
Changes in CO2 Adsorption Affinity Related to Ni Doping in FeS Surfaces: A DFT-D3 Study

 , , ,
, , ,  and
and
Abstract
:1. Introduction
- (1)
- What is the energetic cost to form Ni defects in otherwise pristine surfaces of FeS?
- (2)
- How does substitutionally incorporated Ni distribute itself across the surfaces of FeS?
- (3)
- What influence does Ni exert on the adsorption of CO2 onto the surfaces of FeS?
2. Computational Details
2.1. Defect Calculations
2.2. Surface Calculations
3. Results and Discussion
3.1. Bulk and Pristine FeS Surfaces
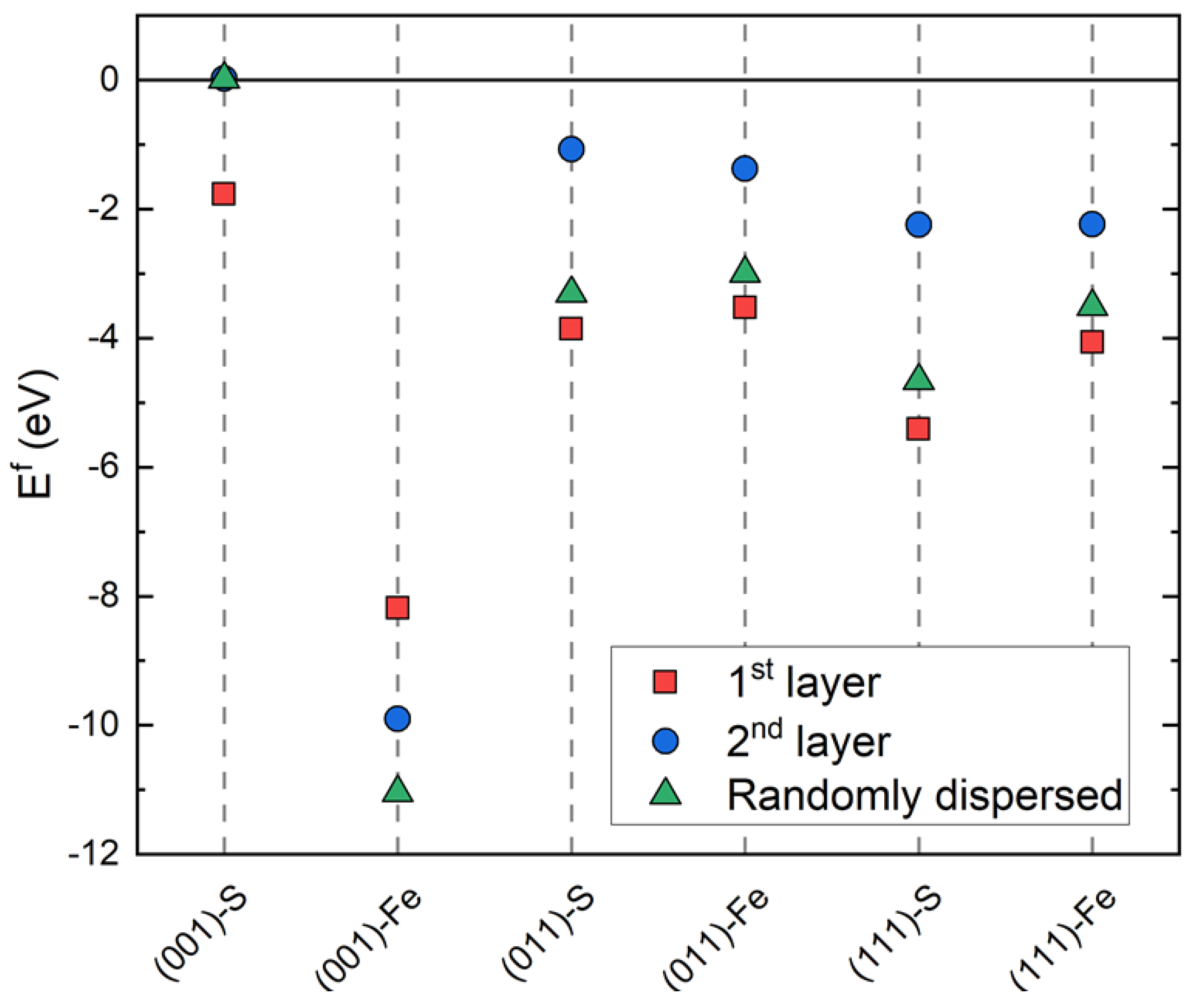
3.2. Ni-Doped FeS Surfaces
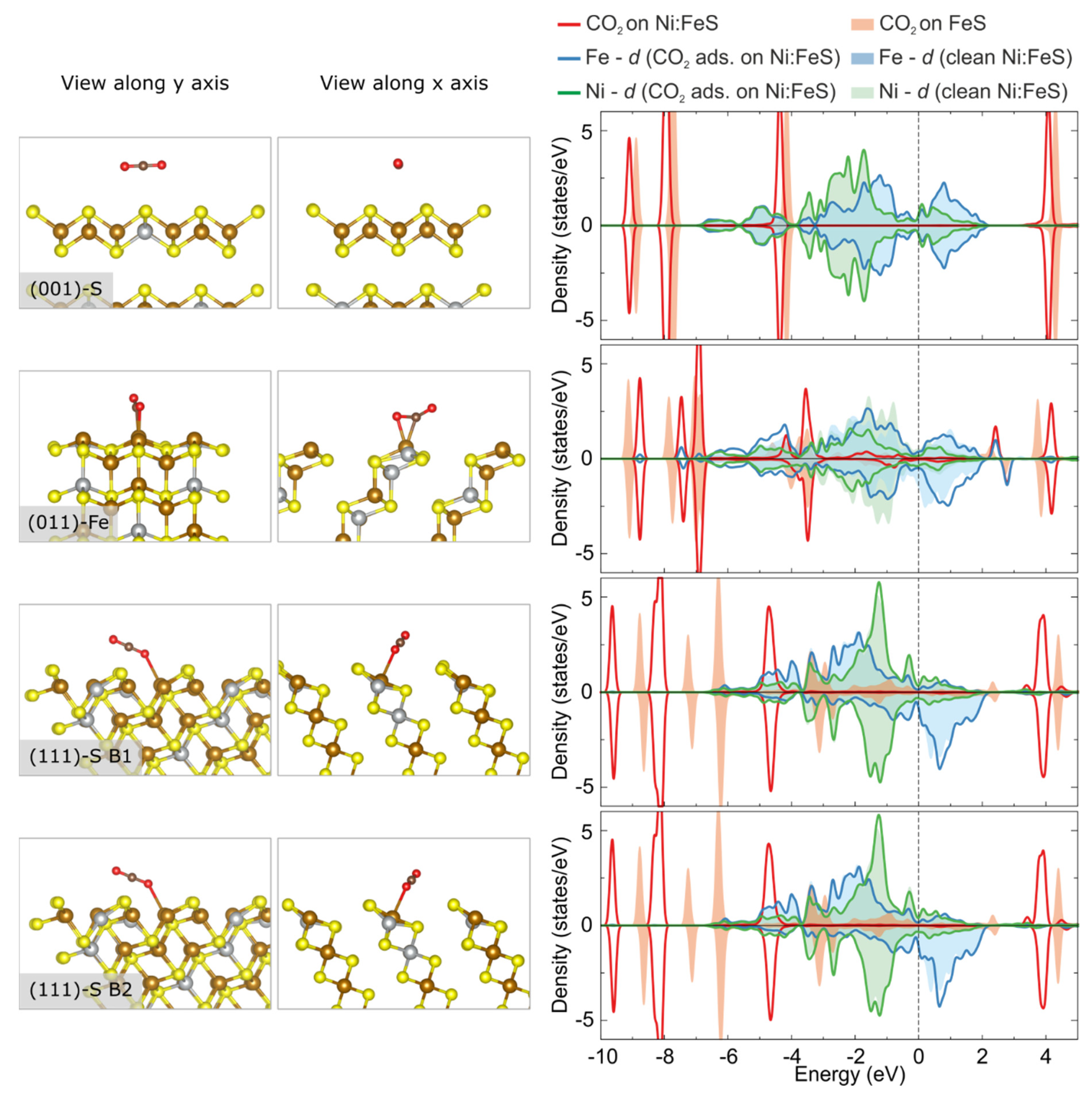
3.3. Adsorption and Activation of CO2 on Pristine vs. Ni-Doped FeS Surfaces
3.4. CO2 Dissociation on Ni-Doped FeS Surfaces
4. Conclusions
- a
- Ni is readily incorporated substitutionally at the Fe site into the FeS matrix, where low formation energies indicate that it may be difficult to control the dopant concentration.
