
Chlorodifluoromethane Hydrodechlorination on Carbon-Supported Pd-Pt Catalysts. Beneficial Effect of Catalyst Oxidation

 ,
,  and
and
Abstract
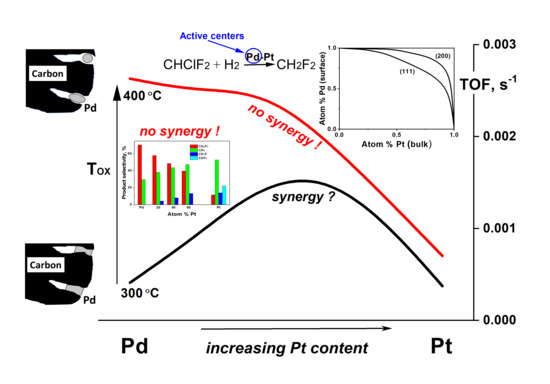
:
1. Introduction
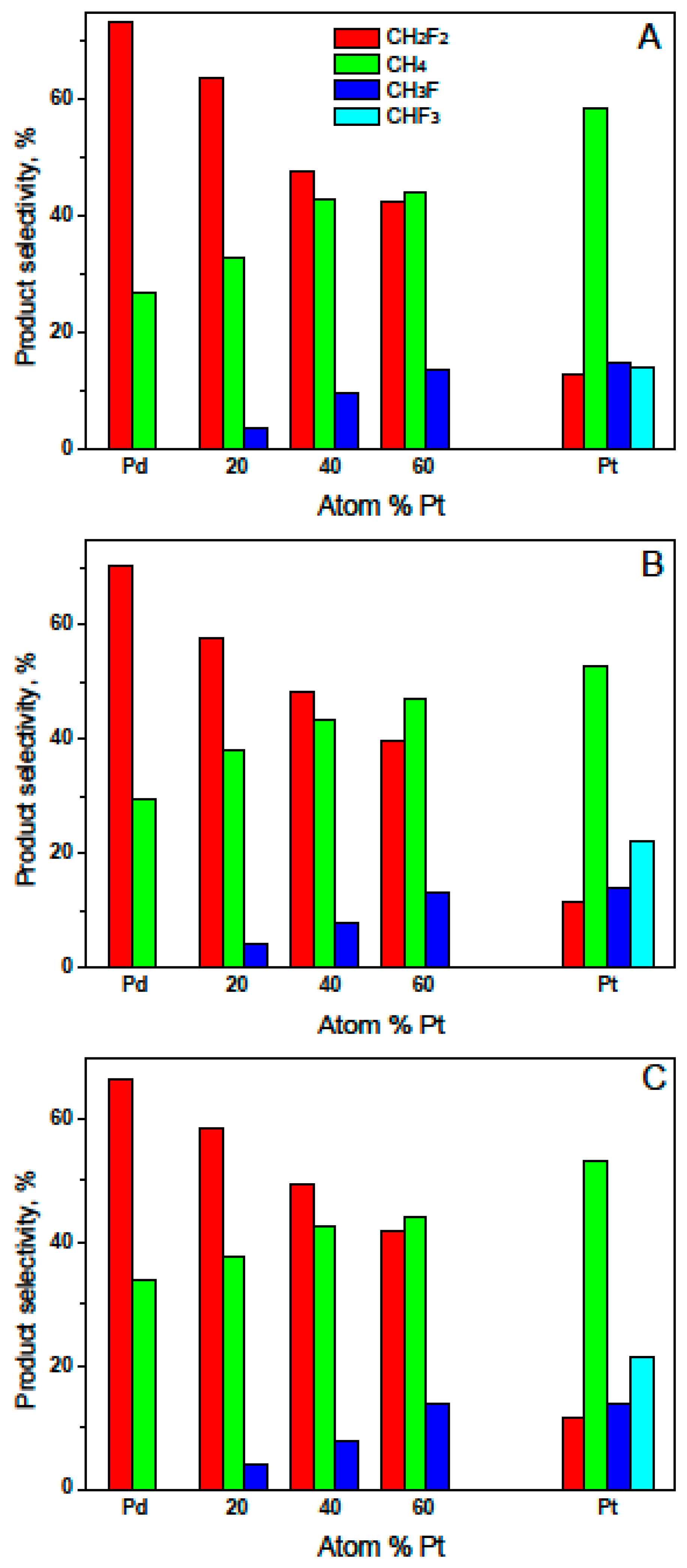
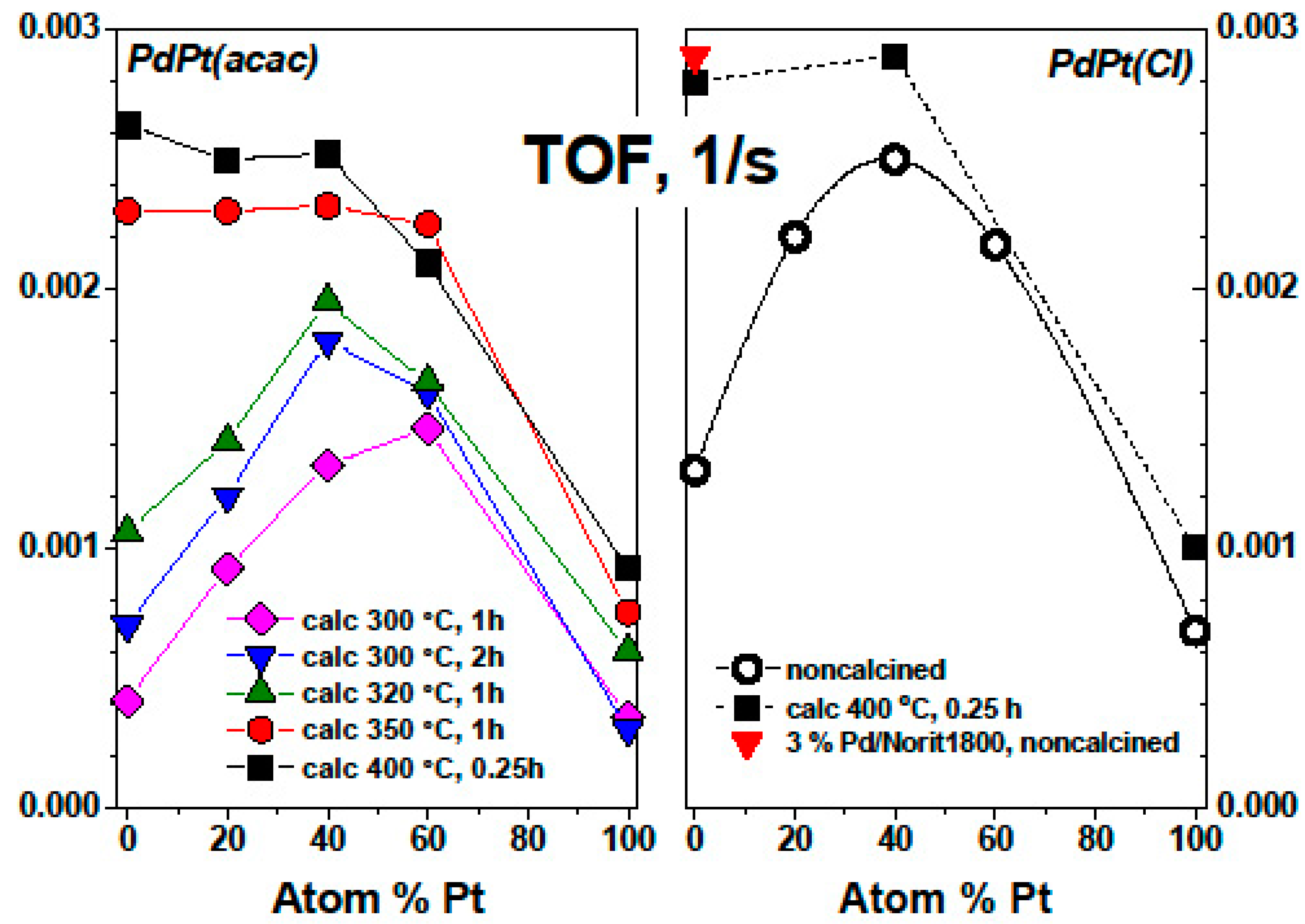
