
Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide



Abstract
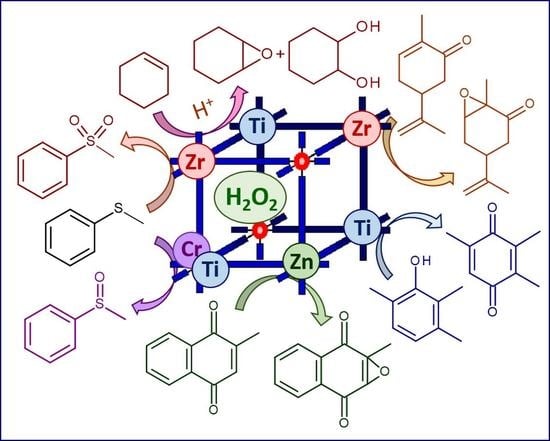
1. Introduction
2. MOFs as Selective Oxidation Catalysts
2.1. Oxidation of S-Compounds
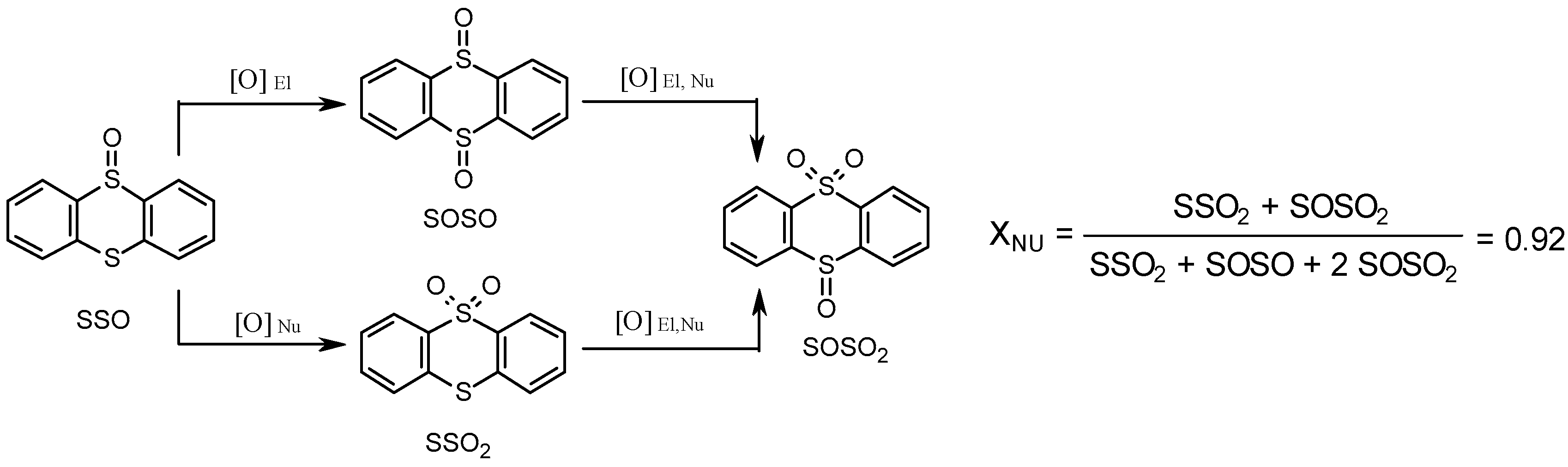
2.1.1. Electrophilic Oxidation of Thioethers to Sulfoxides over Zn- and Cr-MOFs
2.1.2. Oxidative Desulfurization
2.1.3. Unprecedented Selectivity of Zr-MOFs toward Sulfones
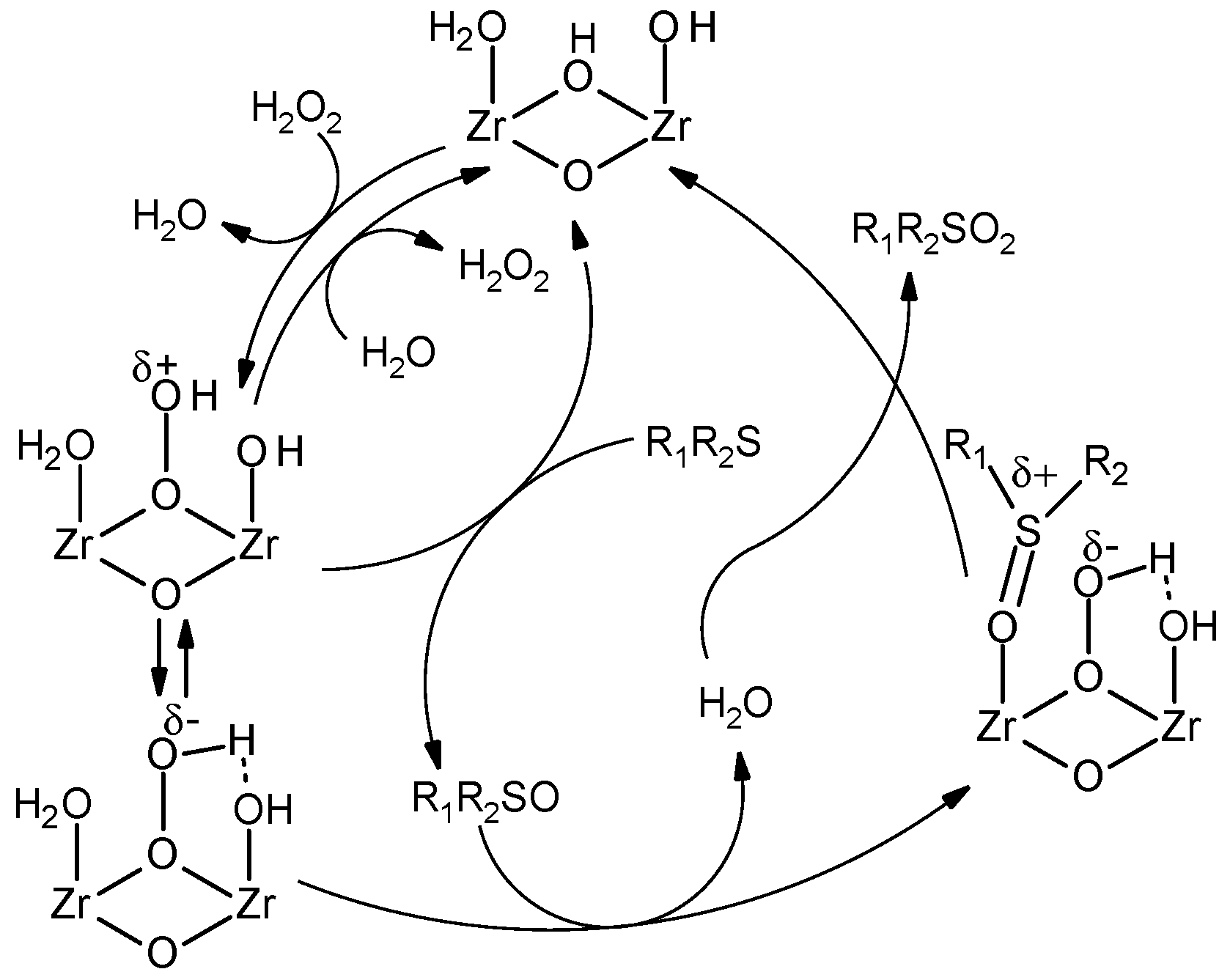
2.1.4. Nucleophilic Activation of H2O2 and Mechanism of Thioether Oxidation
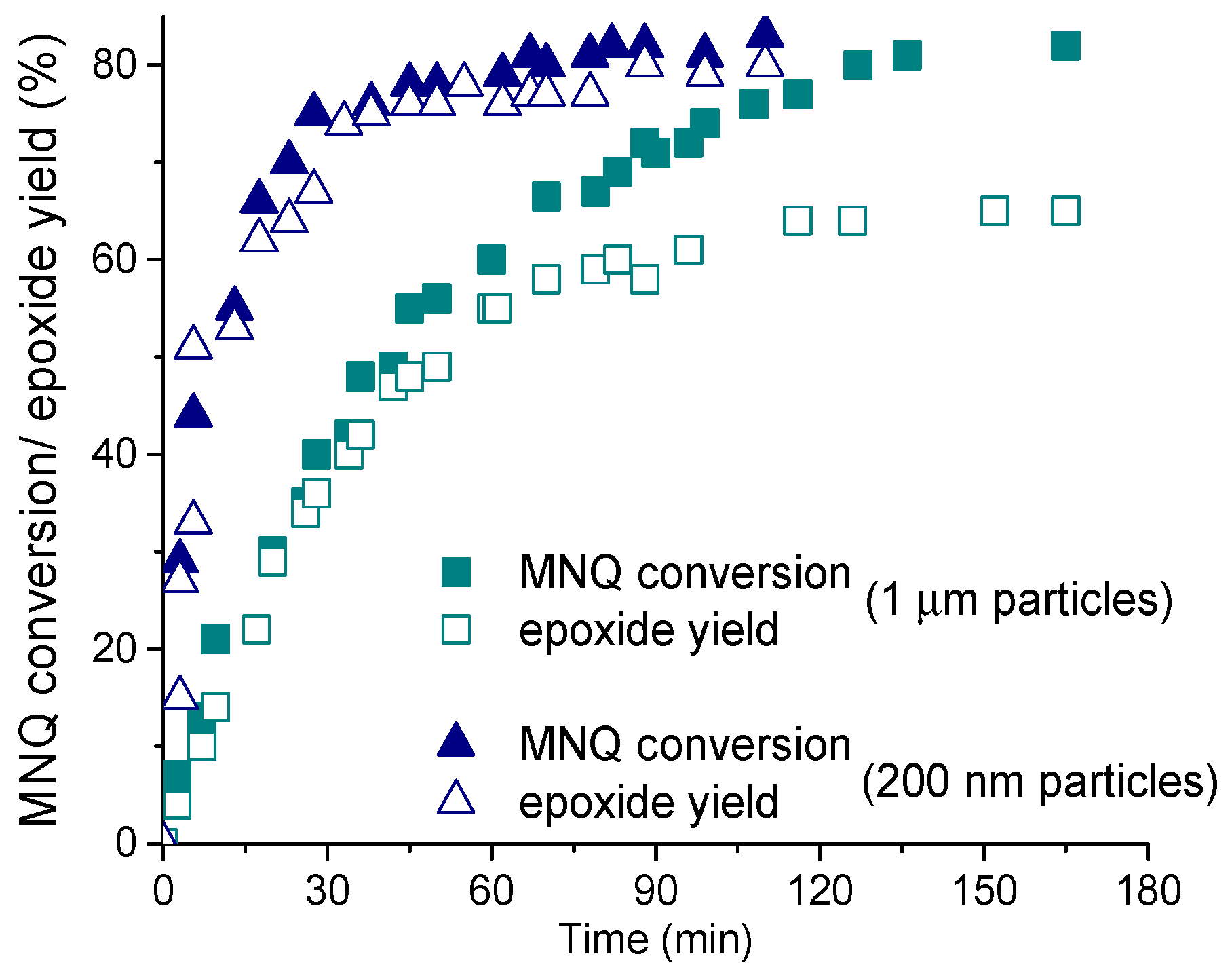
2.2. Epoxidation of C=C Bonds
2.2.1. Nucleophilic Oxidation of Electron-Deficient C=C Bonds
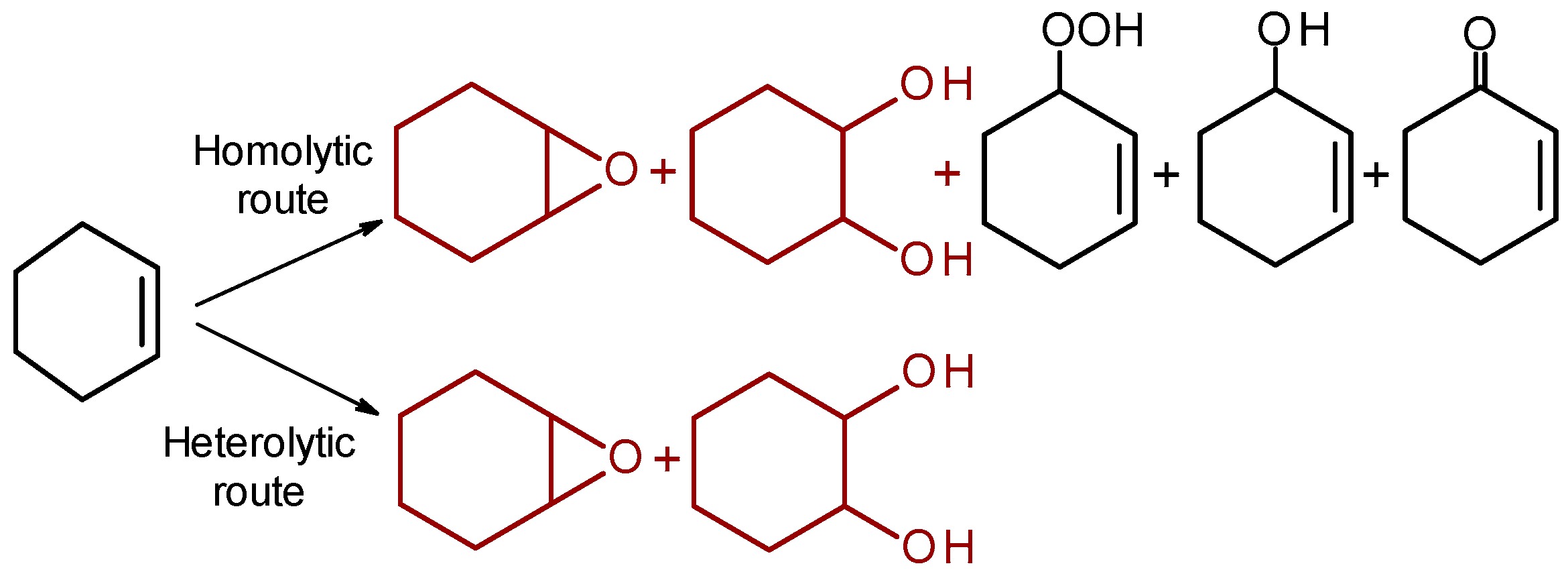
2.2.2. Oxidation of Electron-Rich C=C Bonds: Effect of Protons on Heterolytic Pathway Selectivity
2.3. Oxidation of Alkylphenols
2.4. Oxidation of Alcohols and Diols
2.4.1. Alcohol Oxidation
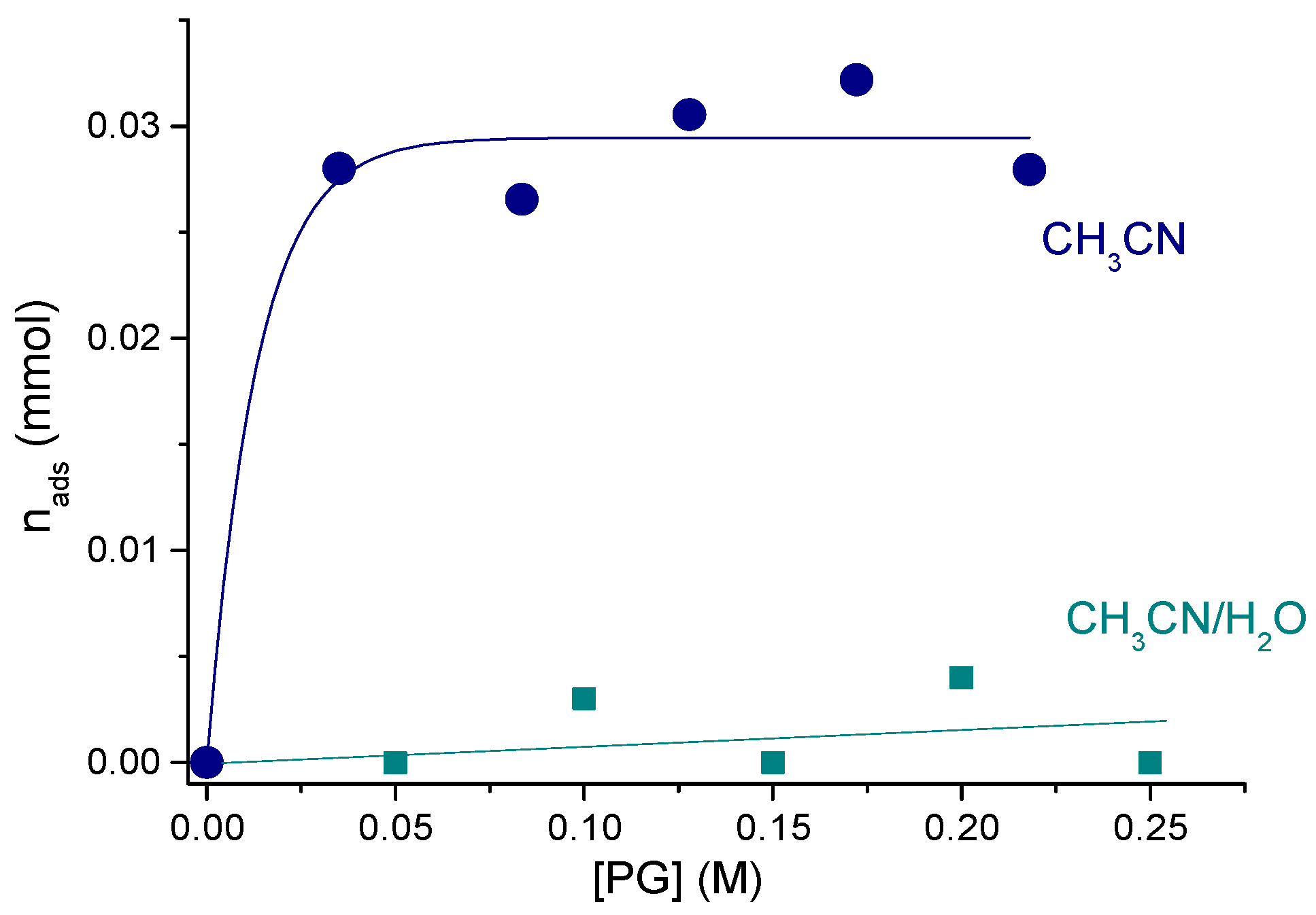
2.4.2. Propylene Glycol Oxidation
2.5. Oxidation of Arenes and Alkanes
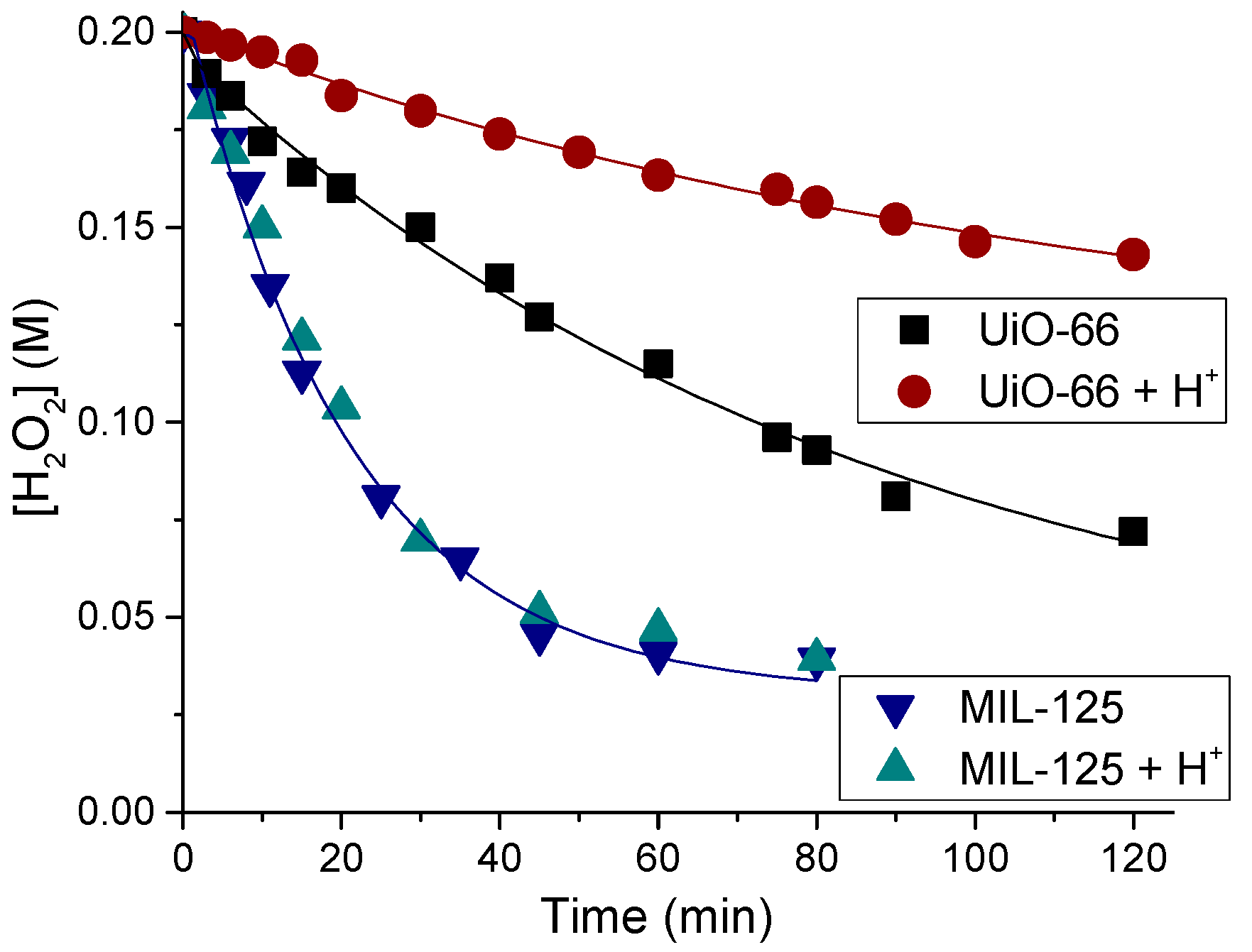
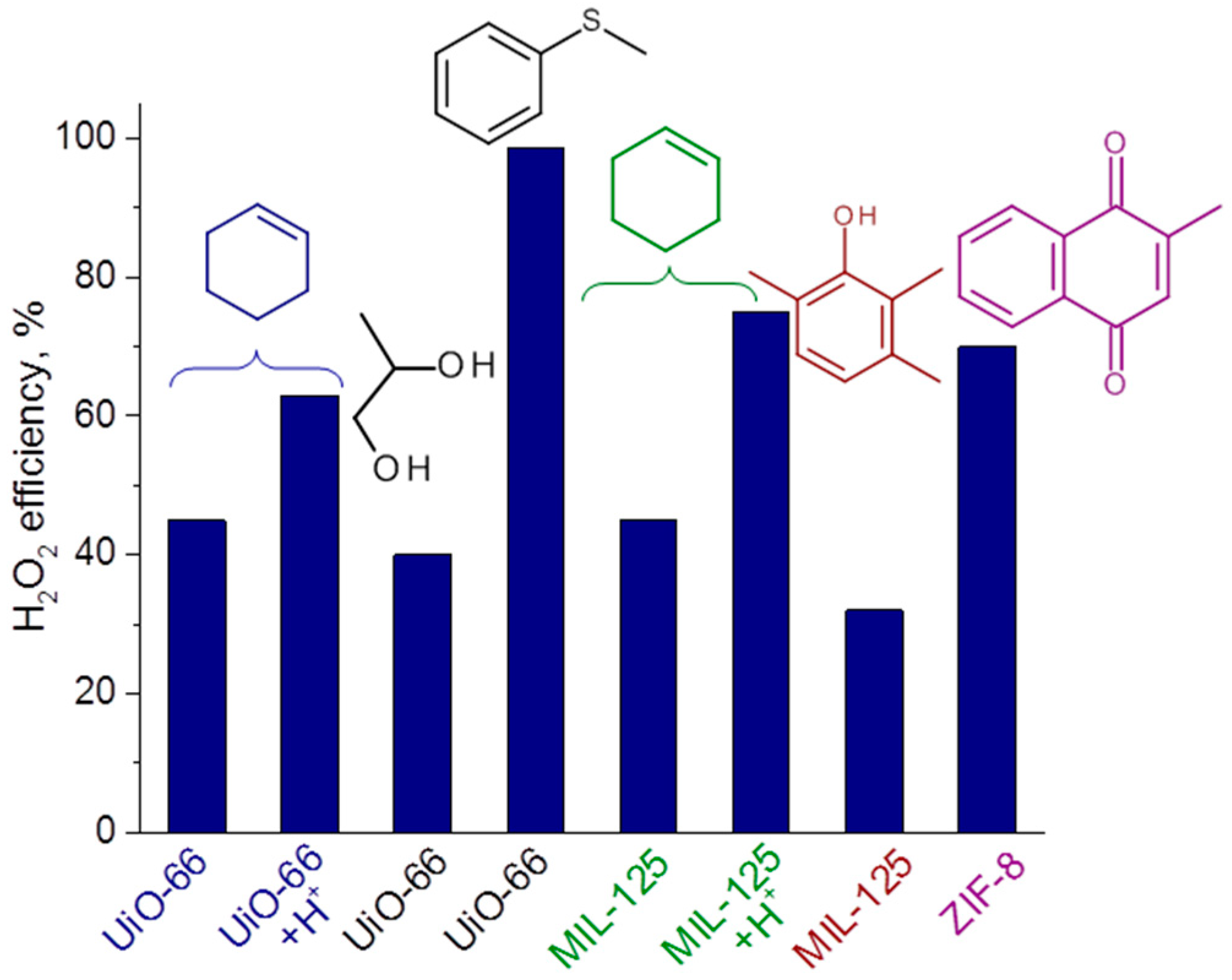
2.6. H2O2 Decomposition and Oxidant Utilization Efficiency
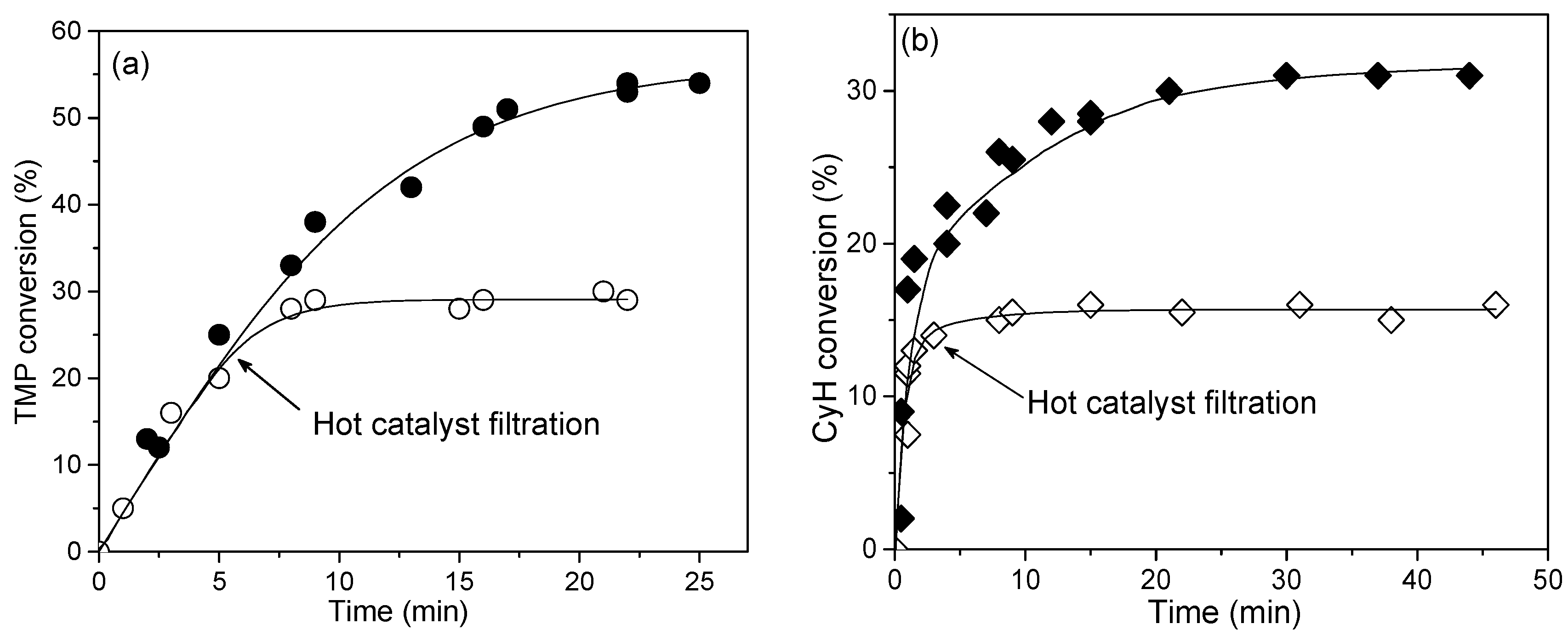
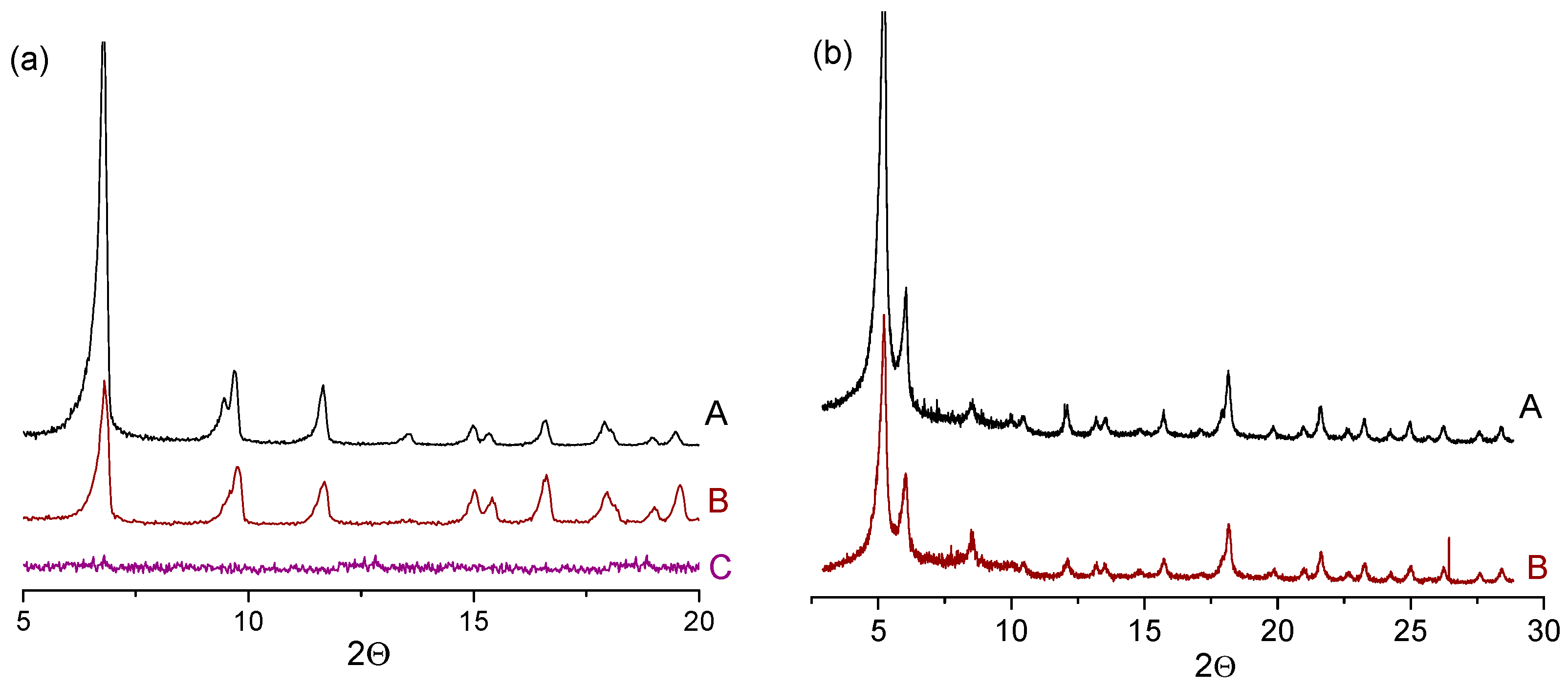
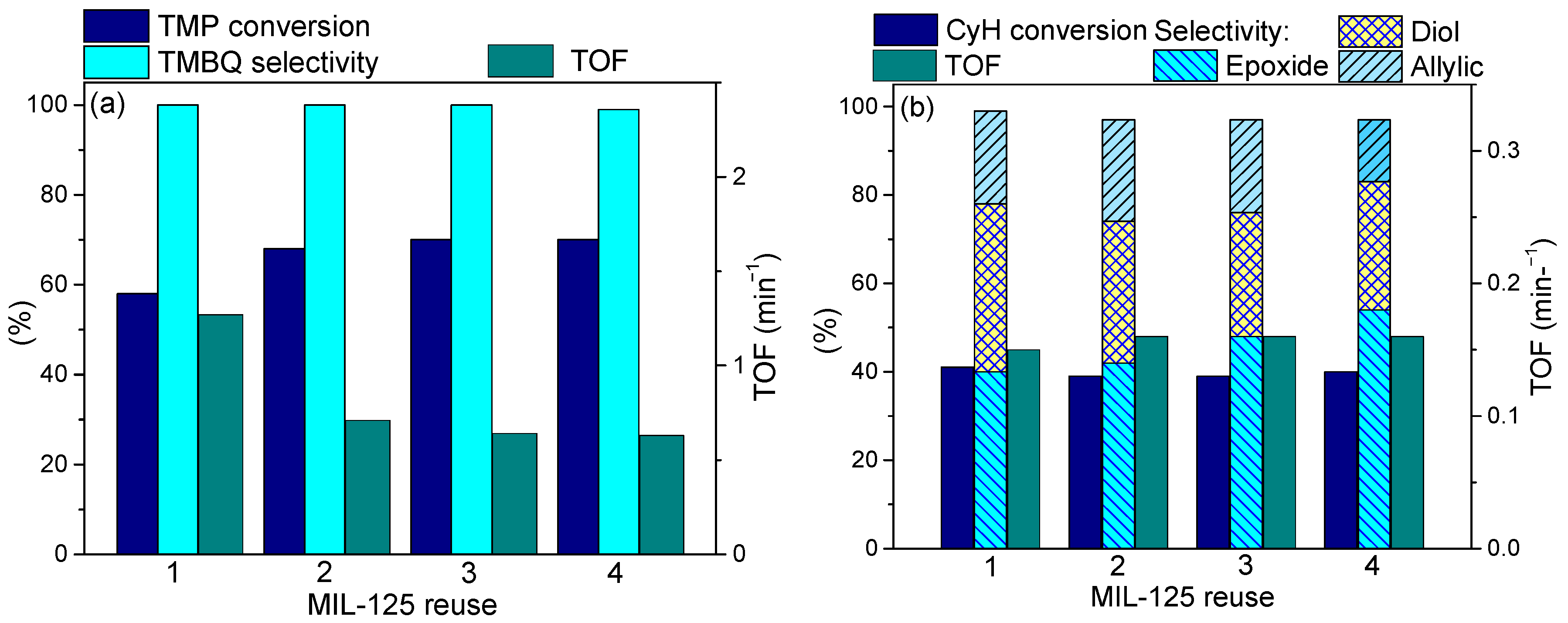
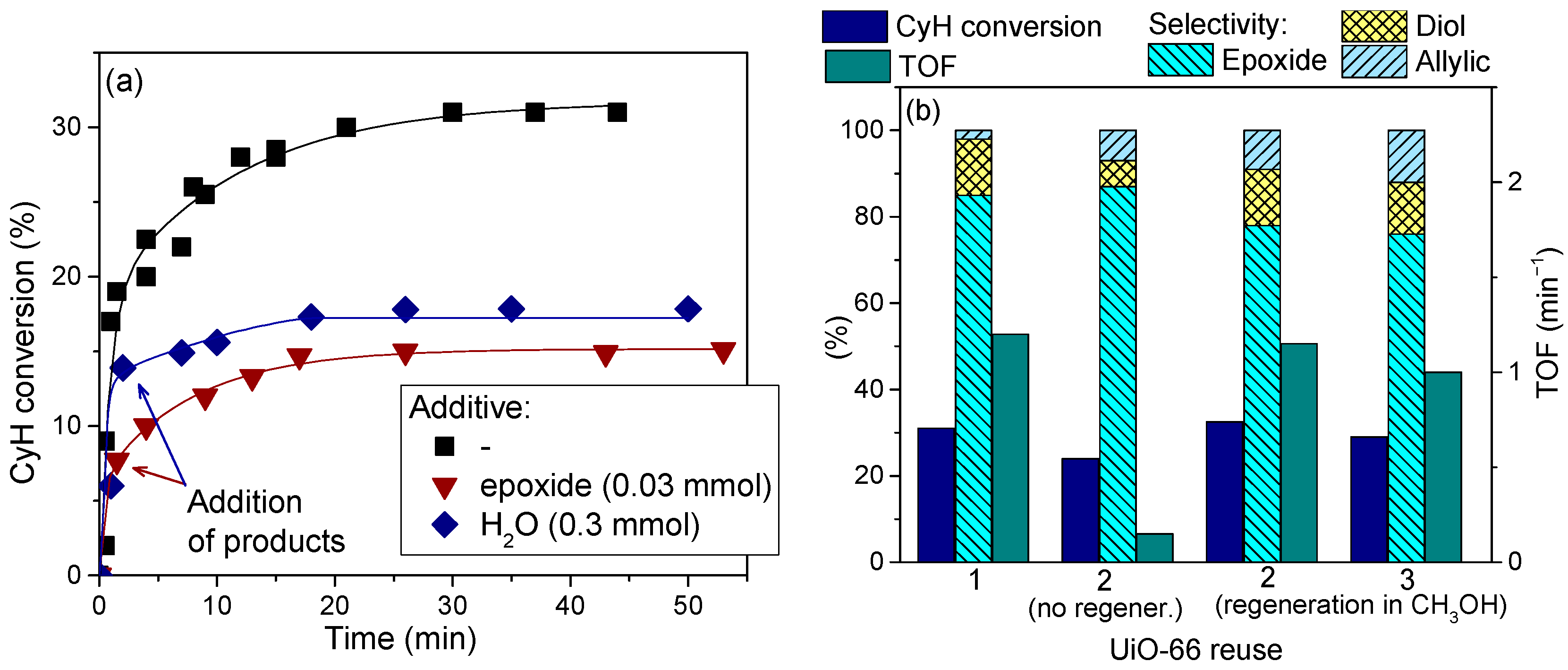
2.7. Catalyst Stability and Reusability
3. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sheldon, R.; Arends, I.W.C.E.; Hanefeld, U. Green Chemistry and Catalysis; Wiley VCH: Weinheim, Germany, 2007. [Google Scholar]
- Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G., Kholdeeva, O.A., Eds.; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D., Cavani, F., Eds.; Imperial College Press: London, UK, 2014. [Google Scholar]
- Jones, C.W. Application of Hydrogen Peroxide and Derivatives; Royal Society of Chemistry: Cambridge, UK, 1999. [Google Scholar]
- Catalytic Oxidations with Hydrogen Peroxide as Oxidant; Strukul, G., Ed.; Kluwer: Dordrecht, The Netherlands, 1992. [Google Scholar]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Edwards, J.K.; Freakley, S.J.; Lewis, R.J.; Pritchard, J.C.; Hutchings, G.J. Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Catal. Today 2015, 248, 3–9. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Ghedini, E.; Strukul, G. Looking for the “dream catalyst” for hydrogen peroxide production from hydrogen and oxygen. Catalysts 2019, 9, 251. [Google Scholar] [CrossRef]
- Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis; Schröder, M., Ed.; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Metal–Organic Frameworks as Heterogeneous Catalysts; Xamena, F.L.I., Gascon, J., Eds.; RCS Publishing: Cambridge, UK, 2013. [Google Scholar]
- Metal-Organic Frameworks: Applications in Separations and Catalysis; García, H., Navalón, S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018. [Google Scholar]
- Elaboration and Applications of Metal-Organic Frameworks, Series on Chemistry, Energy and the Environment; Ma, S., Perman, J.A., Eds.; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2018; Volume 2. [Google Scholar]
- Levason, B.; Bradshaw, D. Special Issue on Chemistry and Applications of Metal Organic Frameworks. Coord. Chem Rev. 2016, 307, 105–424. [Google Scholar] [CrossRef]
- Dietzel, P.; Kitagawa, H. Cluster Issue “Metal–Organic Frameworks Heading towards Application”. Eur. J. Inorg. Chem. 2016, 2016, 4265–4529. [Google Scholar]
- Batten, S.R.; Chen, B.; Vittal, J.J. Special Issue on Coordination Polymers/MOFs. ChemPlusChem 2016, 81, 666–898. [Google Scholar]
- Maurin, G.; Serre, C.; Cooper, A.; Férey, G. Themed issue Metal-organic frameworks and porous polymers—Current and future challenges. Chem. Soc. Rev. 2017, 46, 3104–3107. [Google Scholar] [CrossRef]
- Moon, H.R.; Lim, D.-W.; Suh, M.P. Fabrication of metal nanoparticles in metal–organic frameworks. Chem. Soc. Rev. 2013, 42, 1807–1824. [Google Scholar] [CrossRef]
- Silva, P.; Vilela, S.M.F.; Tomé, J.P.C.; Paz, F.A.A. Multifunctional metal–organic frameworks: From academia to industrial applications. Chem. Soc. Rev. 2015, 44, 6774–6803. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Li, B.; He, H.; Zhou, W.; Chen, B.; Qian, G. Metal–organic frameworks as platforms for functional materials. Acc. Chem. Res. 2016, 49, 483–493. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, Q.; Jiang, H.-L. Metal–organic frameworks meet metal nanoparticles: Synergistic effect for enhanced catalysis. Chem. Soc. Rev. 2017, 46, 4774–4808. [Google Scholar] [CrossRef] [PubMed]
