hand, Lig-Ac (Selective) showed single peaks at 168.2 and 20.6 ppm. The absence of the peaks in the Lig-Ac (Full) spectrum that might correspond to C=O and CH3– in Ar–OAc groups implies that the selective modification of R–OH groups in Lig-Ac (Selective) was achieved.
2.7. Solubility in Common Solvents
Considering the future applications of Kraft lignin derivatives, their solubility in common solvents is often necessary. Table 2 shows the measured solubilities of Lig-Ac (Selective), Lig-Ac (Full), and Kraft lignin in several solvents (see Supplementary Materials for the pictures in Figure S6). The original Kraft lignin hardly dissolved in common solvents except for DMSO, while Lig-Ac (Full) was also soluble in acetone and chloroform. Interestingly, Lig-Ac (Selective) dissolved in a chloroform/methanol mixed solvent (8:2, v/v), although it was insoluble in chloroform. Therefore, selective R–OH modification improved the limited solubility of the original Kraft lignin despite the lower substitution degree than that of Lig-Ac (Full).
Table 2.
Solubilities of Kraft lignin and acetyl derivatives.
2.8. Thermal Properties
To investigate the effects of selective R–OH modification in Kraft lignin on the thermal properties, the degradation temperature (Td), Tg, and the temperature at which heat flow begins (Tflow) were determined by thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and heat-flow tester, respectively, as shown in Figure 6. As shown in the TG curves in Figure 6a, Kraft lignin gradually degraded with heating, and the 1%-weight-loss-temperature (Td-1%) was 186 °C because the original Kraft lignin contains easily volatized impurities. On the other hand, Td-1% of Lig-Ac (Selective) increased to 215 °C, indicating its purity and acceptable thermal stability. Furthermore, the Td-1% of Lig-Ac (Selective) was higher than that of Lig-Ac (Full) at 201 °C. The higher Td could be attributable to the absence of Ar–OAc groups in Lig-Ac (Selective).
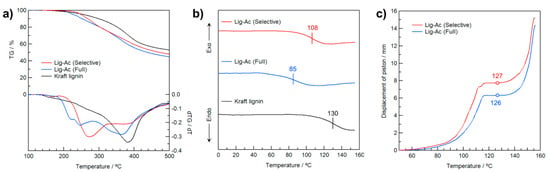
Figure 6.
(a) TG-DTG and (b) DSC curves of Lig-Ac (Selective), Lig-Ac (Full), and Kraft lignin and (c) thermal flow curves of the acetyl derivatives expressed as displacement of a piston in a capillary rheometer.
The residual weight of Kraft lignin (53 wt.%) after heating to 500 °C under an N2 atmosphere gradually decreased with increasing substituted Ac groups, with weights of 48 and 45 wt.% for Lig-Ac (Selective) and Lig-Ac (Full), respectively. The slightly higher residual weight of Lig-Ac (Selective) might be due to its fewer Ac groups than Lig-Ac (Full) because the elimination of Ac groups and subsequent volatilization are much easier than the thermal degradation of the aromatic backbone of lignin itself.
Figure 6b shows the DSC curves of Lig-Ac (Selective), Lig-Ac (Full), and Kraft lignin in the 2nd heating. The DSC curve of the original Kraft lignin showed a step in the baseline corresponding to Tg at 130 °C. This broad transition with increasing temperature is consist with previous results [41] Tg decreased with increasing substitution degree of Ac groups, reaching 108 and 85 °C for Lig-Ac (Selective) and Lig-Ac (Full), respectively. Considering Td and Tg, Lig-Ac (Selective) is advantageous in terms of thermal stability compared with Lig-Ac (Full) and has the potential for further functionalization utilizing the rich Ar–OH groups.