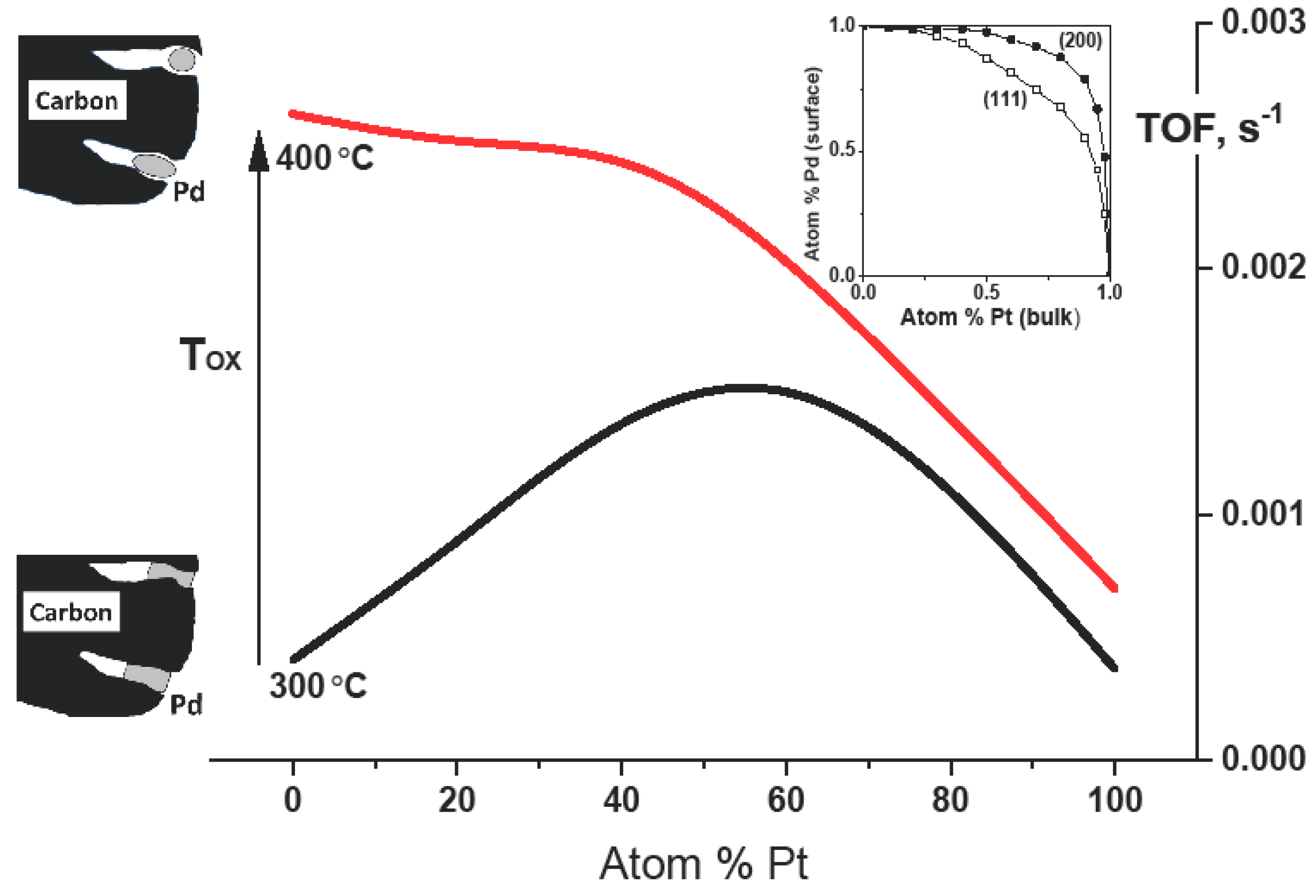
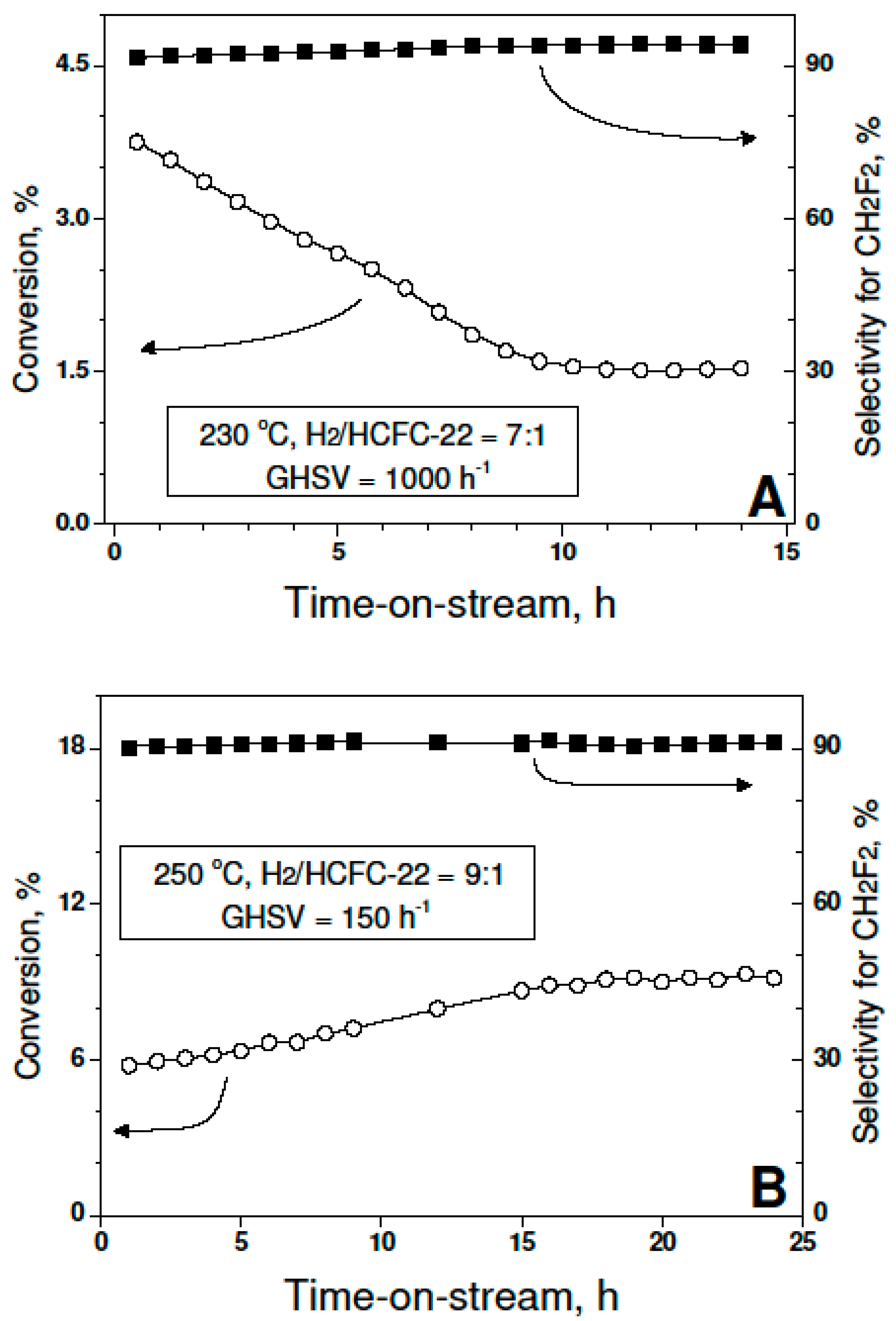
2. Results and Discussion
- (1)
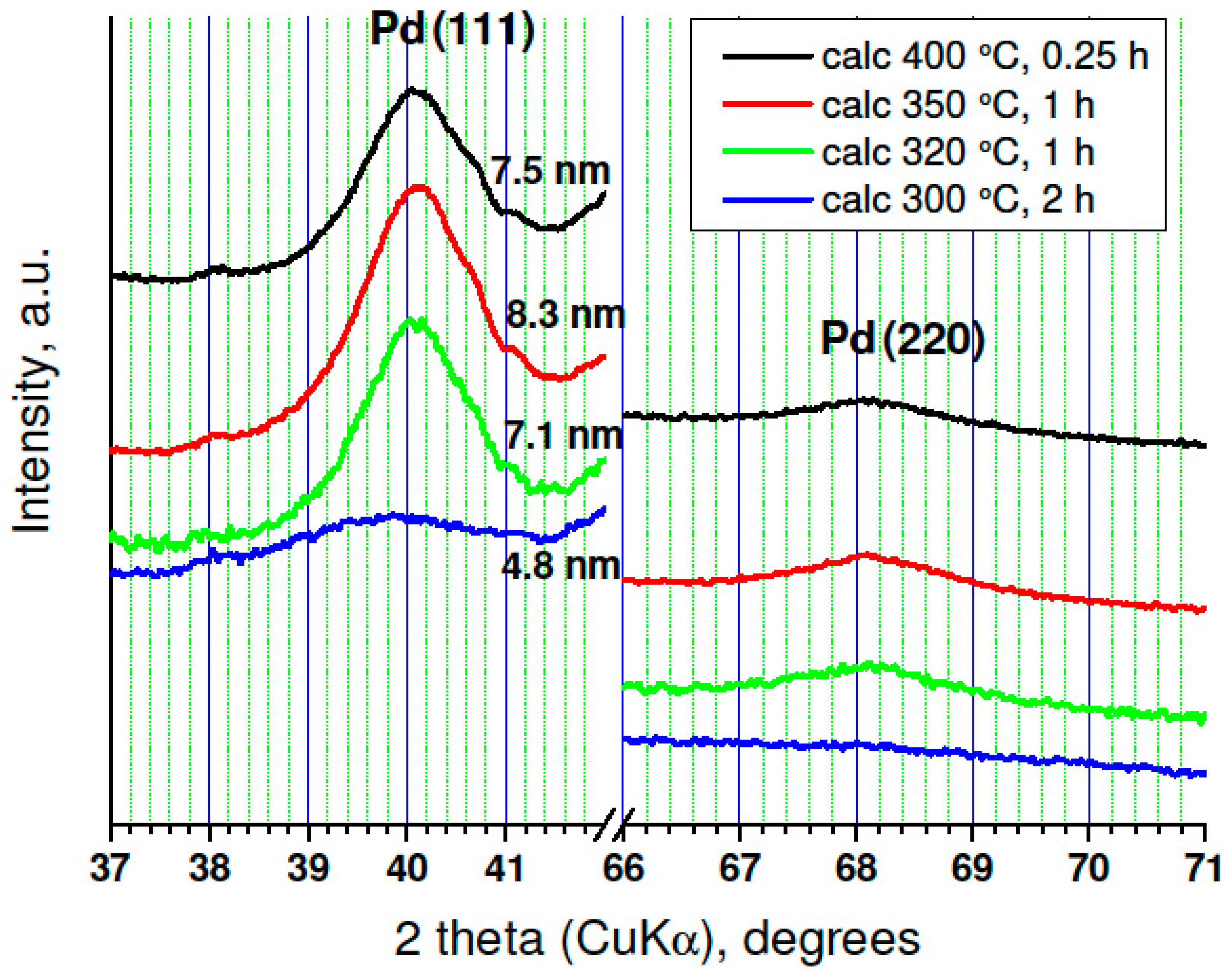
- Oxidation of Pd-on-highly preheated Norit catalyst, which led to nearly an order of magnitude increase in catalytic