- Islamoglu, T.; Chen, Z.; Wasson, M.C.; Buru, C.T.; Kirlikovali, K.O.; Afrin, U.; Mian, M.R.; Farha, O.K. Metal−organic frameworks against toxic chemicals. Chem. Rev. 2020, 120, 8130–8160. [Google Scholar]
- Ryu, U.J.; Jee, S.; Rao, P.C.; Shin, J.; Ko, C.; Yoon, M.; Park, K.S.; Choi, K.M. Recent advances in process engineering and upcoming applications of metal–organic frameworks. Coord. Chem. Rev. 2021, 426, 213544. [Google Scholar] [CrossRef]
- Hwang, Y.K.; Férey, G.; Lee, U.-H.; Chang, J.-S. Liquid phase oxidation of organic compounds by metal-organic frameworks. In Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G., Kholdeeva, O.A., Eds.; Wiley: Hoboken, NJ, USA, 2013; pp. 371–409. [Google Scholar]
- Maksimchuk, N.V.; Zalomaeva, O.V.; Skobelev, I.Y.; Kovalenko, K.A.; Fedin, V.P.; Kholdeeva, O.A. Metal-organic frameworks of the MIL-101 family as heterogeneous single-site catalysts. Proc. R. Soc. A 2012, 468, 2017–2034. [Google Scholar] [CrossRef]
- Valvekens, P.; Vermoortele, F.; De Vos, D. Metal–organic frameworks as catalysts: The role of metal active sites. Catal. Sci. Technol. 2013, 3, 1435–1445. [Google Scholar] [CrossRef]
- Zhao, M.; Ou, S.; Wu, C.-D. Porous Metal–organic frameworks for heterogeneous biomimetic catalysis. Acc. Chem. Res. 2014, 47, 1199–1207. [Google Scholar] [CrossRef]
- Gascon, J.; Corma, A.; Kapteijn, F.; Llabrés i Xamena, F.X. Metal organic framework catalysis: Quo vadis? ACS Catal. 2014, 4, 361–378. [Google Scholar] [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.-Y. Applications of metal–organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.-Y.; Park, J.; Raiff, A.; Wei, Z.; Zhou, H.-C. Metal–organic frameworks as biomimetic catalysts. ChemCatChem 2014, 6, 67–75. [Google Scholar] [CrossRef]
- Leus, K.; Liu, Y.-Y.; Van Der Voort, P. Metal-organic frameworks as selective or chiral oxidation catalysts. Catal. Rev. 2014, 56, 1–56. [Google Scholar] [CrossRef]
- Chughtai, A.H.; Ahmad, N.; Younus, H.A.; Laypkov, A.; Verpoort, F. Metal–organic frameworks: Versatile heterogeneous catalysts for efficient catalytic organic transformations. Chem. Soc. Rev. 2015, 44, 6804–6849. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, A.; Asiri, A.M.; García, H. Metal–organic frameworks as catalysts for oxidation reactions. Chem. Eur. J. 2016, 22, 8012–8024. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Liquid-phase selective oxidation catalysis with metal-organic frameworks. Catal. Today 2016, 278, 22–29. [Google Scholar] [CrossRef]
- Hu, Z.; Zhao, D. Metal–organic frameworks with Lewis acidity: Synthesis, characterization, and catalytic applications. CrystEngComm 2017, 19, 4066–4081. [Google Scholar] [CrossRef]
- Rogge, S.M.J.; Bavykina, A.; Hajek, J.; Garcia, H.; Olivos-Suarez, A.I.; Sepúlveda-Escribano, A.; Vimont, A.; Clet, G.; Bazin, P.; Kapteijn, F.; et al. Metal–organic and covalent organic frameworks as single-site catalysts. Chem. Soc. Rev. 2017, 46, 3134–3184. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Thapa, K.B.; Ju, Q.; Fang, Z.; Huang, W. Heterogeneous catalysts based on mesoporous metal–organic frameworks. Coord. Chem. Rev. 2018, 373, 199–232. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Asiric, A.M.; Herance, J.R.; García, H. Metal organic frameworks as solid promoters for aerobic autoxidations. Catal. Today 2018, 306, 2–8. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Li, Z.; García, H. Catalysis and photocatalysis by metal organic frameworks. Chem. Soc. Rev. 2018, 47, 8134–8172. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-S.; Yuan, S.; Lollar, C.; Pang, J.; Alsalme, A.; Zhou, H.-C. Stable metal–organic frameworks as a host platform for catalysis and biomimetics. Chem. Commun. 2018, 54, 4231–4249. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Gates, B.C. Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research. ACS Catal. 2019, 9, 1779–1798. [Google Scholar] [CrossRef]
- Liu, M.; Wu, J.; Hou, H. Metal–organic framework (MOF)-based materials as heterogeneous catalysts for C-H Bond activation. Chem. Eur. J. 2019, 25, 2935–2948. [Google Scholar] [CrossRef]
- Kang, Y.-S.; Lu, Y.; Chen, K.; Zhao, Y.; Wang, P.; Sun, W.-Y. Metal–organic frameworks with catalytic centers: From synthesis to catalytic application. Coord. Chem. Rev. 2019, 378, 262–280. [Google Scholar] [CrossRef]