- b
- FeS surfaces doped with Ni exhibit weaker binding as well as deactivation of adsorbed CO2 molecules, when compared to the same process on undoped mackinawite surfaces.
- c
- The (average) position of the d-band centre of the Ni-doped surfaces of FeS mackinawite is found at a consistently lower position than it is at the pristine surfaces. This is linked to the electronic configuration of Ni atoms, which is closer to a closed-shell system than that of the open d-orbitals of Fe atoms.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Handoko, A.D.; Wei, F.; Yeo, B.S.; Seh, Z.W. Understanding heterogeneous electrocatalytic carbon dioxide reduction through operando techniques. Nat. Catal. 2018, 1, 922–934. [Google Scholar] [CrossRef]
- Wansleben, M.; Vinson, J.; Wählisch, A.; Bzheumikhova, K.; Hönicke, P.; Beckhoff, B.; Kayser, Y. Speciation of iron sulfide compounds by means of X-ray emission spectroscopy using a compact full-cylinder von Hamos spectrometer. J. Anal. At. Spectrom. 2020, 35, 2679–2685. [Google Scholar] [CrossRef]
- Yang, X.; Wang, D. From Fundamental Principles to Materials and Applications. ACS Appl. Energy Mater. 2018, 1, 6657–6693. [Google Scholar] [CrossRef]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.; Lackner, K.S. Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew. Sustain. Energy Rev. 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Xu, S.; Carter, E.A. Theoretical Insights into Heterogeneous (Photo)electrochemical CO2 Reduction. Chem. Rev. 2019, 119, 6631–6669. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, A.; Khalilpour, K.R.; Milani, D.; Panahi, M. Trends in CO2 conversion and utilization: A review from process systems perspective. J. Environ. Chem. Eng. 2018, 6, 5771–5794. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, D.J.; Becker, U.; Wright, K. Sulphide mineral surfaces: Theory and experiment. Int. J. Miner. Process. 1997, 51, 1–14. [Google Scholar] [CrossRef]
- Vaughan, D.J. Sulfide Mineralogy and Geochemistry: Introduction and Overview. Rev. Mineral. Geochem. 2006, 61, 1–5. [Google Scholar] [CrossRef]
- Inosov, D.S. Quantum magnetism in minerals. Adv. Phys. 2018, 67, 149–252. [Google Scholar] [CrossRef] [Green Version]
- Rickard, D.; Luther, G.W. Chemistry of Iron Sulfides. Chem. Rev. 2007, 107, 514–562. [Google Scholar] [CrossRef] [PubMed]
- Huber, C. Peptides by Activation of Amino Acids with CO on (Ni,Fe)S Surfaces: Implications for the Origin of Life. Science 1998, 281, 670–672. [Google Scholar] [CrossRef] [Green Version]
- Drobner, E.; Huber, H.; Wächtershäuser, G.; Rose, D.; Stetter, K.O. Pyrite formation linked with hydrogen evolution under anaerobic conditions. Nature 1990, 346, 742–744. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.J.; Daniel, R.M.; Hall, A.J.; Sherringham, J.A. A hydrothermally precipitated catalytic iron sulphide membrane as a first step toward life. J. Mol. Evol. 1994, 39, 231–243. [Google Scholar] [CrossRef]