To further investigate the effect of the selective modification of Kraft lignin on its thermal processability, Lig-Ac (Selective) and Lig-Ac (Full) were subjected to heat-flow testing in a capillary rheometer. In Figure 6c, the heat-flow curve of each sample is depicted as the change in displacement of a piston on the resin packed into a cylinder and gradually heated (see Supplementary Materials for heat-flow tester diagrams in Figure S7). Since the original Kraft lignin is not heat-meltable, this measurement was not applicable. On the other hand, Lig-Ac (Full) began heat flow at 126 °C due to the sufficiently high degree of acetylation. Surprisingly, Lig-Ac (Selective) showed a similar heat-flow behavior, with Tflow of 127 °C. Based on these results, selective R–OH modification in Kraft lignin provides thermal fluidity with an excellent processing window (i.e., difference between Tflow and Td), and the lower degree of substitution of Lig-Ac (Selective) contributes to a higher Tg.
3. Materials and Methods
3.1. Materials
1-Ethyl-3-methyl-imidazolium acetate (EmimOAc) was obtained from Kanto Chemical Co., Inc. (Tokyo, Japan) used without further purification. Kraft lignin ([R–OH]: 2.7 mmol g−1, [Ar–OH]: 3.4 mmol g−1) was procured from Sigma-Aldrich Co., LLC. (St. Louis, MO, USA) and dried under vacuum at 70 °C until a constant weight before use. Isopropenyl acetate (IPAc) and anhydrous dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich Co., LLC. (St. Louis, MO, USA) and used as received. DMSO-d6 (99.9 atom% D with 0.03 vol% TMS) was purchased from Kanto Chemical Co., Inc. (Tokyo, Japan), and phenol (PhOH), 2-phenylethanol (Ph–EtOH), guaiacol (GuOH), 2,6-dimethoxyphenol (syringol, SyOH), phenyl acetate (PhOAc), and 2-phenethyl acetate (Ph–EtOAc) were purchased from Tokyo Chemical Industry Co., LTD. (Tokyo, Japan) and always stored and used in the grove box. Other chemicals were commercially available and used as received unless otherwise stated.
3.2. In Situ 1H NMR Analysis in Transesterification of Model Compounds
EmimOAc was previously vacuum dried at 80 °C for over 24 h and transferred into the glove box filled with Ar gas and less than 10 ppm of water. A determined amount of the dried EmimOAc (1 or 10 Equations/[R–OH]) was weighted into a 9 mL vial and dissolved in DMSO-d6 (1.5 mL). The low molecular weight model compounds (e.g., Ph–EtOH, PhOH; 0.6 mmol) and IPAc (1 or 3 Equations/[OH]) were added into the vial. After shaking the reaction solution by hand, it was transferred into an NMR test tube. The tube was closed with a plastic cap and tightly wrapped by a parafilm. Then, it was taken out from the glove box, and 1H NMR of the resultant solution was immediately measured at room temperature (r.t., 22 °C) in 80 scans. The obtained 1H NMR spectrum was regarded as the reaction time: 0 min. After the first measurement, the test tube was heated to 80 °C in the NMR spectrometer. After the prove tuning and re-gradient shimming, 1H NMR was measured again at 80 °C in 80 scans, and the obtained spectrum was regarded as the reaction time: 10 min. Subsequently, the 1H NMR spectra were repeatedly measured in the predetermined period in the similar manner. The concentration of each model compound and product was estimated by the integration ratio of the corresponded peaks of the proton.
3.3. Selective R–OH Modification in Kraft Lignin
Kraft lignin (1.8 g, 6 wt.%/EmimOAc]) in EmimOAc (30 g) was dried under vacuum at 80 °C for 16 h to remove moisture. After DMSO (45 mL) was added to the solution under an Ar atmosphere, the resultant mixture was stirred at 110 °C for 2 h for complete dissolution of the Kraft lignin. The obtained clear blackish-brown solution was moved to another oil bath at 80 °C and stirred for 30 min. It was further stirred for 1 h at 80 °C with a determined amount of IPAc (1–12 Equations/[R–OH]), which was performed as a step (i) for acetylation of both R–OH and Ar–OH groups of Kraft lignin. Then, a small amount of distilled water (0.22 mL, 2 Equations/[Ar–OH]) was added to the reacted solution by a micro syringe, and it was further stirred for 1 h at 80 °C. The latter reaction was regarded as a step (ii) for selective deacetylation of the generated Ar–OAc groups during the step (i). Notably, the amount of Ar–OH groups (3.4 mmol g−1) in Kraft lignin was higher than that of R–OH groups (2.7 mmol g−1), implying that the proton source was insufficient for deacetylation of all the Ar–OAc groups. Then, the water was subsequently added in the step (ii). Carefully, excess water can inhibit the catalytic activity of EmimOAc in transesterification of OH groups and IPAc, and so the acetylation of OH groups and selective deacetylation of the Ar–OAc groups were performed in two steps with adding IPAc and water, in order. After quickly cooling of the reaction solution using ice bath to quench the reaction, it was poured into 1.6 L of distilled water to precipitate the light brown particles. The precipitated polymer was collected by vacuum filtration, and repeatedly washed with distilled water. After freeze-drying for 2 days, it was vacuum dried at 70 °C for over 24 h to afford selectively modified Kraft lignin, Lig-Ac (Selective).