- Bavykina, A.; Kolobov, N.; Khan, I.S.; Bau, J.A.; Ramirez, A.; Gascon, J. Metal−organic frameworks in heterogeneous catalysis: Recent progress, new trends, and future perspectives. Chem. Rev. 2020, 120, 8468–8535. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Dou, Y.; Xie, L.-H.; Rutledge, W.; Li, J.-R.; Zhou, H.-C. Zr-Based metal–organic frameworks: Design, synthesis, structure, and applications. Chem. Soc. Rev. 2016, 45, 2327–2367. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Feng, L.; Wang, K.; Pang, J.; Bosch, M.; Lollar, C.; Sun, Y.; Qin, J.; Yang, X.; Zhang, P.; et al. Stable Metal–Organic Frameworks: Design, Synthesis, and Applications. Adv. Mater. 2018, 30, 1704303. [Google Scholar]
- Du, D.Y.; Qin, J.S.; Li, S.L.; Su, Z.M.; Lan, Y.Q. Recent advances in porous polyoxometalate based metal–organic framework materials. Chem. Soc. Rev. 2014, 43, 4615–4632. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Recent progress in selective oxidations with hydrogen peroxide catalyzed by polyoxometalates. In Frontiers of Green Catalytic Selective Oxidations; Bryliakov, K.P., Ed.; Springer Nature Singapore Pte Ltd.: Singapore, 2019; pp. 61–92. [Google Scholar]
- Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surble, S.; Margiolaki, I. A Chromium terephthalate-based solid with unusually large pore volumes and surface area. Science 2005, 309, 2040–2042. [Google Scholar] [CrossRef]
- Dan-Hardi, M.; Serre, C.; Frot, T.; Rozes, L.; Maurin, G.; Sanchez, C.; Ferey, G. A new photoactive crystalline highly porous titanium (IV) dicarboxylate. J. Am. Chem. Soc. 2009, 131, 10857–10859. [Google Scholar] [CrossRef]
- Shearer, G.C.; Chavan, S.; Bordiga, S.; Svelle, S.; Olsbye, U.; Lillerud, K.P. Defect engineering: Tuning the porosity and composition of the metal−organic framework UiO-66 via modulated synthesis. Chem. Mater. 2016, 28, 3749–3761. [Google Scholar] [CrossRef]
- DeCoste, J.B.; Peterson, G.W.; Jasuja, H.; Glover, T.G.; Huang, Y.-g.; Walton, K.S. Stability and degradation mechanisms of metal–organic frameworks containing the Zr6O4(OH)4 secondary building unit. J. Mater. Chem. A 2013, 1, 5642–5650. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Wißmann, G.; Schaate, A.; Lilienthal, S.; Bremer, I.; Schneider, A.M.; Behrens, P. Modulated synthesis of Zr-fumarate MOF. Micropor. Mesopor. Mater. 2012, 152, 64–70. [Google Scholar] [CrossRef]
- Wang, H.; Dong, X.; Lin, J.; Teat, S.J.; Jensen, S.; Cure, J.; Alexandrov, E.V.; Xia, Q.; Tan, K.; Wang, Q.; et al. Topologically guided tuning of Zr-MOF pore structures for highly selective separation of C6 alkane isomers. Nat. Commun. 2018, 9, 1745. [Google Scholar] [CrossRef]
- Wang, S.; Lee, J.S.; Wahiduzzaman, M.; Park, J.; Muschi, M.; Martineau-Corcos, C.; Tissot, A.; Cho, K.H.; Marrot, J.; Shepard, W.; et al. A Robust large–pore zirconium carboxylate metal–organic framework for energy-efficient water–sorption–driven refrigeration. Nat. Energy 2018, 3, 985–993. [Google Scholar] [CrossRef]
- Huang, X.-C.; Lin, Y.-Y.; Zhang, J.-P.; Chen, X.-M. Ligand-directed strategy for zeolite-type metal–organic frameworks: Zinc(II) imidazolates with unusual zeolitic topologies. Angew. Chem. Int. Ed. 2006, 45, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Ni, Z.; Côté, A.P.; Choi, J.Y.; Huang, R.; Uribe-Romo, F.J.; Chae, H.K.; O’Keeffe, M.; Yaghi, O.M. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc. Natl. Acad. Sci. USA 2006, 103, 10186–10191. [Google Scholar] [CrossRef]
- Di Furia, F.; Modena, G. Mechanism of oxygen transfer from peroxo species. Pure Appl. Chem. 1982, 54, 1853–1866. [Google Scholar] [CrossRef]
- Bonchio, M.; Campestrini, S.; Conte, V.; Di Furia, F.; Moro, S. A Theoretical and experimental investigation of the electrophilic oxidation of thioethers and sulfoxides by peroxides. Tetrahedron 1995, 51, 12363–12372. [Google Scholar] [CrossRef]
- Gomez-Lor, B.; Gutiérrez-Puebla, E.; Iglesias, M.; Monge, M.A.; Ruiz-Valero, C.; Snejko, N. In2(OH)3(BDC)1.5 (BDC) = 1,4-Benzendicarboxylate): An In(III) supramolecular 3d framework with catalytic activity. Inorg. Chem. 2002, 41, 2428–2432. [Google Scholar] [CrossRef]
- Perles, J.; Iglesias, M.; Martin-Luengo, M.-A.; Monge, M.A.; Ruiz-Valero, C.; Snejko, N. Metal-organic scandium framework: Useful material for hydrogen storage and catalysis. Chem. Mater. 2005, 17, 5837–5842. [Google Scholar] [CrossRef]
