Acidity as Descriptor for Methanol Desorption in B-, Ga- and Ti-MFI Zeotypes

 ,
,  , , and
, , and
Abstract
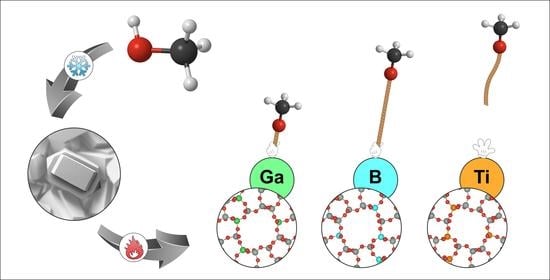
1. Introduction
2. Results and Discussion
2.1. Materials Characterization
2.2. Temperature Programmed Desorption of Methanol
2.3. Further Considerations on the Zeotypes’ Acidity and Their Applications
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| XRD | X-ray diffraction |
| SEM | Scanning electron microscopy |
| DRIFTS | Diffuse reflectance Fourier transform spectroscopy |
| B-S | B-silicalite |
| Ga-S | Ga-silicalite |
| Ti-S | Ti-silicalite |
References
- Kondratenko, E.V.; Peppel, T.; Seeburg, D.; Kondratenko, V.A.; Kalevaru, N.; Martin, A.; Wohlrab, S. Methane conversion into different hydrocarbons or oxygenates: Current status and future perspectives in catalyst development and reactor operation. Catal. Sci. Technol. 2017, 7, 366–381. [Google Scholar] [CrossRef]
- Wang, X.; Martin, N.M.; Nilsson, J.; Carlson, S.; Gustafson, J.; Skoglundh, M.; Carlsson, P.A. Copper-modified zeolites and silica for conversion of methane to methanol. Catalysts 2018, 8, 545. [Google Scholar] [CrossRef]
- Dyballa, M.; Thorshaug, K.; Pappas, D.K.; Borfecchia, E.; Kvande, K.; Bordiga, S.; Berlier, G.; Lazzarini, A.; Olsbye, U.; Beato, P.; et al. Zeolite Surface Methoxy Groups as Key Intermediates in the Stepwise Conversion of Methane to Methanol. ChemCatChem 2019, 11, 5022–5026. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Starokon, E.V.; Parfenov, M.V.; Pirutko, L.V.; Abornev, S.I.; Panov, G.I. Room-temperature oxidation of methane by ɑ-oxygen and extraction of products from the FeZSM-5 surface. J. Phys. Chem. C 2011, 115, 2155–2161. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Kulkarni, A.; Vilella, L.; Nørskov, J.K.; Studt, F. Theoretical Insights into the Selective Oxidation of Methane to Methanol in Copper-Exchanged Mordenite. ACS Catal. 2016, 6, 3760–3766. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef]
- Alayon, E.M.C.; Nachtegaal, M.; Bodi, A.; Van Bokhoven, J.A. Reaction conditions of methane-to-methanol conversion affect the structure of active copper sites. ACS Catal. 2014, 4, 16–22. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Román-Leshkov, Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, T.; Hamill, C.D.; Goguet, A.; Rooney, D.W.; Thompson, J.M. A low temperature, isothermal gas-phase system for conversion of methane to methanol over Cu-ZSM-5. Chem. Commun. 2014, 50, 11053–11055. [Google Scholar] [CrossRef] [PubMed]
- Pappas, D.K.; Martini, A.; Dyballa, M.; Kvande, K.; Teketel, S.; Lomachenko, K.A.; Baran, R.; Glatzel, P.; Arstad, B.; Berlier, G.; et al. The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment. J. Am. Chem. Soc. 2018, 140, 15270–15278. [Google Scholar] [CrossRef] [PubMed]
- Wulfers, M.J.; Lobo, R.F.; Ipek, B.; Teketel, S. Conversion of Methane to Methanol on Copper-Containing Small-Pore Zeolites and Zeotypes. Chem. Commun. 2015, 51, 4447–4450. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.R.; Reimer, J.A.; Bell, A.T.; Janicke, M.T.; Ott, K.C. Methanol formation on Fe/Al-MFI via the oxidation of methane by nitrous oxide. J. Catal. 2004, 225, 300–306. [Google Scholar] [CrossRef]