3.4. Complete Acetylation Kraft Lignin
Kraft lignin (1.8 g, 6 wt.%/EmimOAc) in EmimOAc (30 g) was dried under vacuum at 80 °C for 16 h to remove moisture. After DMSO (45 mL) was added to the solution under an Ar atmosphere, the resultant mixture was stirred at 110 °C for 2 h for complete dissolution of the Kraft lignin. The obtained clear blackish-brown solution was moved to another oil bath at 80 °C and stirred for 30 min. An excess amount of IPAc (50 Equations/[Total OH]) was applied to complete acetylation of both R–OH and Ar–OH groups within Kraft lignin, and the reaction was performed at 80 °C for 1 h. After quickly cooling of the reaction solution using ice bath to quench the reaction, it was poured into 1.6 L of distilled water to precipitate the light brown particles. The precipitated polymer was collected by vacuum filtration, and repeatedly washed with distilled water. After freeze-drying for 2 days, it was vacuum dried at 70 °C for over 24 h to afford fully acetylated Kraft lignin, Lig-Ac (Full).
3.5. Nuclear Magnetic Resonance (NMR) Spectroscopy
The 1H, 13C, and 31P NMR spectra in solution were recorded using a JNM-ECA 600 spectrometer (JEOL Ltd., Tokyo, Japan) in deuterated solvents at Advanced Research Center in Kanazawa University. All NMR spectra were analyzed using Delta NMR software (JEOL Ltd., Tokyo, Japan), and the chemical shifts (δ, ppm) were referenced to tetramethylsilane (TMS, δ = 0 ppm) as an internal standard.
The contents of OH groups in the Kraft lignin and the acetyl derivatives were estimated by quantitative 31P NMR analysis by the phosphitylation of the samples, according to previous studies [37,38]. The used NMR spectrometer was the same JNM-ECA 600 spectrometer, but different internal standards (ISs) of N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (IS-1, 10 g L−1) and cyclohexanol (IS-2, 10 g L−1) were applied with a relaxation reagent of tris(2,4-pentanedionato)-chromium (III) (5 g L−1) in CDCl3/pyridine mixed solvent. The inverse gated 1H decoupling sequence was employed with a recycle delay of 25 s. The free induction decays of 128 were collected and averaged to obtain each 31P spectrum. The phosphitylation reaction was conducted in the same manner at room temperature for 15 min as our previous report [29] using 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholate (TMDP) which is a common phosphitylation reagent.
3.6. Size Exclusion Chromatography (SEC)
The molecular weights of the Kraft lignin and the acetyl derivatives were determined by SEC (UFLC system, Shimadzu Co., Kyoto, Japan) based on polystyrene standards. All the SEC measurements were carried out at 40 °C using TSK gel a-M (Tosoh Co., Tokyo, Japan), and 0.01 mol L−1 LiBr in dimethylformamide (DMF, HPLC grade, Kanto Chemicals Co., Inc., Tokyo, Japan) was used as an eluent at the flow rate of 1.0 mL min−1.
3.7. Fourier Transform Infrared (FT-IR)
FT-IR spectra were recorded on a Thermo Fisher Scientific Nicolet IS10 spectrometer (Thermo Fisher Scientific Inc., Tokyo, Japan) equipped with an attenuated total reflection (ATR) unit: Measurement range, 4000–400 cm−1, number of scans, 64, resolution, 4 cm−1, S/N, 35,000:1.