- Gándara, F.; de Andrés, A.; Gómez-Lor, B.; Gutiérrez-Puebla, E.; Iglesias, M.; Monge, M.A.; Proserpio, D.M.; Snejko, N. Rare-earth MOF series: Fascinating structure, efficient light emitters, and promising catalyst. Cryst. Growth Des. 2008, 8, 378–380. [Google Scholar] [CrossRef]
- Gándara, F.; Gutierrez Puebla, E.; Iglesias, M.; Proserpio, D.M.; Snejko, N.; Angeles, M.M. Controlling the structure of arenedisulfonates toward catalytically active materials. Chem. Mater. 2009, 21, 655–661. [Google Scholar] [CrossRef]
- Perles, J.; Snejko, N.; Iglesias, M.; Monge, M.A. 3D scandium and yttrium arenedisulfonate MOF materials as highly thermally stable bifunctional heterogeneous catalysts. J. Mater. Chem. 2009, 19, 6504–6511. [Google Scholar] [CrossRef]
- Dybtsev, D.N.; Nuzhdin, A.L.; Chun, H.; Bryliakov, K.P.; Talsi, E.P.; Fedin, V.P.; Kim, K. A Homochiral metal–organic material with permanent porosity, enantioselective sorption properties, and catalytic activity. Angew. Chem. Int. Ed. 2006, 45, 916–920. [Google Scholar] [CrossRef]
- Hwang, Y.K.; Hong, D.-Y.; Chang, J.-S.; Seo, H.; Yoon, M.; Kim, J.; Jhung, S.H.; Serre, C.; Férey, G. Selective sulfoxidation of aryl sulfides by coordinatively unsaturated metal centers in chromium carboxylate MIL-101. Appl. Catal. A Gen. 2009, 358, 249–253. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Kovalenko, K.A.; Fedin, V.P.; Kholdeeva, O.A. Heterogeneous selective oxidation of alkenes to α,β-unsaturated ketones over coordination polymer MIL-101. Adv. Synth. Catal. 2010, 352, 2943–2948. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Kovalenko, K.A.; Fedin, V.P.; Kholdeeva, O.A. Cyclohexane selective oxidation over metal–organic frameworks of MIL-101 family: Superior catalytic activity and selectivity. Chem. Commun. 2012, 48, 6812–6814. [Google Scholar] [CrossRef] [PubMed]
- Campos-Martin, J.M.; Capel-Sanchez, M.C.; Perez-Presas, P.; Fierro, J.L.G. Oxidative processes of desulfurization of liquid fuels. J. Chem. Technol. Biotechnol. 2010, 85, 879–890. [Google Scholar] [CrossRef]
- Zlotea, C.; Phanon, D.; Mazaj, M.; Heurtaux, D.; Guillerm, V.; Serre, C.; Horcajada, P.; Devic, T.; Magnier, E.; Cuevas, F.; et al. Effect of NH2 and CF3 functionalization on the hydrogen sorption properties of MOFs. Dalton Trans. 2011, 40, 4879–4881. [Google Scholar] [CrossRef]
- Bhadra, B.N.; Song, J.Y.; Khan, N.A.; Jhung, S.H. TiO2-containing carbon derived from a metal–organic framework composite: A highly active catalyst for oxidative desulfurization. ACS Appl. Mater. Interfaces 2017, 9, 31192–31202. [Google Scholar] [CrossRef]
- Zhang, Y.; Lia, G.; Kong, L.; Lu, H. Deep oxidative desulfurization catalyzed by Ti-based metal-organic frameworks. Fuel 2018, 219, 103–110. [Google Scholar] [CrossRef]
- Mondloch, J.E.; Bury, W.; Fairen-Jimenez, D.; Kwon, S.; DeMarco, E.J.; Weston, M.H.; Sarjeant, A.A.; Nguyen, S.T.; Stair, P.C.; Snurr, R.Q.; et al. Vapor-phase metalation by atomic layer deposition in a metal–organic framework. J. Am. Chem. Soc. 2013, 135, 10294–10297. [Google Scholar] [CrossRef] [PubMed]
- Devic, T.; Serre, C. High Valence 3p and transition metal based MOFs. Chem. Soc. Rev. 2014, 43, 6097–6115. [Google Scholar] [CrossRef]
- Howarth, A.J.; Liu, Y.; Li, P.; Li, Z.; Wang, T.C.; Hupp, J.T.; Farha, O.K. Chemical, thermal and mechanical stabilities of metal–organic frameworks. Nat. Rev. Mater. 2016, 1, 15018. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Johnson, J.A.; Chen, Y.-S.; Zhang, J. Highly porous zirconium metal−organic frameworks with β-UH3-like topology based on elongated tetrahedral linkers. J. Am. Chem. Soc. 2016, 138, 8380–8383. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, A.; Santiago-Portillo, A.; Asiri, A.M.; Garcia, H. Engineering UiO-66 metal organic framework for heterogeneous catalysis. ChemCatChem 2019, 11, 899–923. [Google Scholar] [CrossRef]
- Granadeiro, C.M.; Ribeiro, S.O.; Karmaoui, M.; Valenca, R.; Ribeiro, J.C.; de Castro, B.; Cunha-Silva, L.; Balula, S.S. Production of ultra–deep sulfur–free diesels using a sustainable catalytic system based on UiO-66(Zr). Chem. Commun. 2015, 51, 13818–13821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, P.; Liu, A.; Zhu, M. A Metal–Organic Framework for Oxidative Desulfurization: UIO-66(Zr) as a Catalyst. Fuel 2017, 209, 417–423. [Google Scholar] [CrossRef]
- Ye, G.; Zhang, D.; Li, X.; Leng, K.; Zhang, W.; Ma, J.; Sun, Y.; Xu, W.; Ma, S. Boosting catalytic performance of metal–organic framework by increasing the defects via a facile and green approach. ACS Appl. Mater. Interfaces 2017, 9, 34937–34943. [Google Scholar] [CrossRef]