- Zakaria, K. Direct conversion technologies of methane to methanol: An overview. Renew. Sustain. Energy Rev. 2016, 65, 250–261. [Google Scholar] [CrossRef]
- Lawton, T.J.; Rosenzweig, A.C. Methane-oxidizing enzymes: An upstream problem in biological gas-to-liquids conversion. J. Am. Chem. Soc. 2016, 138, 9327–9340. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites. ACS Catal. 2016, 6, 6531–6536. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Cation-exchanged zeolites for the selective oxidation of methane to methanol. Catal. Sci. Technol. 2018, 8, 114–123. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Jenkins, R.L.; Lopez-Sanchez, J.A.; Kondrat, S.A.; Hasbi Ab Rahim, M.; Forde, M.M.; Thetford, A.; Taylor, S.H.; Hagen, H.; et al. Elucidation and evolution of the active component within Cu/Fe/ZSM-5 for catalytic methane oxidation: From synthesis to catalysis. ACS Catal. 2013, 3, 689–699. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Lopez-Sanchez, J.A.; Jenkins, R.L.; Whiting, G.; Kondrat, S.A.; Ab Rahim, M.H.; Forde, M.M.; Thetford, A.; Hagen, H.; et al. Aqueous-phase methane oxidation over Fe-MFI zeolites; Promotion through isomorphous framework substitution. ACS Catal. 2013, 3, 1835–1844. [Google Scholar] [CrossRef]
- Wang, X.; Arvidsson, A.A.; Cichocka, M.O.; Zou, X.; Martin, N.M.; Nilsson, J.; Carlson, S.; Gustafson, J.; Skoglundh, M.; Hellman, A.; et al. Methanol Desorption from Cu-ZSM-5 Studied by in Situ Infrared Spectroscopy and First-Principles Calculations. J. Phys. Chem. C 2017, 121, 27389–27398. [Google Scholar] [CrossRef]
- Wang, X.; Shishkin, A.; Hemmingsson, F.; Skoglundh, M.; Martinez-Casado, F.J.; Bock, L.; Idström, A.; Nordstierna, L.; Härelind, H.; Carlsson, P.A. Methane adsorption and methanol desorption of copper modified boron silicate. RSC Adv. 2018, 8, 36369–36374. [Google Scholar] [CrossRef]
- Arvidsson, A.A.; Plessow, P.N.; Studt, F.; Hellman, A. Influence of Acidity on the Methanol-to-DME Reaction in Zeotypes: A First, Principles-Based Microkinetic Study. J. Phys. Chem. C 2020, 124, 14658–14663. [Google Scholar] [CrossRef]
- Berndt, H.; Martin, A.; Kosslick, H.; Lücke, B. Comparison of the acidic properties of ZSM-5 zeolites isomorphously substituted by Ga, In, B and Fe. Microporous Mater. 1994, 2, 197–204. [Google Scholar] [CrossRef]
- Borade, R.B. Synthesis and characterization of ferrisilicate zeolite of pentasil group. Zeolites 1987, 7, 398–403. [Google Scholar] [CrossRef]
- Chu, C.T.W.; Kuehl, G.H.; Lago, R.M.; Chang, C.D. Isomorphous substitution in zeolite frameworks. II. Catalytic properties of [B]ZSM-5. J. Catal. 1985, 93, 451–458. [Google Scholar] [CrossRef]
- Creci, S.; Carlsson, P.A.; Wang, X.; Skoglundh, M. Tuned Acidity for Catalytic Reactions: Synthesis and Characterization of Fe- and Al-MFI Zeotypes. Top. Catal. 2019, 62, 689–698. [Google Scholar] [CrossRef]