3.8. Solubility Test
The solubility of the Kraft lignin and the acetyl derivatives in common solvents were investigated by a brief test. All samples were vacuum dried at 70 °C for 24 h before the test. The dried sample (10 mg) was weighted into a 1 mL vial, and various solvents were added, respectively. The solubility was observed by visual confirmation after standing at room temperature for 24 h.
3.9. Thermogravimetric Analysis (TGA)
TGA was performed by a DTG-60AH (Shimadzu Co., Kyoto, Japan) equipped with a gas-flow controller (FC-60A, Shimadzu Co., Kyoto, Japan) and an analysis system (TA-60, Shimadzu Co., Kyoto, Japan). All samples were vacuum dried at 70 °C for 24 h prior to use, and the measurement temperature range was settled from 50 to 500 °C at a heating rate of 10 °C min−1 in a N2-flow rate of 50 mL min-1. A sample of about 10 mg was used with pre-drying at 120 °C for 2 h. The thermal decomposition temperature (Td) was taken as the onset of 1%-weight-loss (Td-1%).
3.10. Differential Scanning Calorimetry (DSC)
DSC was performed using a DSC-60A plus (Shimadzu Co., Kyoto, Japan) apparatus. Prior to DSC measurements, all samples were prepared in a glove box under Ar atmosphere after vacuum drying at 70 °C for 24 h. Each sample was first cooled to −100 °C and then heated at a scanning rate of 20 °C min−1 to +170 °C, which was maintained at 170 °C for 3 min (first heating scan) and immediately quenched to −100 °C at a rate of −50 °C min−1. The second heating scans were run from −100 °C to +170 °C at a scanning rate of 20 °C min−1 to record stable thermograms.
3.11. Heat Flow Tester
Heat-flow tester, a capillary rheometer (CFT-500EX, Shimadzu Corporation, Kyoto, Japan), was used to measure the temperature at which heat flow begins (Tflow). Prior to the measurement, all samples were vacuum dried at 70 °C for 24 h. The dried sample (1 g) was loaded into the barrel and roughly pressed with a piston (diameter, 10 mm). After preheating at 50 °C for 300 s, a constant pressure of 4.9 MPa was applied on the piston and heating was started at a heating rate of 3 °C min−1. The temperature where the sample was extruded from an equipped die (diameter, 1.0 mm and length, 10 mm) under the barrel was defined as Tflow.
4. Conclusions
In situ 1H NMR analyses of the transesterification of several model compounds with R–OH and Ar–OH groups revealed that an ionic liquid, EmimOAc, catalyzes the acetylation of both types of OH groups with isopropenyl acetate (IPAc) as an acyl donor and the selective deacetylation of Ar–OAc groups. The selective deacetylation was found to arise from the transesterification of Ar–OAc groups and remaining R–OH groups, and the reaction was faster than the transesterification of R–OH groups with IPAc. Analyses suggested that these reactions enabled the facile selective modification of R–OH groups depending on the combined amount of IPAc and EmimOAc. Applying the optimized conditions determined using model reactions considering the effects of electron-donating OMe groups at the o-positions of Ar–OH groups within the lignin structure, the R–OH groups in Kraft lignin were selectively modified with Ac groups. The process was performed in a one-pot two-step reaction at 80 °C for a total of 2 h using IPAc followed by the addition of water as a proton source to promote Ar–OAc deacetylation. The resulting selectively modified Kraft lignin, Lig-Ac (Selective), was found to dissolve in DMSO and a chloroform/methanol mixed solvent and had a higher Tg at 108 °C and Td-1% at 215 °C than those of a reference sample of fully modified Kraft lignin, Lig-Ac (Full). Furthermore, despite the lower substitution degree, Lig-Ac (Selective) showed a similar heat-flowing temperature of 126 °C to that of Lig-Ac (Full). The selective modification of R–OH groups in Kraft lignin and resulting solubility, thermal stability, and heat-meltability warrant further functionalization, such as polymerization via the rich Ar–OH groups for lignin-derived polymer applications. Therefore, this facile and sustainable method for selective R–OH modification paves the way for the valorization of Kraft lignin as a functional polymeric material.