- Xiao, W.; Dong, Q.; Wang, Y.; Li, Y.; Deng, S.; Zhang, N. Time modulation of defects in UiO-66 and application in oxidative desulfurization. CrystEngComm 2018, 20, 5658–5662. [Google Scholar] [CrossRef]
- Ye, G.; Qi, H.; Zhou, W.; Xu, W.; Sun, Y. Green and scalable synthesis of nitro- and amino-functionalized UiO-66(Zr) and the effect of functional groups on the oxidative desulfurization performance. Inorg. Chem. Front. 2019, 6, 1267–1274. [Google Scholar] [CrossRef]
- Viana, A.M.; Ribeiro, S.O.; de Castro, B.; Balula, S.S.; Cunha-Silva, L. Influence of UiO-66(Zr) Preparation strategies in its catalytic efficiency for desulfurization process. Materials 2019, 12, 3009. [Google Scholar] [CrossRef]
- Zheng, H.-Q.; Zeng, Y.-N.; Chen, J.; Lin, R.-G.; Zhuang, W.-E.; Cao, R.; Lin, Z.-J. Zr-Based metal–organic frameworks with intrinsic peroxidase-like activity for ultradeep oxidative desulfurization: Mechanism of H2O2 decomposition. Inorg. Chem. 2019, 58, 6983–6992. [Google Scholar] [CrossRef]
- Gu, Y.; Xu, W.; Sun, Y. Enhancement of catalytic performance over MOF-808(Zr) by acid treatment for oxidative desulfurization of dibenzothiophene. Catal. Today 2020. [Google Scholar] [CrossRef]
- Ye, G.; Qi, H.; Li, X.; Leng, K.; Sun, Y.; Xu, W. Enhancement of oxidative desulfurization performance over UiO-66(Zr) by titanium ion exchange. ChemPhysChem 2017, 18, 1903–1908. [Google Scholar] [CrossRef]
- Piscopo, C.G.; Voellinger, L.; Schwarzer, M.; Polyzoidis, A.; Bošković, D.; Loebbecke, S. Continuous flow desulfurization of a model fuel catalysed by titanium functionalized UiO-66ю. Chem. Sel. 2019, 4, 2806–2809. [Google Scholar]
- Limvorapitux, R.; Chen, H.; Mendonca, M.L.; Liu, M.; Snurr, R.Q.; Nguyen, S.B.T. Elucidating the mechanism of the UiO-66−catalyzed sulfide oxidation: Activity and selectivity enhancements through changes in the node coordination environment and solvent. Catal. Sci. Technol. 2019, 9, 327–335. [Google Scholar] [CrossRef]
- Trickett, C.A.; Gagnon, K.J.; Lee, S.; Gándara, F.; Bürgi, H.-B.; Yaghi, O.M. Definitive molecular level characterization of defects in UiO-66 crystals. Angew. Chem. Int. Ed. 2015, 54, 11162–11167. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Slater, B. Dynamic acidity in defective UiO-66. Chem. Sci. 2016, 7, 4706–4712. [Google Scholar] [CrossRef]
- Zalomaeva, O.V.; Evtushok, V.Y.; Ivanchikova, I.D.; Glazneva, T.S.; Chesalov, Y.A.; Larionov, K.P.; Kholdeeva, O.A. Nucleophilic vs. electrophilic activation of hydrogen peroxide over Zr-based metal-organic frameworks. Inorg. Chem. 2020, 59, 10634–10649. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Ivanchikova, I.D.; Cho, K.H.; Zalomaeva, O.V.; Evtushok, V.Y.; Larionov, K.P.; Glazneva, T.S.; Chang, J.-S.; Kholdeeva, O.A. Catalytic performance of Zr-based metal-organic frameworks Zr-abtc and MIP-200 in selective oxidations with H2O2. Chem. Eur. J. 2021. [Google Scholar] [CrossRef]
- Zalomaeeva, O.V. Boreskov Institute of Catalysis, Novosibirsk, Russia, Unpublished Results.
- Gall, R.D.; Faraj, M.; Hill, C.L. Role of water in polyoxometalate-catalyzed oxidations in nonaqueous media. Scope, kinetics, and mechanism of oxidation of thioether mustard (HD) analogs by tert-butyl hydroperoxide catalyzed by H5PV2Mo10040. Inorg. Chem. 1994, 33, 5015–5021. [Google Scholar] [CrossRef]
- Adam, W.; Haas, W.; Sieker, G. Thianthrene 5-oxide as mechanistic probe in oxygen-transfer reactions: The case of carbonyl oxides vs. dioxiranes. J. Am. Chem. Soc. 1984, 106, 5020–5022. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Fischer, R.W.; Correia, J.D.G. Multiple bonds between main-group elements and transition-metals. Part 133. Methyltrioxorhenium as a catalyst of the Baeyer-Villiger oxidation. J. Mol. Catal. 1994, 94, 213–223. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Tomaselli, G.A.; Toscano, R.M.; Conte, V.; Di Furia, F. Application of the thianthrene 5-oxide mechanistic probe to peroxometal complexes. J. Am. Chem. Soc. 1991, 113, 6209–6212. [Google Scholar] [CrossRef]
- Caratelli, C.; Hajek, J.; Cirujano, F.G.; Waroquier, M.; Llabrés i Xamena, F.X.; Van Speybroeck, V. Nature of active sites on UiO-66 and beneficial influence of water in the catalysis of Fischer esterification. J. Catal. 2017, 352, 401–414. [Google Scholar] [CrossRef]