- Strodel, P.; Neyman, K.M.; Knözinger, H.; Rösch, N. Acidic properties of [Al], [Ga] and [Fe] isomorphously substituted zeolites. Density functional model cluster study of the complexes with a probe CO molecule. Chem. Phys. Lett. 1995, 240, 547–552. [Google Scholar] [CrossRef]
- Stave, M.S.; Nicholas, J.B. Density functional studies of zeolites. 2. Structure and acidity of [T]-ZSM-5 models (T = B, Al, Ga, and Fe). J. Phys. Chem. 1995, 99, 15046–15061. [Google Scholar] [CrossRef]
- Parrillo, D.J.; Lee, C.; Gorte, R.J.; White, D.; Farneth, W.E. Comparison of the acidic properties of H-[Al]ZSM-5, H-[Fe]ZSM-5, and H-[Ga]ZSM-5 using microcalorimetry, hexane cracking, and propene oligomerization. J. Phys. Chem. 1995, 99, 8745–8749. [Google Scholar] [CrossRef]
- Creci, S.; Wang, X.; Carlsson, P.A.; Martinelli, A.; Skoglundh, M. Methoxy ad-species in MFI zeotypes during methane exposure and methanol desorption followed by in situ IR spectroscopy. Catal. Today 2020, in press. [Google Scholar] [CrossRef]
- Bordiga, S.; Lamberti, C.; Bonino, F.; Travert, A.; Thibault-Starzyk, F. Probing zeolites by vibrational spectroscopies. Chem. Soc. Rev. 2015, 44, 7262–7341. [Google Scholar] [CrossRef] [PubMed]
- Flanigen, E.M.; Bennett, J.M.; Grose, R.W.; Cohen, J.P.; Patton, R.L.; Kirchner, R.M.; Smith, J.V. Silicalite, a new hydrophobic crystalline silica molecular sieve. Nature 1978, 271, 512–516. [Google Scholar] [CrossRef]
- Baerlocher, C.; McCusker, L. Database of Zeolite Structures. Available online: http://www.iza-structure.org (accessed on 11 January 2021).
- Perego, G.; Bellussi, G.; Millini, R.; Alberti, A.; Zanardi, S. B-containing molecular sieves crystallized in the presence of ethylenediamine. Part II: Crystal structure of as-synthesized B-MFI. Microporous Mesoporous Mater. 2003, 58, 213–223. [Google Scholar] [CrossRef]
- Hudec, P.; Smieskova, A.; Zidek, Z.; Zubek, M.; Schneider, P.; Kocirik, M.; Kozankova, J. Adsorption Properties of ZSM-5 Zeolites. Collect. Czech. Chem. Commun. 1998, 63, 141–154. [Google Scholar] [CrossRef]
- Shannon, B.Y.R.D.; Baur, N.H.; Gibbs, O.H.; Eu, M.; Cu, V. Revised Effective Ionic Radii and Systematic Studies of Interatomie Distances in Halides and Chaleogenides Central Research and Development Department, Experimental Station, E . L Du Pont de Nemours The effective ionic radii of Shannon & Prewitt. Acta Cryst. 1976, A32, 751–767. [Google Scholar]
- Ko, Y.S.; Ahn, W.S. Characterization and catalytic properties of titanium silicalite-1 catalyst. Korean J. Chem. Eng. 1998, 15, 182–191. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Alothman, Z.A. A review: Fundamental aspects of silicate mesoporous materials. Materials 2012, 5, 2874–2902. [Google Scholar] [CrossRef]
- Thommes, M. Physical adsorption characterization of nanoporous materials. Chem. Ing. Tech. 2010, 82, 1059–1073. [Google Scholar] [CrossRef]
- Yang, Y.; Xia, Q.; Feng, M.; Zhang, P. Temperature dependence of IR absorption of OH species in clinopyroxene. Am. Mineral. 2010, 95, 1439–1443. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook. Available online: http://webbook.nist.gov/chemistry/ (accessed on 11 January 2021).