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/11/1/120/s1, Figure S1: Conversions of Ph–EtOH (0.38 mol L−1) in transesterification reaction with different acyl donors of PhOAc or IPAc (each 0.38 mol L−1) for 12 h at 80 °C catalyzed by EmimOAc (0.38 mol L−1), as determined by in situ 1H NMR in DMSO-d6, Figure S2: Changes in the relative contents of PhOH and Ph–EtOH (each 0.38 mol L−1) in transesterification of the mixture with excess IPAc (1.14 mol L−1) for 10 h at 80 °C catalyzed by EmimOAc (0.38 mol L−1), compared with those in transesterification of individual PhOH or Ph–EtOH under the same condition, as determined by in situ 1H NMR in DMSO-d6, Figure S3: OH contents of R–OH and Ar–OH (H/G/S-types and C5-Sub. OH) groups in Kraft lignin before and after acetylation using IPAc (12 Equation/[R–OH]) at 80 °C for 1 h and subsequent selective deacetylation with added water (2 Equation/[Ar–OH]) at 80 °C for 0−24 h in an EmimOAc/DMSO mixed system, Figure S4: Quantitative 31P NMR spectra of the phosphitylated Lig-Ac (Selective), Lig-Ac (Full), and Kraft lignin measured in CDCl3/pyridine mixed solvent using two kinds of ISs and a relaxation reagent, Figure S5: Molecular weight distributions of Lig-Ac (Selective), Lig-Ac (Full), and Kraft lignin measured by SEC in 0.01 mol L−1 LiBr/DMF solution, Figure S6: Pictures of solutions of Lig-Ac (Selective), Lig-Ac (Full), and Kraft lignin in chloroform/methanol (8:2, v/v) solution, Figure S7: Heat-flow tester diagrams, Table S1: OH contents of the used Kraft lignin and the assignments in quantitative 31P NMR.
Author Contributions
S.S. designed the experiments and wrote the manuscript; K.T. supervised study; S.S. and S.K. performed the experiments; S.S., N.W., K.T. contributed to scientific discussions. All authors have read and agreed to the published version of the manuscript.
Funding
The authors gratefully thank the financial support received through the Center of Innovation Science and Technology based Radical Innovation and Entrepreneurship Program (COI stream, (JPMJCE1315)) aimed at the “Construction of next generation infrastructure using innovative materials: Realization of a safe and secure society that can coexist with the Earth for centuries”, supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Japan Science and Technology Agency (JST). Also, this work was carried out as part of a Grant-in-Aid for the Japan Society for the Promotion of Science (JSPS) Research Fellow (No. 19J00642).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no competing financial interest.
References
- Stewart, D. Lignin as a base material for materials applications: Chemistry, application and economics. Ind. Crops Prod. 2008, 27, 202–207. [Google Scholar] [CrossRef]
- Norgren, M.; Edlund, H. Lignin: Recent advances and emerging applications. Curr. Opin. Colloid Interface Sci. 2014, 19, 409–416. [Google Scholar] [CrossRef]
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crops Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Nishimura, H.; Kamiya, A.; Nagata, T.; Katahira, M.; Watanabe, T. Direct evidence for α ether linkage between lignin and carbohydrates in wood cell walls. Sci. Rep. 2018, 8, 6538. [Google Scholar] [CrossRef]
- Dessbesell, L.; Paleologou, M.; Leitch, M.; Pulkki, R.; Xu, C. Global lignin supply overview and kraft lignin potential as an alternative for petroleum-based polymers. Renew. Sustain. Energy Rev. 2020, 123, 109768. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef]