- Caratelli, C.; Hajek, J.; Rogge, S.M.J.; Vandenbrande, S.; Meijer, E.J.; Waroquier, M.; Van Speybroeck, V. Influence of a confined methanol solvent on the reactivity of active sites in UiO-66. ChemPhysChem 2018, 19, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.B.; Deming, P.H.; Williams, P.H. Reactions of hydrogen peroxide. VII. Alkali-Catalyzed epoxidation and oxidation using a nitrile as co-reactant. J. Org. Chem. 1961, 26, 659–663. [Google Scholar] [CrossRef]
- Mak, K.K.W.; Lai, Y.M.; Siu, Y.-H. Regiospecific epoxidation of carvone: A Discovery-oriented experiment for understanding the selectivity and mechanism of epoxidation reactions. J. Chem. Educ. 2006, 83, 1058–1061. [Google Scholar] [CrossRef]
- Ivanchikova, I.D.; Evtushok, V.Y.; Zalomaeva, O.V.; Kolokolov, D.I.; Stepanov, A.G.; Kholdeeva, O.A. Heterogeneous epoxidation of menadione with hydrogen peroxide over zeolite imidazolate framework ZIF-8. Dalton Trans. 2020, 49, 12546–12549. [Google Scholar] [CrossRef]
- Peralta, D.; Chaplais, G.; Paillaud, J.L.; Simon-Masseron, A.; Barthelet, K.; Pirngruber, G.D. The separation of xylene isomers by ZIF-8: A demonstration of the extraordinary flexibility of the ZIF-8 framework. Micropor. Mesopor. Mater. 2013, 173, 1–5. [Google Scholar] [CrossRef]
- Kolokolov, D.I.; Diestel, L.; Caro, J.; Freude, D.; Stepanov, A.G. Rotational and translational motion of benzene in ZIF-8 studied by 2H NMR: Estimation of microscopic self-diffusivity and its Comparison with macroscopic measurements. J. Phys. Chem. C 2014, 118, 12873–12879. [Google Scholar] [CrossRef]
- Khudozhitkov, A.E.; Arzumanov, S.S.; Kolokolov, D.I.; Stepanov, A.G. Mobility of aromatic guests and isobutane in ZIF-8 metal–organic framework studied by 2H solid state NMR spectroscopy. J. Phys. Chem. C 2019, 123, 13765–13774. [Google Scholar] [CrossRef]
- Chizallet, C.; Lazare, S.; Bazer-Bachi, D.; Bonnier, F.; Lecocq, V.; Soyer, E.; Quoineaud, A.-A.; Bats, N. Catalysis of transesterification by a nonfunctionalized metal−organic Framework: Acido-basicity at the external surface of ZIF-8 probed by FTIR and ab initio calculations. J. Am. Chem. Soc. 2010, 132, 12365–12377. [Google Scholar] [CrossRef]
- Nguyen, H.G.T.; Mao, L.; Peters, A.W.; Audu, C.O.; Brown, Z.J.O.; Farha, K.; Hupp, J.T.; Nguyen, S.B.T. Comparative study of titanium-functionalized UiO-66: Support effect on the oxidation of cyclohexene using hydrogen peroxide. Catal. Sci. Technol. 2015, 5, 4444–4451. [Google Scholar] [CrossRef]
- Ahn, S.; Thornburg, N.E.; Li, Z.; Wang, T.C.; Gallington, L.C.; Chapman, K.W.; Notestein, J.M.; Hupp, J.T.; Farha, O.K. Stable metal−organic framework-supported niobium catalysts. Inorg. Chem. 2016, 55, 11954–11961. [Google Scholar] [CrossRef] [PubMed]
- Kholdeeva, O.A.; Maksimov, G.M.; Maksimovskaya, R.I.; Kovaleva, L.A.; Fedotov, M.A. Role of protons in methyl phenyl sulfide oxidation with hydrogen peroxide catalyzed by Ti (IV)-monosubstituted heteropolytungstates. React. Kinet. Catal. Lett. 1999, 66, 311–317. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Trubitsina, T.A.; Timofeeva, M.N.; Maksimov, G.M.; Maksimovskaya, R.I.; Rogov, V.A. The role of protons in cyclohexene oxidation with H2O2 catalysed by Ti (IV)-monosubstituted Keggin polyoxometalate. J. Mol. Catal. A Chem. 2005, 232, 173–178. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Maksimovskaya, R.I. Titanium- and zirconium-monosubstituted polyoxometalates as molecular models for studying mechanisms of oxidation catalysis. J. Mol. Catal. A Chem. 2007, 262, 7–24. [Google Scholar] [CrossRef]
- Jiménez-Lozano, P.; Ivanchikova, I.D.; Kholdeeva, O.A.; Poblet, J.M.; Carbó, J.J. Alkene oxidation by Ti-containing polyoxometalates. Unambiguous characterization of the role of the protonation state. Chem. Commun. 2012, 48, 9266–9268. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Hydrogen peroxide activation over TiIV: What have we learned from studies on Ti-containing polyoxometalates? Eur. J. Inorg. Chem. 2013, 2013, 1595–1605. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Maksimov, G.M.; Evtushok, V.Y.; Ivanchikova, I.D.; Chesalov, Y.A.; Maksimovskaya, R.I.; Kholdeeva, O.A.; Solé-Daura, A.; Poblet, J.M.; Carbó, J.J. Relevance of protons in heterolytic activation of H2O2 over Nb(V): Insights from model studies on nb-substituted polyoxometalates. ACS Catal. 2018, 8, 9722–9737. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Ivanchikova, I.D.; Maksimov, G.M.; Eltsov, I.V.; Evtushok, V.Y.; Kholdeeva, O.A.; Lebbie, D.; Errington, R.J.; Solé-Daura, A.; Poblet, J.M.; et al. Why does Nb(V) show higher heterolytic pathway selectivity than Ti(IV) in epoxidation with H2O2? Answers from model studies on Nb- and Ti-substituted Lindqvist tungstates. ACS Catal. 2019, 9, 6262–6275. [Google Scholar] [CrossRef]
