Gas-Phase TiO2 Photosensitized Mineralization of Some VOCs: Mechanistic Suggestions through a Langmuir–Hinshelwood Kinetic Approach


 ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
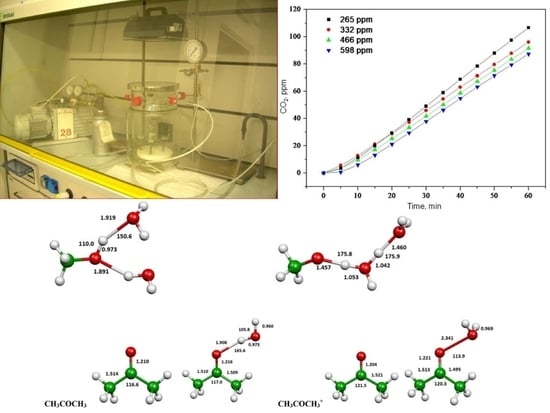
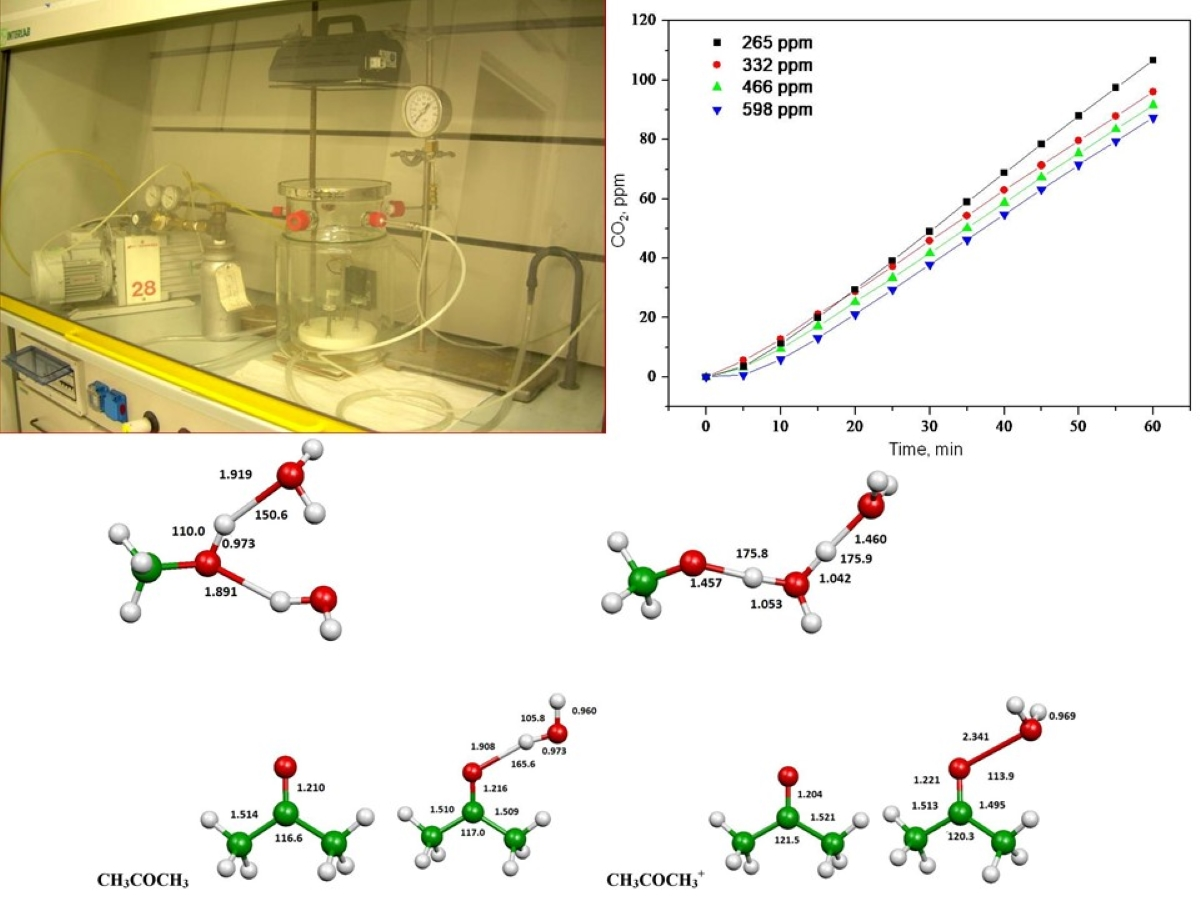
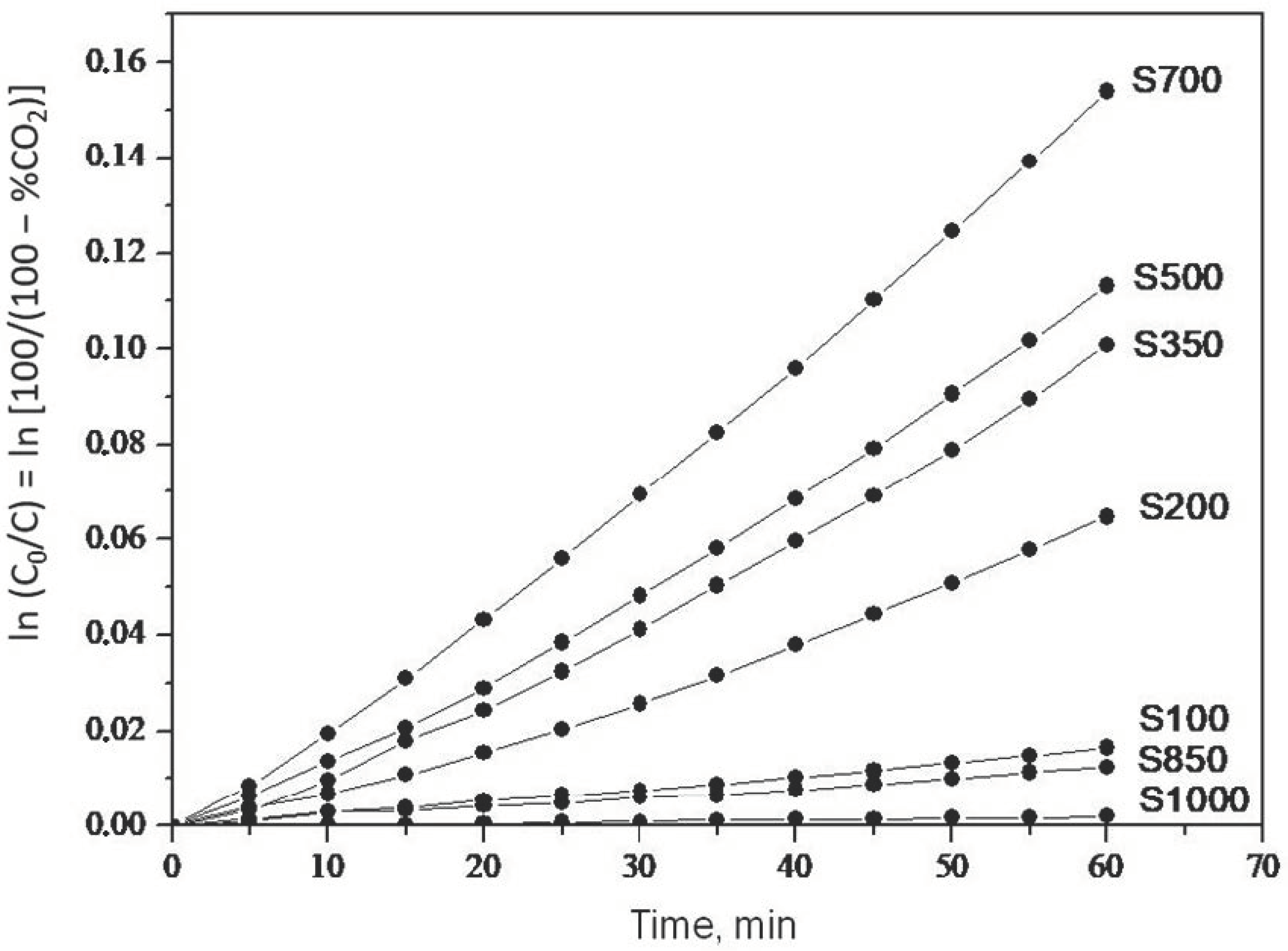
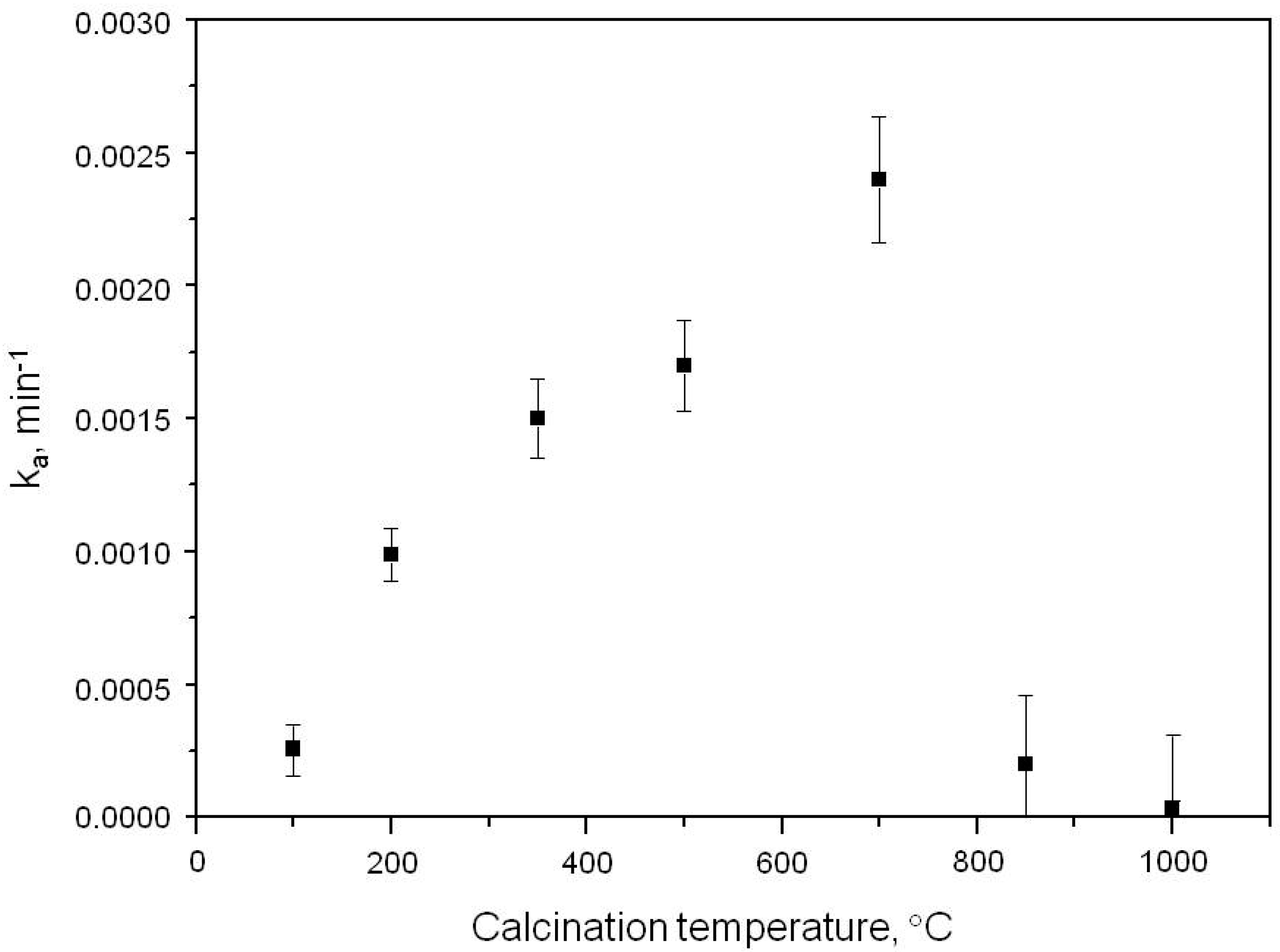
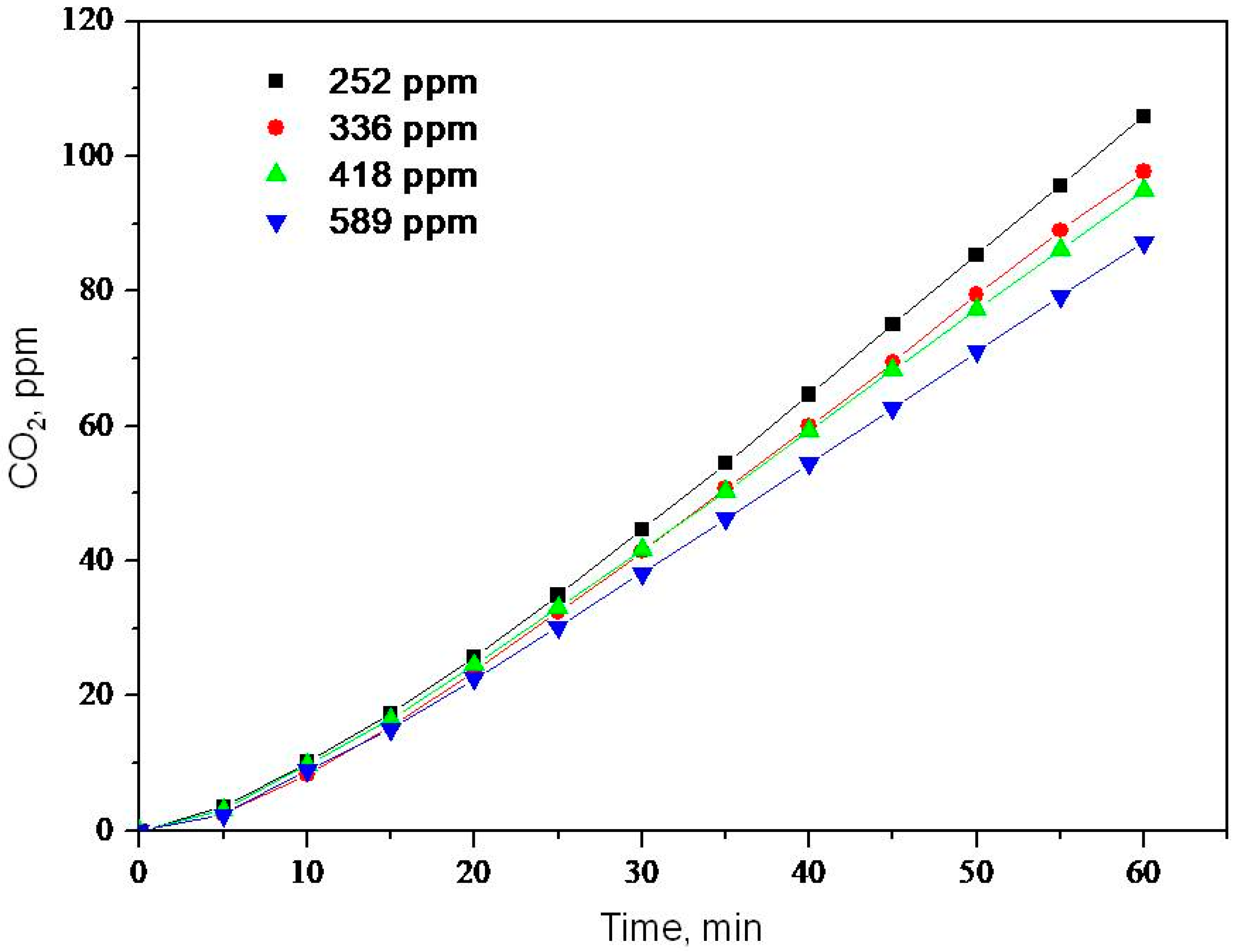
2.1. Photooxidation of Hexane with Different Synthetic TiO2 Powders
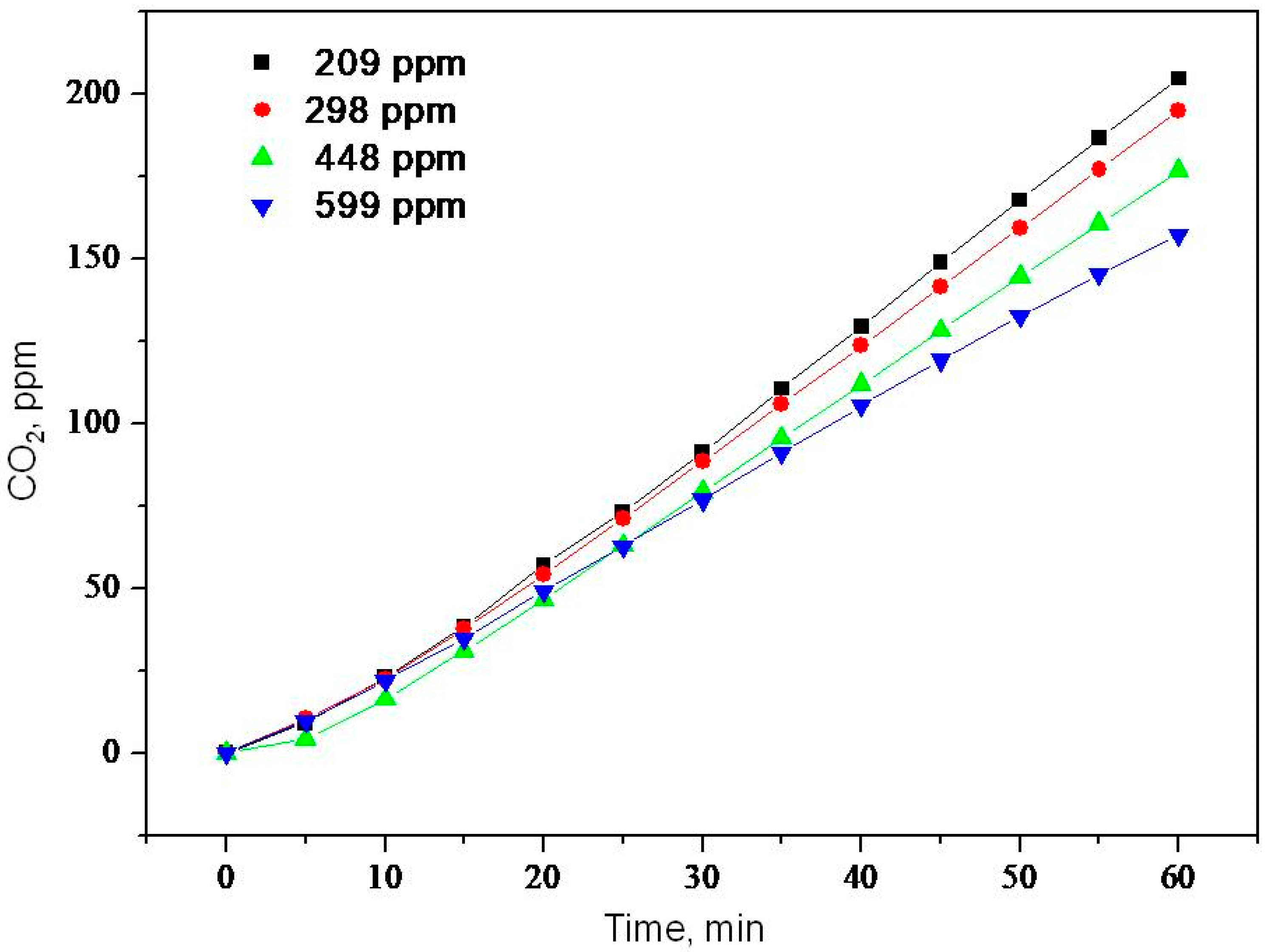
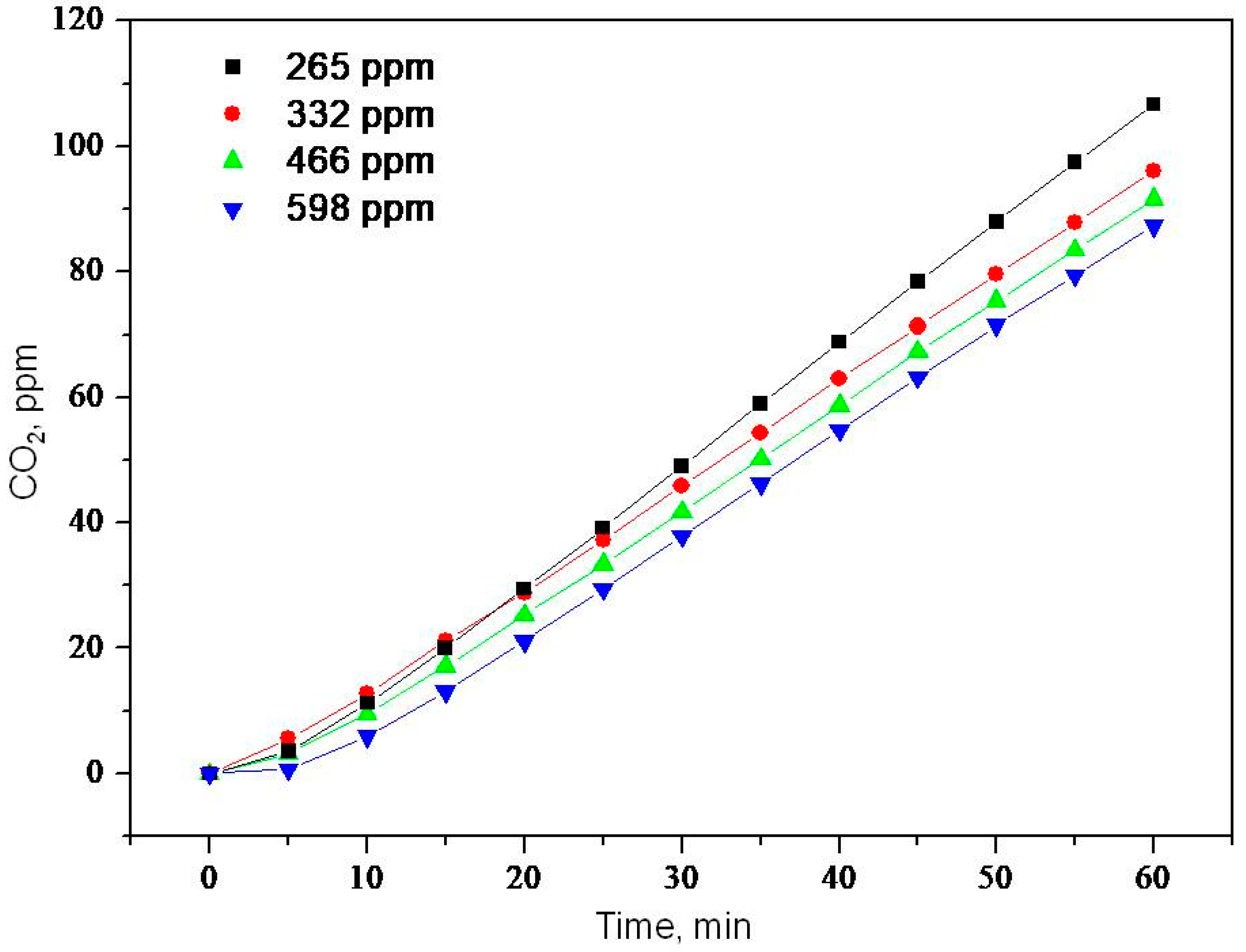
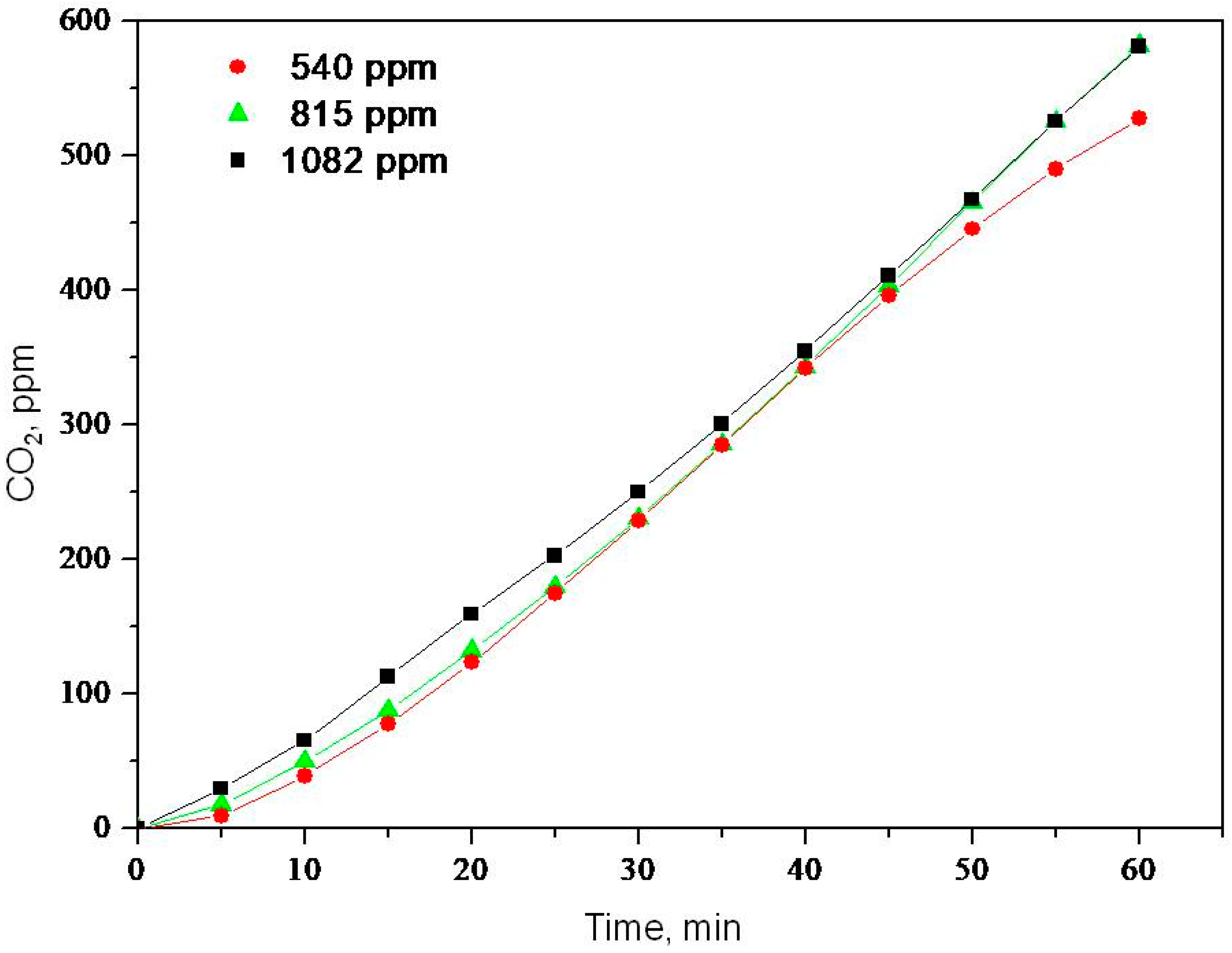
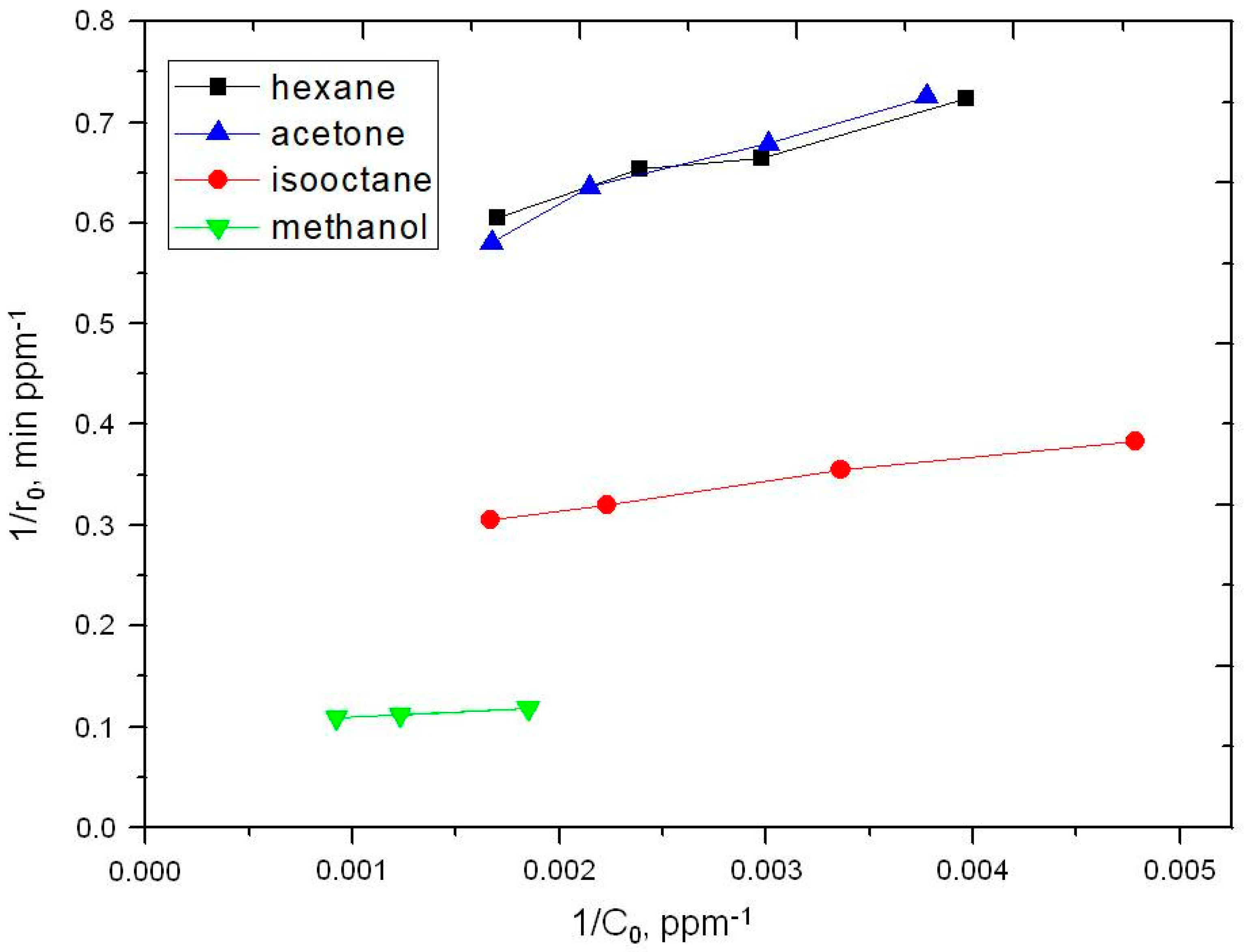
2.2. Photooxidation of Methane, Hexane, Isooctane, Acetone and Methanol with TiO2 P25
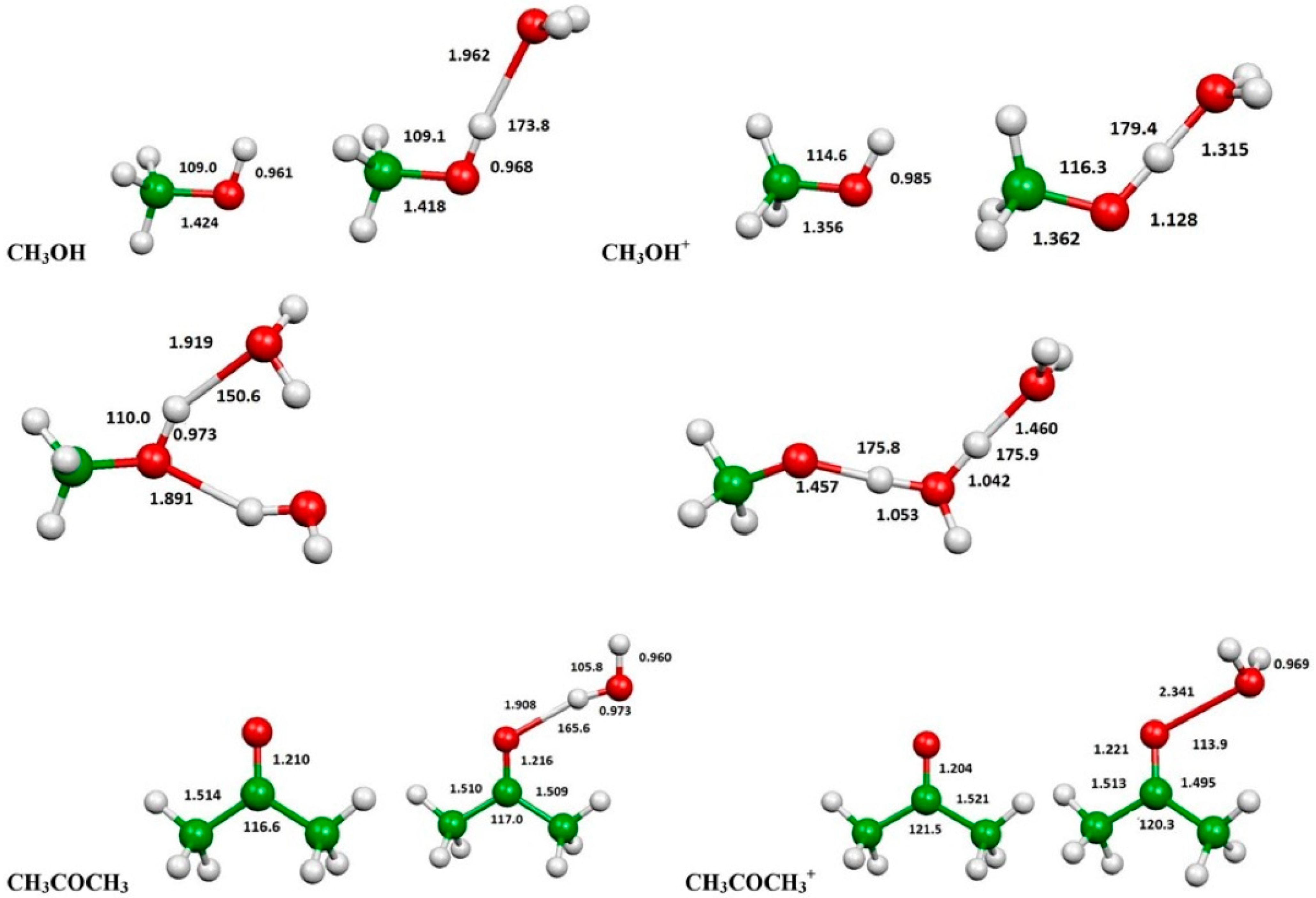
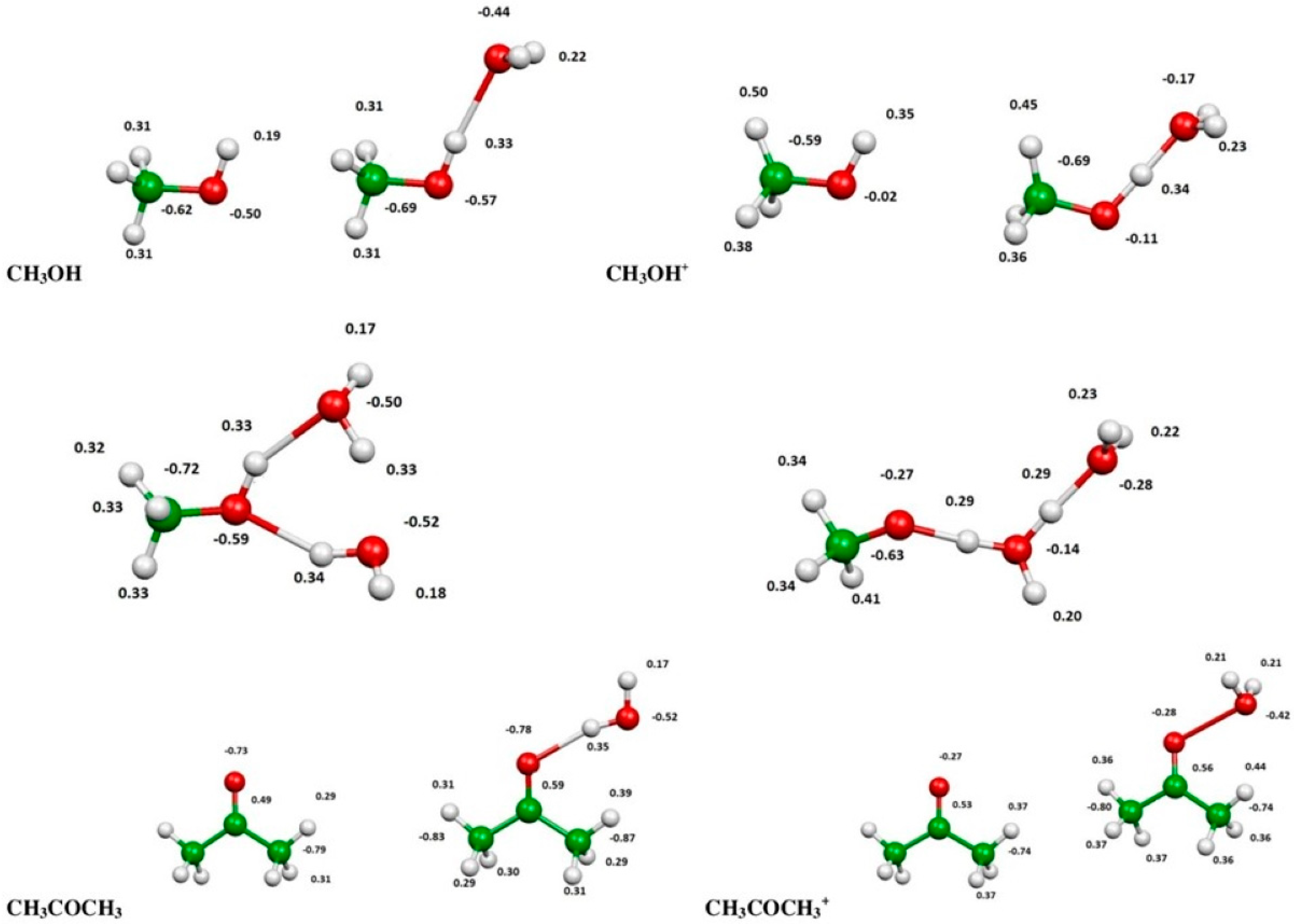
2.3. Evaluation of Ionization Potentials
3. Materials and Methods
3.1. Materials
3.2. Synthesis of TiO2 Powders