- Campbell, S.M.; Jiang, X.Z.; Howe, R.F. Methanol to hydrocarbons: Spectroscopic studies and the significance of extra-framework aluminium. Microporous Mesoporous Mater. 1999, 29, 91–108. [Google Scholar] [CrossRef]
- Pelmenschikov, A.G.; Morosi, G.; Gamba, A.; Zecchina, A.; Bordiga, S.; Paukshtis, E.A. Mechanisms of methanol adsorption on silicalite and silica: IR spectra and ab-initio calculations. J. Phys. Chem. 1993, 97, 11979–11986. [Google Scholar] [CrossRef]
- Kung, M.C.; Lin, S.S.Y.; Kung, H.H. In situ Infrared Spectroscopic Study of CH4 Oxidation Over Co–ZSM-5. Top. Catal. 2012, 55, 108–115. [Google Scholar] [CrossRef]
- Kubelková, L.; Nováková, J.; Nedomová, K. Reactivity of surface species on zeolites in methanol conversion. J. Catal. 1990, 124, 441–450. [Google Scholar] [CrossRef]
- Hunger, B.; Matysik, S.; Heuchel, M.; Einicke, W.D. Adsorption of methanol on ZSM-5 zeolites. Langmuir 1997, 13, 6249–6254. [Google Scholar] [CrossRef]
- Weitkamp, J. Zeolites and catalysis. Solid State Ionics 2000, 131, 175–188. [Google Scholar] [CrossRef]
- Friberg, I.; Sadokhina, N.; Olsson, L. The effect of Si/Al ratio of zeolite supported Pd for complete CH4 oxidation in the presence of water vapor and SO2. Appl. Catal. B 2019, 250, 117–131. [Google Scholar] [CrossRef]
- Feng, G.; Nancy, M.W.; Yilin, W.; Márton, K.; János, S.; Charles, H.P. Effects of Si/Al ratio on Cu/SSZ-13 NH3-SCR catalysts: Implications for the active Cu species and the roles of Brønsted acidity. J. Catal. 2015, 331, 25–38. [Google Scholar] [CrossRef]
- Szostak, R.; Nair, V.; Thomas, T.L. Incorporation and Stability of Iron in Molecular -sieve Structures. J. Chem. Soc. Faraday Trans. 1987, 1, 487–494. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SSA [m g] | Micropore v. [10 cm g] |
|---|---|---|
| B-S | 405 | 78 |
| Ga-S | 335 | 103 |
| Ti-S | 265 | 25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Creci, S.; Martinelli, A.; Vavra, S.; Carlsson, P.-A.; Skoglundh, M. Acidity as Descriptor for Methanol Desorption in B-, Ga- and Ti-MFI Zeotypes. Catalysts 2021, 11, 97. https://doi.org/10.3390/catal11010097
Creci S, Martinelli A, Vavra S, Carlsson P-A, Skoglundh M. Acidity as Descriptor for Methanol Desorption in B-, Ga- and Ti-MFI Zeotypes. Catalysts. 2021; 11(1):97. https://doi.org/10.3390/catal11010097
Chicago/Turabian StyleCreci, Simone, Anna Martinelli, Szilvia Vavra, Per-Anders Carlsson, and Magnus Skoglundh. 2021. "Acidity as Descriptor for Methanol Desorption in B-, Ga- and Ti-MFI Zeotypes" Catalysts 11, no. 1: 97. https://doi.org/10.3390/catal11010097
APA StyleCreci, S., Martinelli, A., Vavra, S., Carlsson, P.-A., & Skoglundh, M. (2021). Acidity as Descriptor for Methanol Desorption in B-, Ga- and Ti-MFI Zeotypes. Catalysts, 11(1), 97. https://doi.org/10.3390/catal11010097