- Maksimchuk, N.; Lee, J.S.; Ayupov, A.; Chang, J.-S.; Kholdeeva, O. Cyclohexene oxidation with H2O2 over metal-organic framework MIL-125(Ti): The effect of protons on the reactivity. Catalysts 2019, 9, 324. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Lee, J.S.; Solovyeva, M.V.; Cho, K.H.; Shmakov, A.N.; Chesalov, Y.A.; Chang, J.-S.; Kholdeeva, O.A. Protons make possible heterolytic activation of hydrogen peroxide over Zr-based metal-organic frameworks. ACS Catal. 2019, 9, 9699–9704. [Google Scholar] [CrossRef]
- Clerici, M.G.; Domine, M.E. Oxidation reactions catalyzed by transitionmetal-substituted zeolites. In Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G., Kholdeeva, O.A., Eds.; Wiley: Hoboke, NJ, USA, 2013; pp. 21–93. [Google Scholar]
- Bordiga, S.; Groppo, E.; Agostini, G.; van Bokhoven, J.A.; Lamberti, C. Reactivity of surface species in heterogeneous catalysts probed by in situ X-ray absorption techniques. Chem. Rev. 2013, 113, 1736–1850. [Google Scholar] [CrossRef]
- Clerici, M.G. The activity of titanium silicalite-1 (TS-1): Some considerations on its origin. Kinet. Catal. 2015, 56, 450–455. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Maksimchuk, N.V.; Skobelev, I.Y. H2O2-based selective epoxidations: Nb-silicates versus Ti-silicates. Catal. Today 2019, 333, 63–70. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Guidotti, M.; Ravasio, N.; Sgobba, M.; Barmatova, M. How to reach 100% selectivity in H2O2-based oxidation of 2,3,6-trimethylphenol to trimethyl-p-benzoquinone over Ti,Si-catalysts. Catal. Today 2009, 141, 330–336. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Guidotti, M.; Pirovano, C.; Ravasio, N.; Barmatova, M.V.; Chesalov, Y.A. Highly selective H2O2-based oxidation of alkylphenols to benzoquinones over silica-supported titanium catalysts: Ti cluster site versus Ti single site. Adv. Synth. Catal. 2009, 351, 1877–1889. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Recent developments in liquid-phase selective oxidation using environmentally benign oxidants and mesoporous metal silicates. Catal. Sci. Technol. 2014, 4, 1869–1889. [Google Scholar] [CrossRef]
- Ivanchikova, I.D.; Lee, J.S.; Maksimchuk, N.V.; Shmakov, A.N.; Chesalov, Y.A.; Ayupov, A.B.; Hwang, Y.K.; Jun, C.-H.; Chang, J.-S.; Kholdeeva, O.A. Highly selective H2O2-based oxidation of alkylphenols to p-benzoquinones over MIL-125 metal–organic frameworks. Eur. J. Inorg. Chem. 2014, 2014, 132–139. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Maksimchuk, N.V.; Mel’gunov, M.S.; Chang, J.-S.; Guidotti, M.; Shutilov, A.A.; Zaikovskii, V.I. Environmentally benign oxidation of alkylphenols to p-benzoquinones: A comparative study of various Ti-containing catalysts. Top. Catal. 2014, 57, 1377–1384. [Google Scholar] [CrossRef]
- Kato, N.; Hasegawa, M.; Sato, T.; Yoshizawa, A.; Inoue, T.; Mori, W. Microporous dinuclear copper(II) trans-1,4-cyclohexanedicarboxylate: Heterogeneous oxidation catalysis with hydrogen peroxide and X-ray powder structure of peroxo copper(II) intermediate. J. Catal. 2005, 230, 226–236. [Google Scholar] [CrossRef]
- Balu, A.M.; Lin, C.S.K.; Liu, H.; Li, Y.; Vargas, C.; Luque, R. Iron oxide functionalised MIL-101 materials in aqueous phase selective oxidations. Appl. Catal. A Gen. 2013, 455, 261–266. [Google Scholar] [CrossRef]
- Fei, H.; Shin, J.W.; Meng, Y.S.; Adelhardt, M.; Sutter, J.; Meyer, K.; Cohen, S.M. Reusable oxidation catalysis using metal-monocatecholato species in a robust metal−organic framework. J. Am. Chem. Soc. 2014, 136, 4965–4973. [Google Scholar] [CrossRef] [PubMed]
- Torbina, V.V.; Nedoseykina, N.S.; Ivanchikova, I.D.; Kholdeeva, O.A.; Vodyankina, O.V. Propylene glycol oxidation with hydrogen peroxide over Zr-containing metal–organic framework UiO-66. Catal. Today 2019, 333, 47–53. [Google Scholar] [CrossRef]
- Meilikhov, M.; Yusenko, K.; Fischer, R.A. Turning MIL-53(Al) redox-active by functionalization of the bridging OH-group with 1,1′-ferrocenediyl-dimethylsilane. J. Am. Chem. Soc. 2009, 131, 9644–9645. [Google Scholar] [CrossRef]
- Wang, D.; Wang, M.; Li, Z. Fe-Based metal–organic frameworks for highly selective photocatalytic benzene hydroxylation to phenol. ACS Catal. 2015, 5, 6852–6857. [Google Scholar] [CrossRef]