- Wells, T., Jr.; Kosa, M.; Ragauskas, A.J. Polymerization of Kraft lignin via ultrasonication for high-molecular-weight applications. Ultrason. Sonochem. 2013, 20, 1463–1469. [Google Scholar] [CrossRef]
- Olivares, M.; Guzmán, J.A.; Natho, A.; Saavedra, A. Kraft lignin utilization in adhesives. Wood Sci. Technol. 1988, 22, 157–165. [Google Scholar] [CrossRef]
- Sakai, H.; Kuroda, K.; Muroyama, S.; Tsukegi, T.; Kakuchi, R.; Takada, K.; Hata, A.; Kojima, R.; Ogoshi, T.; Omichi, M.; et al. Alkylated alkali lignin for compatibilizing agents of carbon fiber-reinforced plastics with polypropylene. Polym. J. 2018, 50, 281–284. [Google Scholar] [CrossRef]
- Zhang, M.; Ogale, A.A. Carbon fibers from dry-spinning of acetylated softwood kraft lignin. Carbon 2014, 69, 626–629. [Google Scholar] [CrossRef]
- Solt, P.; van Herwijnen, H.W.G.; Konnerth, J. Thermoplastic and moisture-dependent behavior of lignin phenol formaldehyde resins. J. Appl. Polym. Sci. 2019, 136, 48011. [Google Scholar] [CrossRef]
- Yoshida, H.; Mörck, R.; Kringstad, K.P.; Hatakeyama, H. Kraft lignin in polyurethanes. II. Effects of the molecular weight of kraft lignin on the properties of polyurethanes from a kraft lignin–polyether triol–polymeric MDI system. J. Appl. Polym. Sci. 1990, 40, 1819–1832. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Wyman, C.A.; Cai, C.M.; Ragauskas, A.J. Lignin-based polyurethanes from unmodified kraft lignin fractionated by sequential precipitation. ACS Appl. Polym. Mater. 2019, 1, 1672–1679. [Google Scholar] [CrossRef]
- Gioia, C.; Colonna, M.; Tagami, A.; Medina, L.; Sevastyanova, O.; Berglund, L.A.; Lawoko, M. Lignin-based epoxy resins: Unravelling the relationship between structure and material properties. Biomacromolecules 2020, 21, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Thanh Binh, N.T.; Luong, N.D.; Kim, D.O.; Lee, S.H.; Kim, B.J.; Lee, Y.S.; Nam, J.D. Synthesis of lignin-based thermoplastic copolyester using kraft lignin as a macromonomer. Compos. Interfaces 2009, 16, 923–935. [Google Scholar] [CrossRef]
- Li, Y.; Sarkanen, S. Alkylated Kraft lignin-based thermoplastic blends with aliphatic polyesters. Macromolecules 2002, 35, 9707–9715. [Google Scholar] [CrossRef]
- Li, Y.; Sarkanen, S. Miscible blends of Kraft lignin derivatives with low-Tg polymers. Macromolecules 2005, 38, 2296–2306. [Google Scholar] [CrossRef]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the structure of softwood Kraft lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
- Liu, L.Y.; Hua, Q.; Renneckar, S. A simple route to synthesize esterified lignin derivatives. Green Chem. 2019, 21, 3682–3692. [Google Scholar] [CrossRef]
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Koivu, K.A.Y.; Sadeghifar, H.; Nousiainen, P.A.; Argyropoulos, D.S.; Sipilä, J. Effect of fatty acid esterification on the thermal properties of softwood Kraft lignin. ACS Sustain. Chem. Eng. 2016, 4, 5238–5247. [Google Scholar] [CrossRef]
- Guo, Z.X.; Gandini, A.; Pla, F. Polyesters from lignin. 1. The reaction of Kraft lignin with dicarboxylic acid chlorides. Polym. Int. 1992, 27, 17–22. [Google Scholar] [CrossRef]
- Han, T.; Sophonrat, N.; Evangelopoulos, P.; Persson, H.; Yang, W.; Jönsson, P. Evolution of sulfur during fast pyrolysis of sulfonated Kraft lignin. J. Anal. Appl. Pyrol. 2018, 133, 162–168. [Google Scholar] [CrossRef]
- Hallett, J.P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Szalaty, T.J.; Klapiszewski, Ł.; Stanisz, M.; Moszyński, D.; Skrzypczak, A.; Jesionowski, T. Catalyst-free activation of kraft lignin in air using hydrogen sulfate ionic liquids. Int. J. Biol. Macromol. 2018, 119, 431–437. [Google Scholar] [CrossRef]