- Roldan, A.; Hollingsworth, N.; Roffey, A.; Islam, H.U.; Goodall, J.B.M.; Catlow, C.R.A.; Darr, J.A.; Bras, W.; Sankar, G.; Holt, K.B.; et al. Bio-inspired CO2 conversion by iron sulfide catalysts under sustainable conditions. Chem. Commun. 2015, 51, 7501–7504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herschy, B.; Whicher, A.; Camprubi, E.; Watson, C.; Dartnell, L.; Ward, J.; Evans, J.R.G.; Lane, N. An Origin-of-Life Reactor to Simulate Alkaline Hydrothermal Vents. J. Mol. Evol. 2014, 79, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Terranova, U.; Mitchell, C.; Sankar, M.; Morgan, D.; De Leeuw, N.H. Initial Oxygen Incorporation in the Prismatic Surfaces of Troilite FeS. J. Phys. Chem. C 2018, 122, 12810–12818. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, G.; Li, R.; Xiao, L.; Zhan, X. Iron sulphides mediated autotrophic denitrification: An emerging bioprocess for nitrate pollution mitigation and sustainable wastewater treatment. Water Res. 2020, 179, 115914. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Liao, Y.; Huang, J.; Dang, Z.; Guo, C. Removal of hexavalent chromium using biogenic mackinawite (FeS)-deposited kaolinite. J. Colloid Interface Sci. 2020, 572, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Vessey, C.J.; Lindsay, M.B.J. Aqueous Vanadate Removal by Iron(II)-Bearing Phases under Anoxic Conditions. Environ. Sci. Technol. 2020, 54, 4006–4015. [Google Scholar] [CrossRef] [PubMed]
- Cody, G.D.; Boctor, N.Z.; Brandes, J.A.; Filley, T.R.; Hazen, R.M.; Yoder, H.S. Assaying the catalytic potential of transition metal sulfides for abiotic carbon fixation. Geochim. Cosmochim. Acta 2004, 68, 2185–2196. [Google Scholar] [CrossRef]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. The surface chemistry of NOx on mackinawite (FeS) surfaces: A DFT-D2 study. Phys. Chem. Chem. Phys. 2014, 16, 15444–15456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. Adsorption of methylamine on mackinawite (FES) surfaces: A density functional theory study. J. Chem. Phys. 2013, 139, 124708. [Google Scholar] [CrossRef] [PubMed]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. Activation and dissociation of CO2 on the (001), (011), and (111) surfaces of mackinawite (FeS): A dispersion-corrected DFT study. J. Chem. Phys. 2015, 143, 094703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. Structures and Properties of As(OH)3 Adsorption Complexes on Hydrated Mackinawite (FeS) Surfaces: A DFT-D2 Study. Environ. Sci. Technol. 2017, 51, 3461–3470. [Google Scholar] [CrossRef] [Green Version]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. Surface and shape modification of mackinawite (FeS) nanocrystals by cysteine adsorption: A first-principles DFT-D2 study. Phys. Chem. Chem. Phys. 2016, 18, 32007–32020. [Google Scholar] [CrossRef] [Green Version]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. DFT-D2 simulations of water adsorption and dissociation on the low-index surfaces of mackinawite (FeS). J. Chem. Phys. 2016, 144, 174704. [Google Scholar] [CrossRef] [Green Version]
- Dzade, N.Y.; Roldan, A.; De Leeuw, N.H. DFT-D2 study of the adsorption and dissociation of water on clean and oxygen-covered {001} and {011} surfaces of mackinawite (FeS). J. Phys. Chem. C 2016, 120, 21441–21450. [Google Scholar] [CrossRef]