3.3. Experimental Apparatus
3.4. Photodegradation of Methane, Hexane, Isooctane, Acetone and Methanol in the Presence of TiO2
3.5. Product Analysis
3.6. Computational Evaluation of Ionization Potentials
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shayegan, Z.; Lee, C.S.; Haghighat, F. TiO2 photocatalyst for removal of volatile organic compounds in gas phase—A review. Chem. Eng. J. 2018, 334, 2408–2439. [Google Scholar] [CrossRef]
- Verbruggen, S.W. TiO2 photocatalysis for the degradation of pollutants in gas phase: From morphological design to plasmonic enhancement. J. Photochem. Photobiol. C Photochem. Rev. 2015, 24, 64–82. [Google Scholar] [CrossRef]
- Paz, Y. Application of TiO2 photocatalysis for air treatment: Patents’ overview. Appl. Catal. B Environ. 2010, 99, 448–460. [Google Scholar] [CrossRef]
- Tseng, T.K.; Lin, Y.S.; Chen, Y.J.; Chu, H. A Review of Photocatalysts Prepared by Sol-Gel Method for VOCs Removal. Int. J. Mol. Sci. 2010, 11, 2336–2361. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Zhang, Y.; Xu, Q.; Lamson, J.J.; Zhao, R. Photocatalytic purification of volatile organic compounds in indoor air: A literature review. Atmos. Environ. 2009, 43, 2229–2246. [Google Scholar] [CrossRef]
- Gaya, U.I.; Abdullah, A.H. Heterogeneous photocatalytic degradation of organic contaminants over titanium dioxide: A review of fundamentals, progress and problems. J. Photochem. Photobiol. C Photochem. Rev. 2008, 9, 1–12. [Google Scholar] [CrossRef]
- Wang, S.; Ang, H.M.; Tade, M.O. Volatile organic compounds in indoor environment and photocatalytic oxidation: State of the art. Eviron. Int. 2007, 33, 694–705. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, X. Photocatalytic oxidation for indoor air purification: A literature review. Build. Environ. 2003, 8, 645–654. [Google Scholar] [CrossRef]
