
Photocatalytic Alkylation of α-(Trifluoromethyl)Styrenes with Potassium Xanthogenates

Abstract
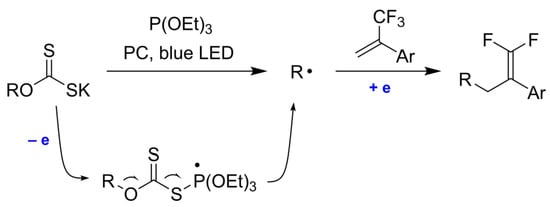
:
1. Introduction
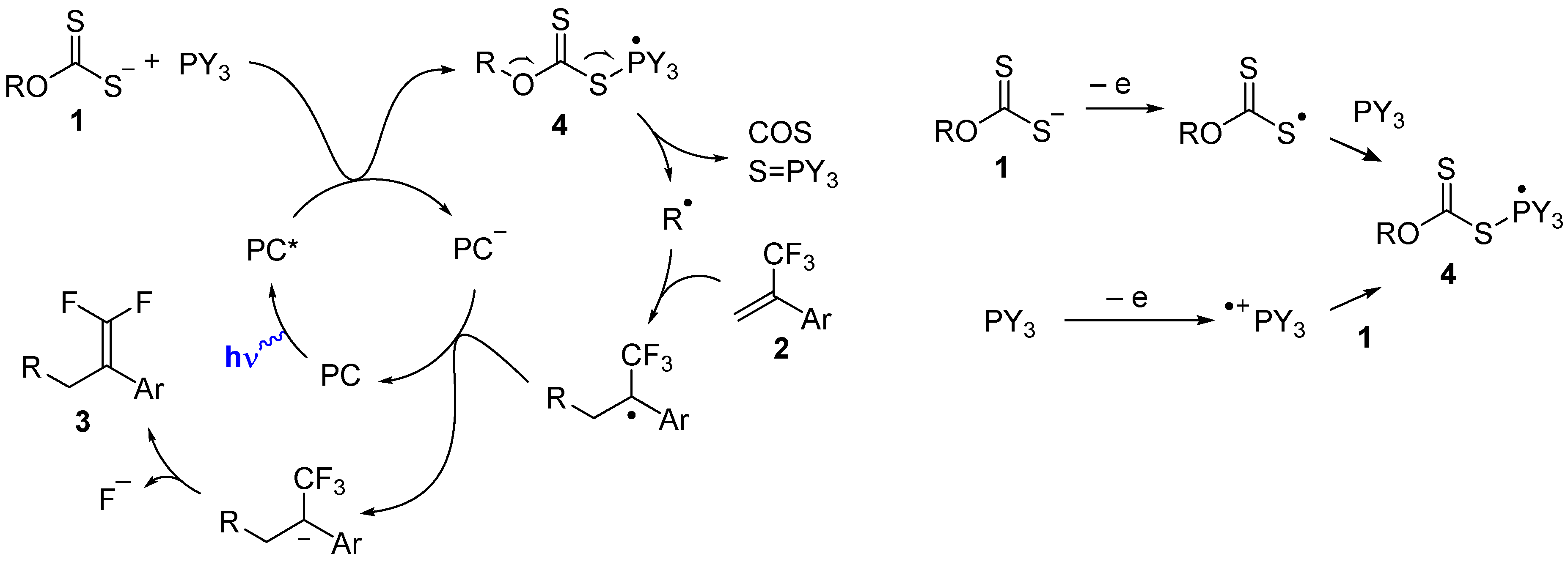
2. Results
3. Materials and Methods
3.1. General Information
3.2. Reaction of Potassium Xanthagenates with α-(Trifluoromethyl)styrenes (General Procedure)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yan, M.; Lo, J.C.; Edwards, J.T.; Baran, P.S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Metal-Free, Cooperative Asymmetric Organophotoredox Catalysis with Visible Light. Angew. Chem. Int. Ed. 2011, 50, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.D.; Murphy, J.A. Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents. Chem. Soc. Rev. 2021, 50, 9540–9685. [Google Scholar] [CrossRef]
- Supranovich, V.I.; Levin, V.V.; Kokorekin, V.A.; Dilman, A.D. Generation of Alkyl Radicals from Thiols via Zinc Thiolates: Application for the Synthesis of gem-Difluorostyrenes. Adv. Synth. Catal. 2021, 363, 2888–2892. [Google Scholar] [CrossRef]
- Zard, S.Z. On the Trail of Xanthates: Some New Chemistry from an Old Functional Group. Angew. Chem. Int. Ed. 1997, 36, 672–685. [Google Scholar] [CrossRef]
- Crich, D.; Quintero, L. Radical chemistry associated with the thiocarbonyl group. Chem. Rev. 1989, 89, 1413–1432. [Google Scholar] [CrossRef]
- Tian, F.; Yan, G.; Yu, J. Recent advances in the synthesis and applications of a-(trifluoromethyl)styrenes in organic synthesis. Chem. Commun. 2019, 55, 13486–13505. [Google Scholar] [CrossRef]
- Yan, G.; Qiu, K.; Guo, M. Recent advance in the C–F bond functionalization of trifluoromethyl-containing compounds. Org. Chem. Front. 2021, 8, 3915–3942. [Google Scholar] [CrossRef]
- Guo, H.-M.; Wu, X. Selective deoxygenative alkylation of alcohols via photocatalytic domino radical fragmentations. Nat. Commun. 2021, 12, 5365. [Google Scholar] [CrossRef] [PubMed]
- Speckmeier, E.; Fischer, T.G.; Zeitler, K. A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [Google Scholar] [CrossRef]
- Bentrude, W.G. Phosphoranyl radicals—Their structure, formation, and reactions. Acc. Chem. Res. 1982, 15, 117–125. [Google Scholar] [CrossRef]
- Bentrude, W.G.; Hansen, E.R.; Khan, W.A.; Min, T.B.; Rogers, P.E. Free-radical chemistry of organophosphorus compounds. III. α vs. β Scission in reactions of alkoxy and thiyl radicals with trivalent organophosphorus derivatives. J. Am. Chem. Soc. 1973, 95, 2286–2293. [Google Scholar] [CrossRef]