activity, could not be the result of marked changes in metal dispersion changes because such changes were not found.
- (2)
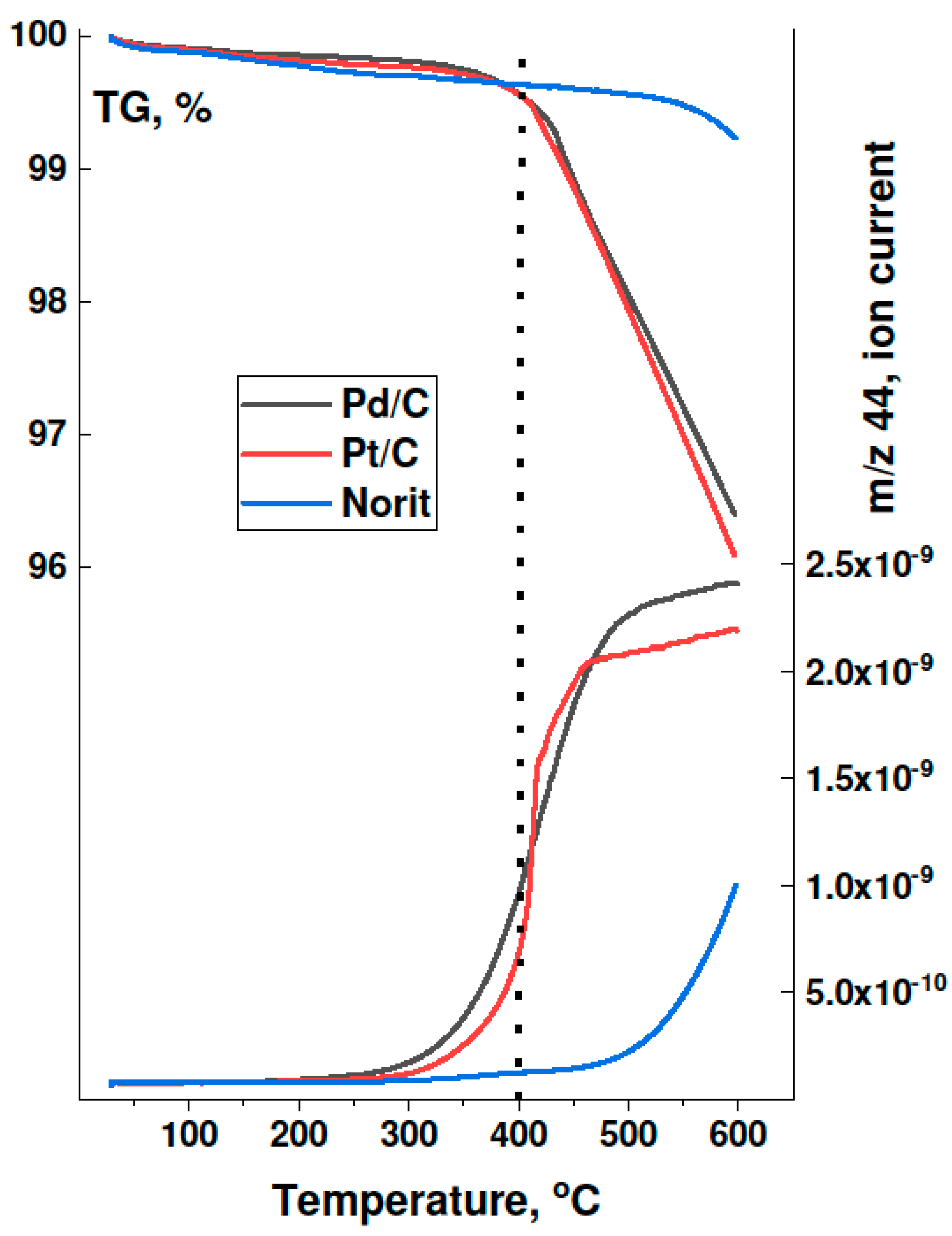
- Possible decontamination of palladium surface from carbon by oxidation at 350–400 °C was also rejected as a basic reason for the activity increase. Preliminary precalcination of 2 wt % Pd100(acac)/Norit1600 samples at 300 °C for 1–2 h should remove the carbon from the metal surface [21,22]. TPO profiles of such catalysts did not contain any signs of presence of such carbon species.