- Peral, J.; Domènech, X.; Ollis, D.F. Heterogeneous Photocatalysis for Purification, Decontamination and Deodorization of Air. J. Chem. Technol. Biotechnol. 1997, 70, 117–140. [Google Scholar] [CrossRef]
- Kim, S.B.; Hong, S.C. Kinetic study for photocatalytic degradation of volatile organic compounds in air using thin film TiO2 photocatalyst. Appl. Catal. B Environ. 2002, 35, 305–315. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Alberici, R.M.; Jardim, W.F. Photocatalytic destruction of VOCs in the gas-phase using titanium dioxide. Appl. Catal. B Environ. 1997, 14, 55–68. [Google Scholar] [CrossRef]
- Deng, X.; Yue, Y.; Gao, Z. Gas-phase photo-oxidation of organic compounds over nanosized TiO2 photocatalysts by various preparations. Appl. Catal. B Environ. 2002, 39, 135–147. [Google Scholar] [CrossRef]
- Boulamanti, A.K.; Philippopoulos, C.J. Photocatalytic degradation of C5-C7 alkanes in the gas-phase. Atmos. Environ. 2009, 43, 3168–3174. [Google Scholar] [CrossRef]
- Stengl, V.; Houskova, V.; Bakardjieva, S.; Murafa, N. Photocatalytic degradation of acetone and butane on mesoporous titania layers. New J. Chem. 2010, 34, 1999–2005. [Google Scholar] [CrossRef]
- Zorn, M.E.; Hay, S.O.; Anderson, M.A. Effect of molecular functionality on the photocatalytic oxidation of gas-phase mixtures. Appl. Catal. B Environ. 2010, 99, 420–427. [Google Scholar] [CrossRef]
- Moulis, F.; Krysa, J. Photocatalytic degradation of several VOCs (n-hexane, n-butyl acetate and toluene) on TiO2 layer in a closed-loop reactor. Catal. Today 2013, 209, 153–158. [Google Scholar] [CrossRef]
- Lichtin, N.N.; Avudaithai, M. TiO2-Photocatalyzed Oxidative Degradation of CH3CN, CH3OH, C2HCl3, and CH2Cl2 Supplied as Vapors and in Aqueous Solution under Similar Conditions. Environ. Sci. Technol. 1996, 30, 2014–2020. [Google Scholar] [CrossRef]