- Kolos, M.; Tunega, D.; Karlický, F. A theoretical study of adsorption on iron sulfides towards nanoparticle modeling. Phys. Chem. Chem. Phys. 2020, 22, 23258–23267. [Google Scholar] [CrossRef]
- Ofili, N.E.R.; Thetford, A.; Kaltsoyannis, N. Adsorption of U(VI) on Stoichiometric and Oxidised Mackinawite: A DFT Study. Environ. Sci. Technol. 2020, 54, 6792–6799. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, C.; Geng, Y.; Zhang, X.; Mei, J.; Yang, S. Remarkable differences between copper-based sulfides and iron-based sulfides for the adsorption of high concentrations of gaseous elemental mercury: Mechanisms, kinetics, and significance. J. Colloid Interface Sci. 2021, 582, 581–590. [Google Scholar] [CrossRef]
- Morse, J.W.; Arakaki, T. Adsorption and coprecipitation of divalent metals with mackinawite (FeS). Geochim. Cosmochim. Acta 1993, 57, 3635–3640. [Google Scholar] [CrossRef]
- Kwon, K.D.; Refson, K.; Sposito, G. Transition metal incorporation into mackinawite (tetragonal FeS). Am. Mineral. 2015, 100, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Wilkin, R.T.; Beak, D.G. Uptake of nickel by synthetic mackinawite. Chem. Geol. 2017, 462, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Ikogou, M.; Ona-Nguema, G.; Juillot, F.; Le Pape, P.; Menguy, N.; Richeux, N.; Guigner, J.M.; Noël, V.; Brest, J.; Baptiste, B.; et al. Long-term sequestration of nickel in mackinawite formed by Desulfovibrio capillatus upon Fe(III)-citrate reduction in the presence of thiosulfate. Appl. Geochemistry 2017, 80, 143–154. [Google Scholar] [CrossRef]
- Swanner, E.D.; Webb, S.M.; Kappler, A. Fate of cobalt and nickel in mackinawite during diagenetic pyrite formation. Am. Mineral. 2019, 104, 917–928. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Yu, M.; Trinkle, D.R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.-T. VASPKIT: A Pre- and Post-Processing Program for VASP code. arXiv 2019, arXiv:1908.08269. [Google Scholar]
- Sheppard, D.; Terrell, R.; Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 2008, 128, 134106. [Google Scholar]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; Van De Walle, C.G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253–305. [Google Scholar] [CrossRef]
- Watson, G.W.; Kelsey, E.T.; De Leeuw, N.H.; Harris, D.J.; Parker, S.C. Atomistic simulation of dislocations, surfaces and interfaces in MgO. J. Chem. Soc. Faraday Trans. 1996, 92, 433. [Google Scholar] [CrossRef]
- Farkaš, B.; Santos-Carballal, D.; Cadi-Essadek, A.; De Leeuw, N.H. A DFT+U study of the oxidation of cobalt nanoparticles: Implications for biomedical applications. Materialia 2019, 7, 100381. [Google Scholar] [CrossRef]
- Wolthers, M.; Van Der Gaast, S.J.; Rickard, D. The structure of disordered mackinawite. Am. Mineral. 2003, 88, 2007–2015. [Google Scholar] [CrossRef]
- Mansor, M.; Winkler, C.; Hochella, M.F.; Xu, J. Nanoparticulate Nickel-Hosting Phases in Sulfidic Environments: Effects of Ferrous Iron and Bacterial Presence on Mineral Formation Mechanism and Solid-Phase Nickel Distribution. Front. Earth Sci. 2019, 7, 151. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Hudson, R.; De Graaf, R.; Rodin, M.S.; Ohno, A.; Lane, N.; McGlynn, S.E.; Yamada, Y.M.A.; Nakamura, R.; Barge, L.M.; Braun, D.; et al. CO2 reduction driven by a pH gradient. Proc. Natl. Acad. Sci. USA 2020, 117, 22873–22879. [Google Scholar] [CrossRef] [PubMed]