- (3)
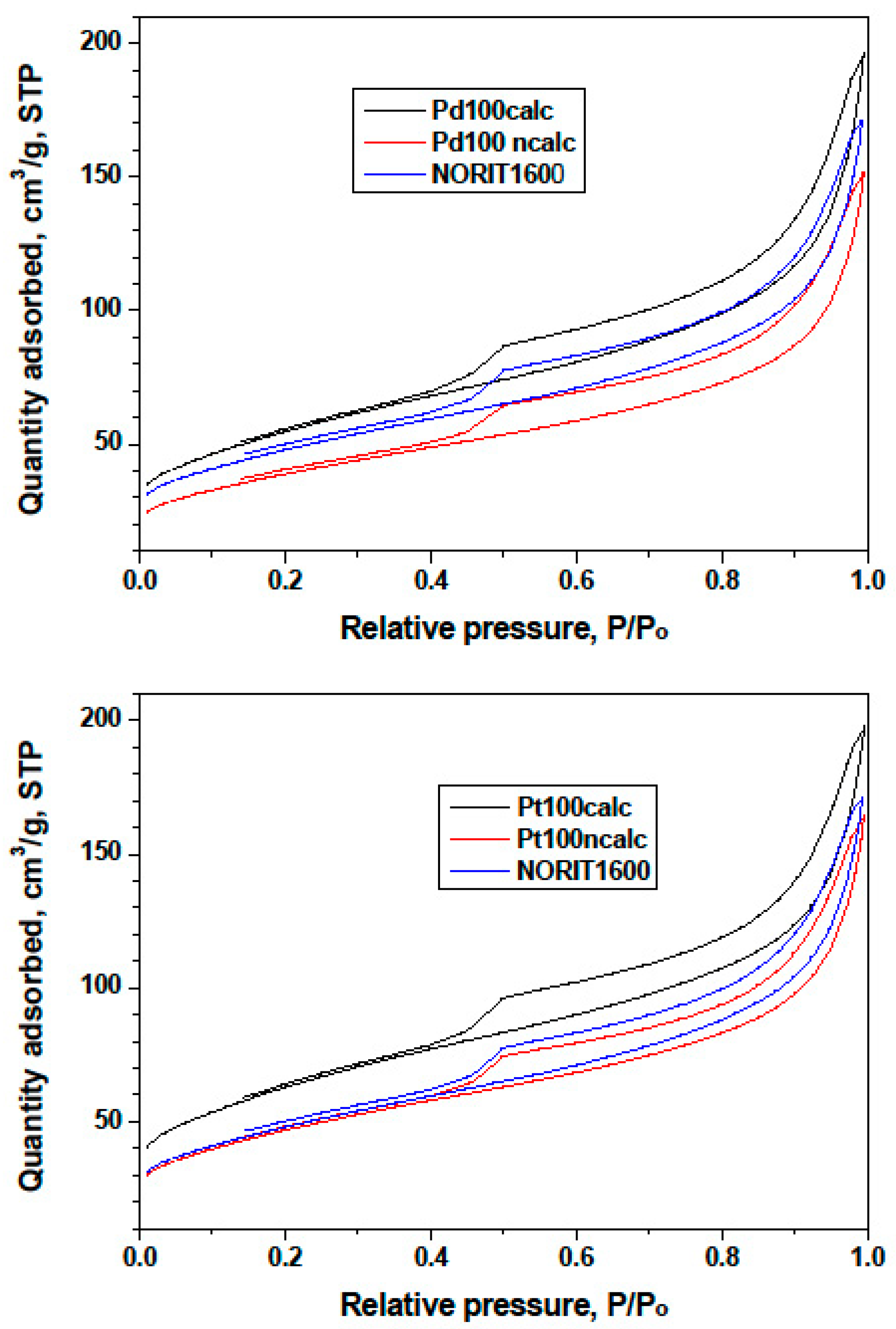
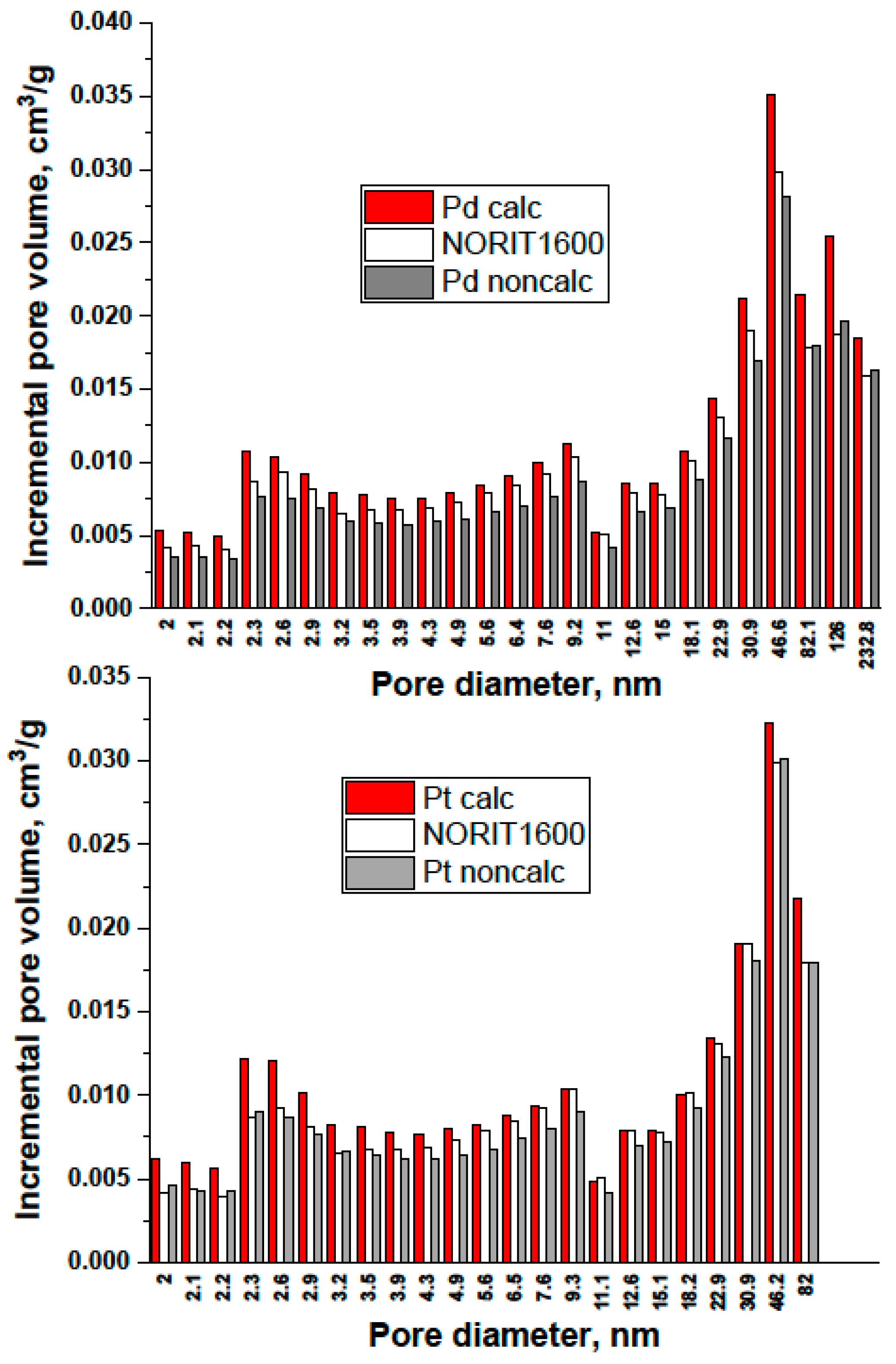
- TGA-MS studies show the beginning of a massive removal of a “proximate” carbon at the temperature 350 °C. Metal-catalyzed burning of carbon support changes the pore structure of Norit1600. In particular, the micropore volume was vastly increased along with catalyst oxidation. Metal nanoparticles, wetting small pores of the support, presumably lose contact with the pore walls as a result of oxidation. Such a catalyst represents enhanced reactivity, proving the accessibility of the active sites to reactants.
3. Methods
3.1. Catalyst Preparation
3.2. Catalytic Tests
3.3. Catalyst Characterization by Hydrogen Chemisorption, XRD, TPO, and Physical Adsorption (BET, Pore Structure)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Department for Environment, Food and Rural Affairs Guidance for Stationary Refrigeration & Air-Conditioning the EU Ozone Regulation: Legislative Update and Strategies for HCFC Phase-Out Information Sheet RAC 8: R22 Phase-Out, April 2012, Guidance for Stationary Refrigeration & Air-Conditioning (publishing.service.gov.uk). Available online: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/394852/fgas-rac8-hcfc-phase-out.pdf (accessed on 30 March 2021).