- Osadchii, D.Y.; Olivos-Suarez, A.I.; Szécsényi, L.G.; Nasalevich, M.A.; Dugulan, I.A.; Crespo, P.S.; Hensen, E.J.M.; Veber, S.L.; Fedin, M.V.; Sankar, G.; et al. Isolated Fe sites in metal organic frameworks catalyze the direct conversion of methane to methanol. ACS Catal. 2018, 8, 5542–5548. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Wallau, M.; Arends, I.W.C.E.; Schuchardt, U. Heterogeneous catalysts for liquid-phase oxidations: Philosophers’ stones or Trojan horses? Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar] [CrossRef]














{kind=link}
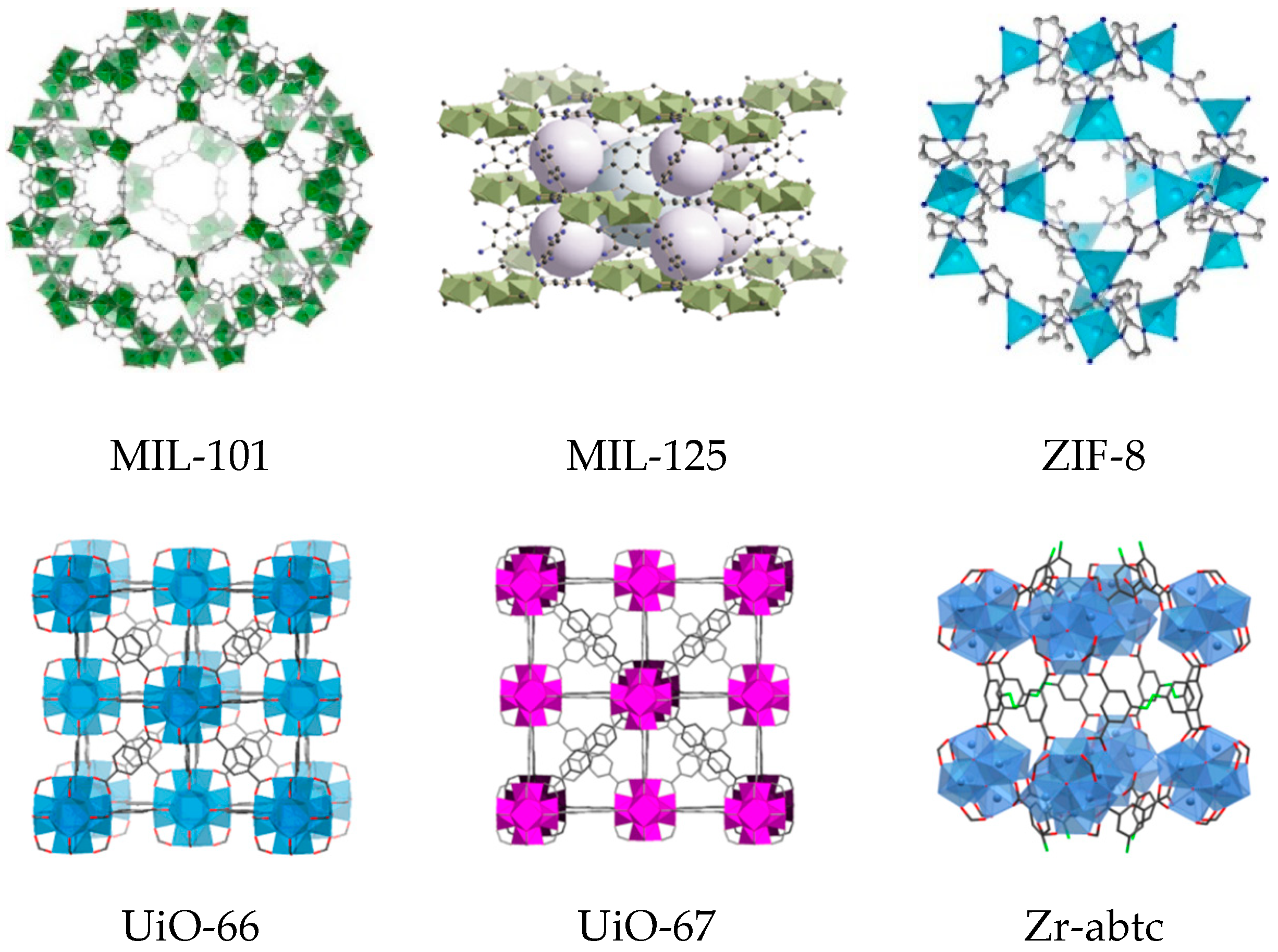{kind=link}
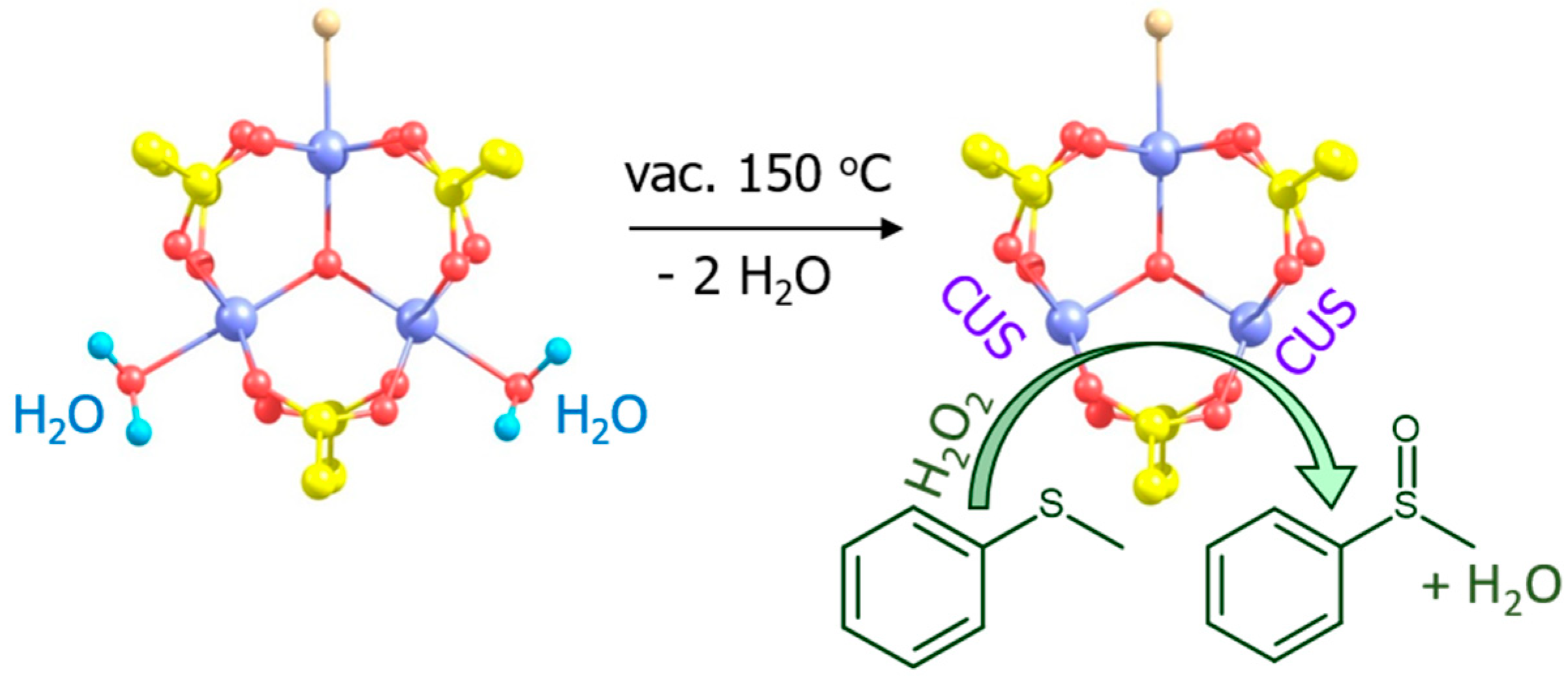{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MOF | Molecular Formula a | SBET | dp | Metal Content | Thermal Stability |
|---|---|---|---|---|---|
| (m2/g) | (Å) | (%) a | (oC) b | ||
| MIL-101 | Cr3OX(BDC)3(H2O) | 3200–3900 | 12 and 16 (windows) | 21.7 | 275 [48] |
| (X = F, OH) | 29 and 34 (cages) | ||||
| MIL-125 | Ti8O8(OH)4(BDC)6 | 1500 | 5–7 (windows) | 24.5 | 360 [49] |
| 6 and 12.5 (cages) | |||||
| UiO-66 | Zr6O4(OH)4(BDC)6 | 1200 | 6 (windows) | 32.9 | 520 [50,51] |
| 8 and 11 (cages) | 540 c [52] | ||||
| UiO-67 | Zr6O4(BPDC)6 | 2300 | 8 (windows) | 25.8 | 520 [51] |
| 11.5 and 18 (cages) | 540 c [52] | ||||
| MOF-801 | Zr6O4(OH)4(fum)6 | 900 | 5 (windows) | 40.1 | 400 [53] |
| 5.5 and 7 (cages) | |||||
| Zr-abtc | Zr6O4(OH)4(abtc)2(OH)4(H2O)4 | 1300 | 7 (1D channels) | 39.5 | 450 c [54] |
| MIP-200 | Zr6O4(OH)4(mdip)2(formate)4 | 1000 | 13 and 6.8 (hexagonal and triangular channels) | 35.5 | 500 [55] |
| ZIF-8 | Zn(MeIM)2 | 1300–1800 | 3.4 (windows) | 28.7 | 400 [56] |
| 11.6 (cages) | 550 c [57] |
| MOF | Time, h | MPS Conv. (%) | Product Selectivity (%) | |
|---|---|---|---|---|
| MPSO | MPSO2 | |||
| - | 24 | 14 | 79 | 21 |
| UiO-66 b | 0.25 | 49 | 1 | 99 |
| UiO-66 c | 0.5 | 51 | 8 | 92 |
| UiO-67 | 0.5 | 51 | 4 | 96 |
| MOF-801 | 2.5 | 47 | 4 | 96 |
| Zr-abtc c | 2 | 52 | 8 | 92 |
| Zr-abtc d | 2 | 46 | 9 | 91 |
| MIP-200 | 4 | 51 | 10 | 90 |
| MIL-125 | 2 | 69 | 80 | 20 |
| Substrate | Catalyst | Time | Substrate Conv. | Epoxide Selectivity | |
|---|---|---|---|---|---|
| (h) | (%) | (%) b | |||
 | - | 4 | 7 |  | 22 |
| Zr-abtc | 1 | 26 | 62 | ||
| Zr-abtc c | 2 | 20 | 55 | ||
| MIP-200 | 2 | 15 | 10 | ||
| UiO-66 | 1 | 20 | 60 | ||
| UiO-67 | 1 | 20 | 55 | ||
| ZIF-8 | 5 | 20 | 50 | ||
 | - | 2 | 4 |  | 75 |
| Zr-abtc | 1.5 | 12 | 92 | ||
| MIP-200 | 2 | 7 | 57 | ||
| UiO-66 | 4 | 10 | 80 | ||
| UiO-67 | 4 | 8 | 88 | ||
| ZIF-8 | 1.5 | 81 | 97 | ||
 | - | 1 | 4 |  | 50 (1:1) d |
| Zr-abtc | 1 | 15 | 87 (6:1) | ||
| MIP-200 | 1 | 5 | 40 (1:1) | ||
| UiO-66 | 0.5 | 8 | 38 (1:2) | ||
| UiO-67 | 1 | 11 | 64 (3:4) | ||
 | UiO-66 | 0.5 | 30 |  | 50 |
| Catalyst | Time, min | CyH conv. b (%) | Product Selectivity c (%) | ||
|---|---|---|---|---|---|
| Epoxide | Diol | Allylic d | |||
| – e | 60 | 6 | 16 | 16 | 65 |
| H+ f | 60 | 8 | 25 | 26 | 47 |
| MIL-125 | 60 | 26 | 25 | 10 | 60 |
| MIL-125 + 1 eq. H+ | 45 | 42 | 48 | 27 | 22 |
| UiO-66 | 60 | 16 | 35 | 10 | 50 |
| UiO-66 + 1 eq. H+ | 20 | 30 | 85 | 13 | 2 |
| UiO-67 | 30 | 17 | 28 | 25 | 43 |
| UiO-67 + 1 eq. H+ | 20 | 33 | 45 | 43 | 11 |
| Zr-abtc | 60 | 10 | 33 | 33 | 30 |
| Zr-abtc + 1 eq. H+ | 20 | 26 | 61 | 25 g | 7 |
| MIP-200 | 60 | 10 | 29 | 43 | 29 |
| MIP-200 + 1 eq. H+ | 20 | 27 | 70 | 19 h | 6 |
| MOF-801 | 60 | 10 | 35 | 20 | 45 |
| MOF-801 + 1 eq. H+ | 20 | 24 | 55 | 35 | 8 |
| Catalyst | Phenol Conversion (%) | Quinone Selectivity (%) | ||
|---|---|---|---|---|
| MIL-125 |  | 56 |  | 100 |
| MIL-125_NH2 | 50 | 100 | ||
| MIL-125 |  | 20 |  | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kholdeeva, O.; Maksimchuk, N. Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide. Catalysts 2021, 11, 283. https://doi.org/10.3390/catal11020283
Kholdeeva O, Maksimchuk N. Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide. Catalysts. 2021; 11(2):283. https://doi.org/10.3390/catal11020283
Chicago/Turabian StyleKholdeeva, Oxana, and Nataliya Maksimchuk. 2021. "Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide" Catalysts 11, no. 2: 283. https://doi.org/10.3390/catal11020283
APA StyleKholdeeva, O., & Maksimchuk, N. (2021). Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide. Catalysts, 11(2), 283. https://doi.org/10.3390/catal11020283