- Neti, N.R.; Parmar, G.R.; Bakardjieva, S.; Subrt, J. Thick film titania on glass supports for vapour phase photocatalytic degradation of toluene, acetone, and ethanol. Chem. Eng. J. 2010, 163, 219–229. [Google Scholar] [CrossRef]
- Bettoni, M.; Candori, P.; Falcinelli, S.; Marmottini, F.; Meniconi, S.; Rol, C.; Sebastiani, G.V. Gas phase photocatalytic efficiency of TiO2 powders evaluated by acetone photodegradation. J. Photochem. Photobiol. A Chem. 2013, 268, 1–6. [Google Scholar] [CrossRef]
- Bettoni, M.; Candori, P.; Marmottini, F.; Perenze, N.; Rol, C.; Sebastiani, G.V.; Vecchiocattivi, F. Photocatalytic Activity in CH3CN Related to the Surface Properties of TiO2 Powders Prepared by Sol-Gel Method. Int. J. Photoenergy 2009, 2009, ID905987. [Google Scholar] [CrossRef]
- Yu, J.C.; Yu, J.; Zhang, L.; Ho, W. Enhancing effects of water content and ultrasonic irradiation on the photocatalytic activity of nano-sized TiO2 powders. J. Photochem. Photobiol. A Chem. 2002, 148, 263–271. [Google Scholar] [CrossRef]
- Yu, H.; Yu, J.; Cheng, B. Photocatalytic activity of the calcined H-titanate nanowires for photocatalytic oxidation of acetone in air. Chemosphere 2007, 66, 2050–2057. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.C.; Leung, M.K.P.; Ho, W.; Cheng, B.; Zhao, X.; Zhao, J. Effects of acidic and basic hydrolysis catalysts on the photocatalytic activity and microstructures of bimodal mesoporous titania. J. Catal. 2003, 217, 69–78. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook. NIST Standard Reference Database Number 69, Gaithersburg MD, 20899. 2005. Available online: http://webbook.nist.gov (accessed on 17 November 2020).