- Montzka, S.A.; McFarland, M.; Andersen, S.O.; Miller, B.R.; Fahey, D.W.; Hall, B.D.; Hu, L.; Siso, C.; Elkins, J.W. Recent Trends in Global Emissions of Hydrochlorofluorocarbons and Hydrofluorocarbons: Reflecting on the 2007 Adjustments to the Montreal Protocol. J. Phys. Chem. A 2015, 119, 4439–4449. [Google Scholar] [CrossRef] [PubMed]
- Montzka, S.A.; Dutton, G.S.; Yu, P.; Ray, E.; Portmann, R.W.; Daniel, J.S.; Kuijpers, L.; Hall, B.D.; Mondeel, D.; Siso, C.; et al. An unexpected and persistent increase in global emissions of ozone-depleting CFC-11. Nat. Cell Biol. 2018, 557, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Prignon, M.; Chabrillat, S.; Minganti, D.; O’Doherty, S.; Servais, C.; Stiller, G.; Toon, G.C.; Vollmer, M.K.; Mahieu, E. Improved FTIR Retrieval Strategy for HCFC-22 (CHClF2), Comparisons with in Situ and Satellite Datasets with the Support of Models, and Determination of its Long-Term Trend Above Jungfraujoch. Atmos. Chem. Phys. Discuss. 2019, 19, 12309–12324. [Google Scholar] [CrossRef] [Green Version]
- Oram, D.E.; Ashfold, M.J.; Laube, J.C.; Gooch, L.J.; Humphrey, S.; Sturges, W.T.; Leedham-Elvidge, E.; Forster, G.L.; Harris, N.R.P.; Mead, M.I.; et al. A growing threat to the ozone layer from short-lived anthropogenic chlorocarbons. Atmos. Chem. Phys. Discuss. 2017, 17, 11929–11941. [Google Scholar] [CrossRef] [Green Version]
- Morato, A.; Alonso, C.; Medina, F.; Cesteros, Y.; Salagre, P.; E Sueiras, J.; Tichit, D.; Coq, B. Palladium Hydrotalcites as Precursors for the Catalytic Hydroconversion of CCl2F2 (CFC-12) and CHClF2 (HCFC-22). Appl. Catal. B Environ. 2001, 32, 167–179. [Google Scholar] [CrossRef]
- Morato, A.; Medina, F.; Sueiras, J.E.; Cesteros, Y.; Salagre, P.; De Menorval, L.-C.; Tichit, D.; Coq, B. Characterization and Catalytic Properties of Several KMg1−xPdxF3 with Perovskite-like Structures for the Hydroconversion of CHClF2. Appl. Catal. B Environ. 2003, 42, 251–264. [Google Scholar] [CrossRef]
- Yu, H.; Kennedy, E.M.; Uddin, A.; Sakata, Y.; Dlugogorski, B.Z. Gas-Phase and Pd-Catalyzed Hydrodehalogenation of CBrClF2, CCl2F2, CHClF2, and CH2F2. Ind. Eng. Chem. Res. 2005, 44, 3442–3452. [Google Scholar] [CrossRef]
- Legawiec-Jarzyna, M.; Śrębowata, A.; Juszczyk, W.; Karpiński, Z. Hydrodechlorination over Pd-Pt/Al2O3 Catalysts A Comparative Study of Chlorine Removal from Dichlorodifluoromethane, Carbon Tetrachloride and 1,2-Dichloroethane. Appl. Catal. A 2004, 271, 61–68. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Bedia, J.; Arevalo-Bastante, A.; Rodriguez, J.J. Enhanced Activity of Carbon-Supported Pd-Pt Catalysts in the Hydrodechlorination of Dichloromethane. Appl. Catal. B 2016, 184, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Bedia, J.; Gómez-Sainero, L.M.; Grau, J.M.; Busto, M.; Martin-Martinez, M.; Rodriguez, J.J. Hydrodechlorination of Dichloromethane with Mono- and Bimetallic Pd-Pt on Sulfated and Tungstated Zirconia Catalysts. J. Catal. 2012, 294, 207–215. [Google Scholar] [CrossRef]
- Garcia, C.M.; Woolfolk, L.G.; Martin, N.; Granados, A.; de los Reyes, J.A. Evaluation of Mono and Bimetallic Catalysts Supported on Al2O3-TiO2 in the Reaction of Hydrodechlorination of 1,2-Dichloroethane. Rev. Mex. Ing. Quim. 2012, 11, 463–468. [Google Scholar]
- Han, W.; Li, X.; Liu, B.; Li, L.; Tang, H.; Li, Y.; Ch, L.; Ch, X.; Li, X. Microwave Assisted Combustion of Phytic Acid for the Preparation of Ni2P@C as a Robust Catalyst for Hydrodechlorination. Chem. Commun. 2019, 55, 9279–9282. [Google Scholar] [CrossRef]
- Schoebrechts, J.-P.; Wilmet, V. Catalytic System Comprising a Hydrogenation Catalyst on a Support and Process for the Hydrodechlorination of Chlorofluorinated Hydrocarbons. US Patent 5561069, 1 October 1996. [Google Scholar]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Palomar, J.; Omar, S.; Rodriguez, J.J. Dechlorination of Dichloromethane by Hydrotreatment with Bimetallic Pd-Pt/C Catalyst. Catal. Lett. 2016, 146, 2614–2621. [Google Scholar] [CrossRef]
- Patil, P.T.; Dimitrov, A.; Kirmse, H.; Neumann, W.; Kemnitz, E. Non-Aqueous Sol–Gel Synthesis, Characterization and Catalytic Properties of Metal Fluoride Supported Palladium Nanoparticles. Appl. Catal. B 2008, 78, 80–91. [Google Scholar] [CrossRef]
- Radlik, M.; Juszczyk, W.; Matus, K.; Raróg-Pilecka, W.; Karpiński, Z. Hydrodechlorination of CHClF2 (HCFC-22) Over Pd–Pt Catalysts Supported on Thermally Modified Activated Carbon. Catalysts 2020, 10, 1291. [Google Scholar] [CrossRef]