- Rossi-Ashton, J.A.; Clarke, A.K.; Unsworth, W.P.; Taylor, R.J.K. Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis. ACS Catal. 2020, 10, 7250–7261. [Google Scholar] [CrossRef]
- Hu, X.-Q.; Hou, Y.-X.; Liu, Z.-K.; Gao, Y. Recent advances in phosphoranyl radical-mediated deoxygenative functionalisation. Org. Chem. Front. 2020, 7, 2319–2324. [Google Scholar] [CrossRef]
- Maddigan-Wyatt, J.; Hooper, J.F. Phosphorus Compounds as Precursors and Catalysts for Radical C–C Bond-Forming Reactions. Adv. Synth. Cat. 2021, 363, 924–936. [Google Scholar] [CrossRef]
- Levin, V.V.; Dilman, A.D. One-pot synthesis of a-trifluoromethylstyrenes from aryl ketones and the Rupper–Prakash reagent. Mendeleev Commun. 2021, 31, 684–685. [Google Scholar] [CrossRef]
- He, W.; Ding, Y.; Tu, J.; Que, C.; Yang, Z.; Xu, J. Thermal conversion of primary alcohols to disulfides via xanthate intermediates: An extension to the Chugaev elimination. Org. Biomol. Chem. 2018, 16, 1659–1666. [Google Scholar] [CrossRef]
- Akram, R.; Khan, M.D.; Zequine, C.; Zhao, C.; Gupta, R.K.; Akhtar, M.; Akhtar, J.; Malik, M.A.; Revaprasadu, N.; Bhatti, M.H. Cobalt sulfide nanoparticles: Synthesis, water splitting and supercapacitance studies. Mater. Sci. Semicond. Process. 2020, 109, 104925. [Google Scholar] [CrossRef]
- Carta, F.; Akdemir, A.; Scozzafava, A.; Masini, E.; Supuran, C.T. Xanthates and Trithiocarbonates Strongly Inhibit Carbonic Anhydrases and Show Antiglaucoma Effects In Vivo. J. Med. Chem. 2013, 56, 4691–4700. [Google Scholar] [CrossRef]
- Allwright, E.; Silber, G.; Crain, J.; Matsushita, M.M.; Awaga, K.; Robertson, N. Electrochemical deposition of highly-conducting metal dithiolene films. Dalton Trans. 2016, 45, 9363–9368. [Google Scholar] [CrossRef] [Green Version]
- Dumur, F.; Mayer, C.R. Unexpected Ritter Reaction During Acid-Promoted 1,3-Dithiol-2-one Formation. Helv. Chim. Acta 2013, 96, 889–896. [Google Scholar] [CrossRef]
- Hu, M.; Ni, C.; Li, L.; Han, Y.; Hu, J. gem-Difluoroolefination of Diazo Compounds with TMSCF3 or TMSCF2Br: Transition-Metal-Free Cross-Coupling of Two Carbene Precursors. J. Am. Chem. Soc. 2015, 137, 14496–14501. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, J.; Sakoda, K.; Wada, Y. The 5-endo-trig Cyclization of gem-Difluoroolefins with sp3 Carbon Nucleophiles: Synthesis of 1-Fluorocyclopentenes. Chem. Lett. 2002, 31, 282–283. [Google Scholar] [CrossRef]
- Dai, W.; Lin, Y.; Wan, Y.; Cao, S. Cu-Catalyzed tertiary alkylation of α-(trifluoromethyl)styrenes with tertiary alkylmagnesium reagents. Org. Chem. Front. 2018, 5, 55–58. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