- Behnajady, M.A.; Eskandarloo, H.; Modirshahla, N.; Shokri, M. Investigation of the effect of sol-gel synthesis variables on structural and photocatalytic properties of TiO2 nanoparticles. Desalination 2011, 278, 10–17. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Field. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Leonori, F.; Petrucci, R.; Balucani, N.; Casavecchia, P.; Rosi, M.; Berteloite, C.; Le Picard, S.D.; Canosa, A.; Sims, I.R. Observation of organosulfur products (thiovinoxy, thioketene and thioformyl) in crossed-beam experiments and low temperature rate coefficients for the reaction S(1D) + C2H4. Phys. Chem. Chem. Phys. 2009, 11, 4701–4706. [Google Scholar] [CrossRef]
- Berteloite, C.; Le Picard, S.D.; Sims, I.R.; Rosi, M.; Leonori, F.; Petrucci, R.; Balucani, N.; Casavecchia, P. Low temperature kinetics, crossed beam dynamics and theoretical studies of the reaction S(1D) + CH4 and low temperature kinetics of S(1D) + C2H2. Phys. Chem. Chem. Phys. 2011, 13, 8485–8501. [Google Scholar] [CrossRef] [PubMed]
- de Petris, G.; Rosi, M.; Troiani, A. SSOH and HSSO radicals: An experimental and theoretical study of [S2OH](0/+/-) species. J. Phys. Chem. A 2007, 111, 6526–6653. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.J. Many-Body Perturbation Theory and Coupled Cluster Theory for Electron Correlation in Molecules. Annu. Rev. Phys. Chem. 1981, 32, 359–401. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Olsen, J.; Jorgensen, P.; Koch, H.; Balkova, A.; Bartlett, R.J. Full configuration-interaction and state of the art correlation calculations on water in a valence double-zeta basis with polarization functions. J. Chem. Phys. 1996, 104, 8007–8015. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Flükiger, P.; Lüthi, H.P.; Portmann, S.; Weber, J. MOLEKEL 4.3; Swiss Center for Scientific Computing: Lugano, Switzerland, 2000–2002.
- Portmann, S.; Lüthi, H.P. MOLEKEL: An interactive molecular graphics tool. CHIMIA Int. J. Chem. 2000, 54, 766–770. [Google Scholar]
- Falcinelli, S.; Vecchiocattivi, F.; Pirani, F. General treatment for stereo-dynamics of state-to-state chemi-ionization reactions. Commun. Chem. 2020, 3, 64. [Google Scholar] [CrossRef]
- Falcinelli, S.; Farrar, J.M.; Vecchiocattivi, F.; Pirani, F. Quantum-State Controlled Reaction Channels in Chemi-ionization Processes: Radiative (Optical-Physical) and Exchange (Oxidative-Chemical) Mechanisms. Acc. Chem. Res. 2020, 53, 2248–2260. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ka × 103, min−1 | R |
|---|---|---|
| S100 | 0.260 | 0.9988 |
| S200 | 0.990 | 0.9907 |
| S350 | 1.500 | 0.9916 |
| S500 | 1.700 | 0.9947 |
| S700 | 2.400 | 0.9973 |
| S850 | 0.200 | 0.9972 |
| S1000 | 0.032 | 0.9908 |
| Substrate | k, ppm min−1 | K × 102, ppm−1 | kK × 102, min−1 |
|---|---|---|---|
| Hexane | 1.9 | 1.05 | 2.0 |
| Acetone | 3.8 | 1.05 | 4.0 |
| Isooctane | 2.1 | 0.74 | 1.5 |
| Methanol | 9.9 | 1.11 | 11.0 |
| Substance | IP(eV) | IP(eV) in the Presence of H2O | IP(eV) in the Presence of 2 H2O | |
|---|---|---|---|---|
| CCSD(T) | Experiment | CCSD(T) | CCSD(T) | |
| Methanol | 10.81 | 10.84 | 9.63 | 9.17 |
| Acetone | 9.65 | 9.703 | 9.54 | --- |
| n-Hexane | 9.96 | 10.13 | --- | --- |
| Isooctane | --- | 9.89 | --- | --- |
| Methane | 12.66 | 12.61 | --- | --- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bettoni, M.; Falcinelli, S.; Rol, C.; Rosi, M.; Sebastiani, G.V. Gas-Phase TiO2 Photosensitized Mineralization of Some VOCs: Mechanistic Suggestions through a Langmuir–Hinshelwood Kinetic Approach. Catalysts 2021, 11, 20. https://doi.org/10.3390/catal11010020
Bettoni M, Falcinelli S, Rol C, Rosi M, Sebastiani GV. Gas-Phase TiO2 Photosensitized Mineralization of Some VOCs: Mechanistic Suggestions through a Langmuir–Hinshelwood Kinetic Approach. Catalysts. 2021; 11(1):20. https://doi.org/10.3390/catal11010020
Chicago/Turabian StyleBettoni, Marta, Stefano Falcinelli, Cesare Rol, Marzio Rosi, and Giovanni Vittorio Sebastiani. 2021. "Gas-Phase TiO2 Photosensitized Mineralization of Some VOCs: Mechanistic Suggestions through a Langmuir–Hinshelwood Kinetic Approach" Catalysts 11, no. 1: 20. https://doi.org/10.3390/catal11010020
APA StyleBettoni, M., Falcinelli, S., Rol, C., Rosi, M., & Sebastiani, G. V. (2021). Gas-Phase TiO2 Photosensitized Mineralization of Some VOCs: Mechanistic Suggestions through a Langmuir–Hinshelwood Kinetic Approach. Catalysts, 11(1), 20. https://doi.org/10.3390/catal11010020