- Chen, N.; Rioux, R.M.; Barbosa, L.A.M.M.; Ribeiro, F.H. Kinetic and Theoretical Study of the Hydrodechlorination of CH4-xClx (x = 1 − 4) Compounds on Palladium. Langmuir 2010, 26, 16615–16624. [Google Scholar] [CrossRef]
- Kumara, L.S.R.; Sakata, O.; Kobayashi, H.; Song, C.; Kohara, S.; Ina, T.; Yoshimoto, T.; Yoshioka, S.; Matsumura, S.; Kitagawa, H. Hydrogen Storage and Stability Properties of Pd–Pt Solid-Solution Nanoparticles Revealed via Atomic and Electronic Structure. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Krishnankutty, N.; Li, J.; Vannice, M.A. The Effect of Pd Precursor and Pretreatment on the Adsorption and Absorption Behavior of Supported Pd Catalysts. Appl. Catal. A Gen. 1998, 173, 137–144. [Google Scholar] [CrossRef]
- Tengco, J.M.M.; Lugo-José, Y.K.; Monnier, J.R.; Regalbuto, J.R. Chemisorption–XRD Particle Size Discrepancy of Carbon Supported Palladium: Carbon Decoration of Pd? Catal. Today 2015, 246, 9–14. [Google Scholar] [CrossRef]
- Banerjee, R.; Regalbuto, J.R. Rectifying the Chemisorption–XRD Discrepancy of Carbon Supported Pd: Residual Chloride and/or Carbon Decoration. Appl. Catal. A Gen. 2020, 595, 117504. [Google Scholar] [CrossRef]
- Bonarowska, M.; Burda, B.; Juszczyk, W.; Pielaszek, J.; Kowalczyk, Z.; Karpinski, Z. Hydrodechlorination of CCl2F2 (CFC-12) over Pd-Au/C catalysts. Appl. Catal. B Environ. 2001, 35, 13–20. [Google Scholar] [CrossRef]
- Ichikawa, S.; Poppa, H.; Boudart, M. Disproportionation of CO on Small Particles of Silica-Supported Palladium. J. Catal. 1985, 91, 1–10. [Google Scholar] [CrossRef]
- Lee, S.; Kahng, S.-J.; Kuk, Y. Nano-Level Wettings of Platinum and Palladium on Single-Walled Carbon Nanotubes. Chem. Phys. Lett. 2010, 500, 82–85. [Google Scholar] [CrossRef]
- Uchisawa, J.O.; Obuchi, A.; Zhao, Z.; Kushiyama, S. Carbon Oxidation with Platinum Supported Catalysts. Appl. Catal. B Environ. 1998, 18, L183–L187. [Google Scholar] [CrossRef]
- Baker, R.T.K.; France, J.A.; Rouse, L.; Waite, R.J. Catalytic Oxidation of Graphite by Platinum and Palladium. J. Catal. 1976, 41, 22–29. [Google Scholar] [CrossRef]
- Rousset, J.L.; Bertolini, J.C.; Miegge, P. Theory of Segregation Using the Equivalent-Medium Approximation and Bond-Strength Modifications at Surfaces: Application to Fcc Pd-X Alloys. Phys. Rev. B 1996, 53, 4947–4957. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Li, J.; Wang, S.; Li, G.; Liu, X. Hydrodechlorination and Deep Hydrogenation on Single-Palladium-Atom-Based Heterogeneous Catalysts. Appl. Catal. B Environ. 2021, 282, 119518. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, S.; Xu, J.; Wei, K. Effect of Thermal and Oxidative Treatments of Activated Carbon on its Surface Structure and Suitability as a Support for Barium-Promoted Ruthenium in Ammonia Synthesis Catalysts. Carbon 2002, 40, 2597–2603. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Hydrogen Adsorption on Supported Cobalt, Iron, and Nickel. Catal. Lett. 1991, 7, 27–51. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Norit1600 | 2 wt % Pd100(acac)/Norit1600 | 2 wt % Pt100(acac)/Norit1600 | |||
|---|---|---|---|---|---|---|
| Noncalcined a | Calcined b | Noncalcined a | Calcined b | |||
| BET surface area, m2/g | 167.8 | 137.3 | 192.6 | 164.1 | 219.7 | |
| BJH pore volume, cm3/g | from adsorption | 0.254 | 0.229 | 0.292 | 0.242 | 0.286 |
| from desorption | 0.259 | 0.247 | 0.302 | 0.250 | 0.294 | |
| tplot micropore volume, cm3/g | 0.00577 | 0.00188 | 0.00335 | 0.00397 | 0.00667 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radlik, M.; Juszczyk, W.; Raróg-Pilecka, W.; Zybert, M.; Karpiński, Z. Chlorodifluoromethane Hydrodechlorination on Carbon-Supported Pd-Pt Catalysts. Beneficial Effect of Catalyst Oxidation. Catalysts 2021, 11, 525. https://doi.org/10.3390/catal11050525
Radlik M, Juszczyk W, Raróg-Pilecka W, Zybert M, Karpiński Z. Chlorodifluoromethane Hydrodechlorination on Carbon-Supported Pd-Pt Catalysts. Beneficial Effect of Catalyst Oxidation. Catalysts. 2021; 11(5):525. https://doi.org/10.3390/catal11050525
Chicago/Turabian StyleRadlik, Monika, Wojciech Juszczyk, Wioletta Raróg-Pilecka, Magdalena Zybert, and Zbigniew Karpiński. 2021. "Chlorodifluoromethane Hydrodechlorination on Carbon-Supported Pd-Pt Catalysts. Beneficial Effect of Catalyst Oxidation" Catalysts 11, no. 5: 525. https://doi.org/10.3390/catal11050525
APA StyleRadlik, M., Juszczyk, W., Raróg-Pilecka, W., Zybert, M., & Karpiński, Z. (2021). Chlorodifluoromethane Hydrodechlorination on Carbon-Supported Pd-Pt Catalysts. Beneficial Effect of Catalyst Oxidation. Catalysts, 11(5), 525. https://doi.org/10.3390/catal11050525