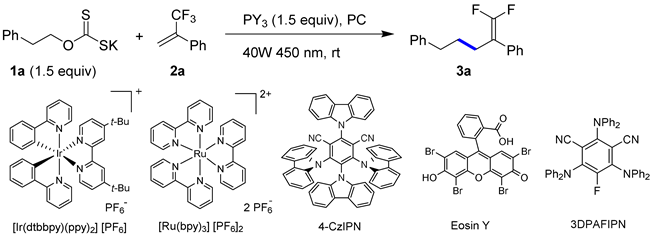 | ||||||
| # | PY3 | PC (mol%) | Time, h | Solv. | 2a (%) 1 | 3a (%) 1 |
| 1 | PPh3 | [Ir(dtbbpy)(ppy)2][PF6] (0.15) | 1.5 | DMF | 91 | 1 |
| 2 | PPh3 | [Ir(dtbbpy)(ppy)2][PF6] (0.15) | 1.5 | MeCN | 67 | 24 |
| 3 | PPh3 | [Ir(dtbbpy)(ppy)2][PF6] (0.15) | 1.5 | CH2Cl2 | 7 | 66 |
| 4 | PPh3 | [Ir(dtbbpy)(ppy)2][PF6] (0.15) | 1.5 | acetone | 91 | 7 |
| 5 | PPh3 | [Ir(dtbbpy)(ppy)2][PF6] (0.15) | 18 | MeCN | 2 | 60 |
| 6 | PPh3 | [Ir(dtbbpy)(ppy)2][PF6] (0.15) | 18 | acetone | 34 | 27 |
| 7 | PPh3 | [Ru(bpy)3][PF6]2 (0.2) | 1.5 | CH2Cl2 | 83 | 15 |
| 8 | PPh3 | 4CzIPN (0.5) | 1.5 | CH2Cl2 | 99 | <1 |
| 9 | PPh3 | Eosin Y (0.6) | 1.5 | CH2Cl2 | 100 | - |
| 10 | PPh3 | 3DPAFIPN (0.6) | 1.5 | CH2Cl2 | 11 | 58 |
| 11 | PPh3 | 3DPAFIPN (0.6) | 16 | MeCN | 5 | 61 |
| 12 | PPh3 | 3DPAFIPN (0.6) | 2.5 | acetone | 74 | 16 |
| 13 | PPh3 | 3DPAFIPN (0.6) | 2.5 | MeCN | 52 | 33 |
| 14 | PBu3 | 3DPAFIPN (0.6) | 16 | MeCN | 20 | 13 |
| 15 | P(OEt)3 | 3DPAFIPN (0.6) | 16 | MeCN | - | 60 |
| 16 | P(OEt)3 | 3DPAFIPN (0.6) | 3 | CH2Cl2 | - | 71 (68 2) |
| 17 3 | P(OEt)3 | 3DPAFIPN (0.6) | 6.5 | MeCN | - | 61 |
| 18 | P(OEt)3 | 3DPAFIPN (0.6) | 17 | EtOAc | - | 58 |
| 19 | P(OEt)3 | 3DPAFIPN (0.6) | 17 | MTBE | 35 | 45 |
| 20 | P(OEt)3 | 3DPAFIPN (0.6) | 3 | dioxane | 30 | 24 |
| 21 | P(OEt)3 | 3DPAFIPN (0.6) | 17 | hexanes | 45 | 26 |
| 22 4 | P(OEt)3 | 3DPAFIPN (0.6) | 17 | MeCN | - | 61 |
| 23 4 | P(OEt)3 | 3DPAFIPN (0.6) | 17 | acetone | - | 63 |
| 24 | P(NMe2)3 | 3DPAFIPN (0.6) | 2.5 | CH2Cl2 | - | 56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Supranovich, V.I.; Dilman, A.D. Photocatalytic Alkylation of α-(Trifluoromethyl)Styrenes with Potassium Xanthogenates. Catalysts 2021, 11, 1555. https://doi.org/10.3390/catal11121555
Supranovich VI, Dilman AD. Photocatalytic Alkylation of α-(Trifluoromethyl)Styrenes with Potassium Xanthogenates. Catalysts. 2021; 11(12):1555. https://doi.org/10.3390/catal11121555
Chicago/Turabian StyleSupranovich, Vyacheslav I., and Alexander D. Dilman. 2021. "Photocatalytic Alkylation of α-(Trifluoromethyl)Styrenes with Potassium Xanthogenates" Catalysts 11, no. 12: 1555. https://doi.org/10.3390/catal11121555
APA StyleSupranovich, V. I., & Dilman, A. D. (2021). Photocatalytic Alkylation of α-(Trifluoromethyl)Styrenes with Potassium Xanthogenates. Catalysts, 11(12), 1555. https://doi.org/10.3390/catal11121555