- Szalaty, T.J.; Klapiszewski, Ł.; Jesionowski, T. Recent developments in modification of lignin using ionic liquids for the fabrication of advanced materials—A review. J. Mol. Liq. 2020, 301, 112417. [Google Scholar] [CrossRef]
- Swatloski, R.P.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Dissolution of cellose with ionic liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. [Google Scholar] [CrossRef]
- Fu, D.; Mazza, G.; Tamaki, Y. Lignin extraction from straw by ionic liquids and enzymatic hydrolysis of the cellulosic residues. J. Agric. Food Chem. 2010, 58, 2915–2922. [Google Scholar] [CrossRef]
- Suzuki, S.; Ishikuro, A.; Hirose, D.; Ninomiya, K.; Takahashi, K. Dual catalytic activity of an ionic liquid in lignin acetylation and deacetylation. Chem. Lett. 2018, 47, 860–863. [Google Scholar] [CrossRef]
- Suzuki, S.; Ishikuro, A.; Hamano, Y.; Hirose, D.; Wada, N.; Takahashi, K. Understanding and suppression of side reaction during transesterification of phenolic hydroxyl groups of lignin with vinyl ester. Chem. Lett. 2020, 49, 900–904. [Google Scholar] [CrossRef]
- Nguyen, Q.V.; Nomura, S.; Hoshino, R.; Ninomiya, K.; Takada, K.; Kakuchi, R.; Takahashi, K. Recyclable and scalable organocatalytic transesterification of polysaccharides in a mixed solvent of 1-ethyl-3-methylimidazolium acetate and dimethyl sulfoxide. Polym. J. 2017, 49, 783–787. [Google Scholar] [CrossRef]
- Nahmany, M.; Melman, A. Chemoselectivity in reactions of esterification. Org. Biomol. Chem. 2004, 2, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Ennis, E.; Handy, S.T. The chemistry of the C2 position of imidazolium room temperature ionic liquids. Curr. Org. Synth. 2007, 4, 381–389. [Google Scholar] [CrossRef]
- Kakuchi, R.; Ito, R.; Nomura, S.; Abroshan, H.; Ninomiya, K.; Ikai, T.; Maeda, K.; Kim, H.J.; Takahashi, K. A mechanistic insight into the organocatalytic properties of imidazolium-based ionic liquids and a positive co-solvent effect on cellulose modification reactions in an ionic liquid. RSC Adv. 2017, 7, 9423–9430. [Google Scholar] [CrossRef]
- Chen, M.J.; Li, R.M.; Zhang, X.Q.; Feng, J.; Feng, J.; Liu, C.F.; Shi, Q.S. Homogeneous transesterification of sugar cane bagasse toward sustainable plastics. ACS Sustain. Chem. Eng. 2017, 5, 360–366. [Google Scholar] [CrossRef]
- Hirose, D.; Wardhana Kusuma, S.B.; Nomura, S.; Yamaguchi, M.; Yasaka, Y.; Kakuchi, R.; Takahashi, K. Effect of anion in carboxylate-based ionic liquids on catalytic activity of transesterification with vinyl esters and the solubility of cellulose. RSC Adv. 2019, 9, 4048–4053. [Google Scholar] [CrossRef]
- Granata, A.; Argyropoulos, D.S. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a reagent for the accurate determination of the uncondensed and condensed phenolic moieties in lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]
- Pu, Y.; Cao, S.; Ragauskas, A.J. Application of quantitative 31P NMR in biomass lignin and biofuel precursors characterization. Energy Environ. Sci. 2011, 4, 3154–3166. [Google Scholar] [CrossRef]
- Tejado, A.; Peña, C.; Labidi, J.; Echeverria, J.M.; Mondragon, I. PhysicO–Chemical characterization of lignins from different sources for use in phenol–formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. [Google Scholar] [CrossRef]
- Schwanninger, M.; Rodrigues, J.C.; Pereira, H.; Hinterstoisser, B. Effects of short-time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose. Vib. Spectrosc. 2004, 36, 23–40. [Google Scholar] [CrossRef]
- Cui, C.; Sadeghifar, H.; Sen, S.; Argyropoulos, D.S. Toward thermoplastic lignin polymers; Part II: Thermal & polymer characteristics of Kraft lignin & derivatives. BioResources 2013, 8, 864–886. [Google Scholar]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).