- Dzade, N.Y.; De Leeuw, N.H. Activating the FeS (001) Surface for CO2 Adsorption and Reduction through the Formation of Sulfur Vacancies: A DFT-D3 Study. Catalysts 2021, 11, 127. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | Termination | Relaxed Surface Energy, γrelaxed (J/m2) | Other Theoretical Works [24] |
|---|---|---|---|
| (001) | S | 0.23 | 0.19 |
| Fe | 3.55 | N/A | |
| (011) | S | 1.14 | 1.47 |
| Fe | 1.15 | 0.95 | |
| (111) | S | 1.27 | 1.51 |
| Fe | 1.66 | 1.69 |
| Pristine Surface | Adsorption Position | Eads (eV) | (O-C-O) (°) | O1-C/O2-C Stretching (%) |
|---|---|---|---|---|
| (001)-S | Top Fe | −0.01 | 180.0 | 0.0/0.0 |
| Top S | −0.01 | 180.0 | 0.0/0.0 | |
| (001)-Fe | Top Fe | No stable configuration/surface reconfiguration observed | ||
| Top S | No stable configuration/surface reconfiguration observed | |||
| (011)-S | Top Fe | −0.23 | 178.6 | 0.7/−0.7 |
| Top S | 0.01 | 180.0 | 0.0/0.0 | |
| (011)-Fe | Top Fe | −0.54 | 138.2 | 6.6/5.9 |
| Top S | −0.17 | 178.9 | −0.5/0.6 | |
| (111)-S | Top Fe | 0.33 | 133.0 | 9.6/2.9 |
| Top S | No stable config | |||
| B1 | −0.99 | 133.4 | 5.6/5.6 | |
| B2 | −1.29 | 139.2 | 3.1/5.6 | |
| (111)-Fe | Top Fe | No stable configuration/surface reconfiguration observed | ||
| Top S | No stable configuration/surface reconfiguration observed | |||
| Ni-Doped Surface | Adsorption Position | Eads (eV) | (O-C-O) (°) | O1-C/O2-C Stretching (%) |
|---|---|---|---|---|
| (001)-S | Top Fe | 0.06 | 180.0 | 0.0/0.0 |
| Top S-1 | 0.07 | 180.0 | 0.0/0.0 | |
| Top S-2 | 0.08 | 180.0 | 0.0/0.0 | |
| Top Ni | 0.06 | 180.0 | 0.0/0.0 | |
| (011)-Fe | Top Fe | −0.14 | 148.1 | 6.2/1.9 |
| Top S | −0.09 | 179.2 | 0.1/−0.1 | |
| (111)-S | B1 | 0.14 | 139.5 | 3.1/6.6 |
| B1-alt | −0.11 | 178.9 | −0.6/0.6 | |
| B2 | −0.53 | 143.9 | 5.7/1.8 | |
| B2-alt | −0.78 | 178.9 | −0.6/0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Živković, A.; Somers, M.; Camprubi, E.; King, H.E.; Wolthers, M.; de Leeuw, N.H. Changes in CO2 Adsorption Affinity Related to Ni Doping in FeS Surfaces: A DFT-D3 Study. Catalysts 2021, 11, 486. https://doi.org/10.3390/catal11040486
Živković A, Somers M, Camprubi E, King HE, Wolthers M, de Leeuw NH. Changes in CO2 Adsorption Affinity Related to Ni Doping in FeS Surfaces: A DFT-D3 Study. Catalysts. 2021; 11(4):486. https://doi.org/10.3390/catal11040486
Chicago/Turabian StyleŽivković, Aleksandar, Michiel Somers, Eloi Camprubi, Helen E. King, Mariette Wolthers, and Nora H. de Leeuw. 2021. "Changes in CO2 Adsorption Affinity Related to Ni Doping in FeS Surfaces: A DFT-D3 Study" Catalysts 11, no. 4: 486. https://doi.org/10.3390/catal11040486
APA StyleŽivković, A., Somers, M., Camprubi, E., King, H. E., Wolthers, M., & de Leeuw, N. H. (2021). Changes in CO2 Adsorption Affinity Related to Ni Doping in FeS Surfaces: A DFT-D3 Study. Catalysts, 11(4), 486. https://doi.org/10.3390/catal11040486