
Enzymatic Polymerization of Dihydroquercetin (Taxifolin) in Betaine-Based Deep Eutectic Solvent and Product Characterization

Abstract
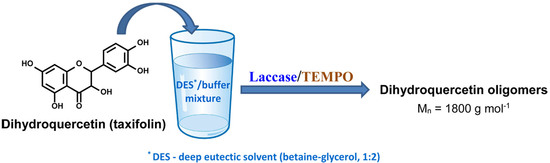
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Enzyme
3.3. Preparation of DESs and DES–Buffer Mixtures
3.4. Determination of the Activity and Stability of Laccase in DESs and DES–Buffer Mixture
3.5. Enzymatic Polymerization of DHQ in DES–Buffer Mixture
3.6. Characterization of DHQ Polymerization Product
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rogers, R.; Seddon, K. Ionic Liquids—Solvents of the Future? Science 2003, 302, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Plechkova, N.; Seddon, K. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Vekariya, R. A review of ionic liquids: Applications towards catalytic organic transformations. J. Mol. Liq. 2017, 227, 44–60. [Google Scholar] [CrossRef]
- Wells, A.; Coombe, V. On the Freshwater Ecotoxicity and Biodegradation Properties of Some Common Ionic Liquids. Org. Process Res. Dev. 2006, 41, 797–828. [Google Scholar] [CrossRef]
- Abbott, A.; Boothby, D.; Capper, G.; Davies, D.; Rasheed, R. Deep Eutectic Solvents Formed between Choline Chloride and Carboxylic Acids: Versatile Alternatives to Ionic Liquids. J. Am. Chem. Soc. 2004, 126, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Abbott, A.; Ryder, K. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [PubMed]
- Wazeer, I.; Hayyan, M.; Hadj-Kali, M. Deep eutectic solvents: Designer fluids for chemical processes. J. Chem. Technol. Biotechnol. 2018, 93, 945–958. [Google Scholar] [CrossRef]
- Hansen, B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.; et al. Deep eutectic solvents: A review of fundamentals and applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y. Tailoring properties of natural deep eutectic solvents with water to facilitate their applications. Food Chem. 2015, 187, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Vanda, H.; Dai, Y.; Wilson, E.; Verpoorte, R.; Choi, Y. Green solvents from ionic liquids and deep eutectic solvents to natural deep eutectic solvents. C. R. Chim. 2018, 21, 628–638. [Google Scholar] [CrossRef]
- Cicco, L.; Dilauro, G.; Perna, F.; Vitale, P.; Capriati, V. Advances in deep eutectic solvents and water: Applications in metal- and biocatalyzed processes, in the synthesis of APIs, and other biologically active compounds. Org. Biomol. Chem. 2021, 19, 2558–2577. [Google Scholar] [CrossRef] [PubMed]
- Tomé, L.; Baião, V.; Silva, W.; Brett, C. Deep eutectic solvents for the production and application of new materials. Appl. Mater. Today 2018, 10, 30–50. [Google Scholar] [CrossRef]
- Mbous, Y.; Hayyan, M.; Hayyan, A.; Wong, W.; Hashim, M.; Looi, C. Applications of deep eutectic solvents in biotechnology and bioengineering—Promises and challenges. Biotechnol. Adv. 2017, 35, 105–134. [Google Scholar] [CrossRef] [PubMed]
- Wescott, C.R.; Klibanov, A.M. The solvent dependence of enzyme specificity. BBA Protein Struct. M. 1994, 1206, 1–9. [Google Scholar] [CrossRef]
- Rasor, J.P.; Voss, E. Enzyme-catalyzed processes in pharmaceutical industry. Appl. Catal. A Gen. 2001, 221, 145–158. [Google Scholar] [CrossRef]
- Juneidi, I.; Hayyan, M.; Hashim, M.A. Intensification of biotransformations using deep eutectic solvents: Overview and outlook. Process Biochem. 2018, 66, 33–60. [Google Scholar] [CrossRef]
- Pätzold, M.; Siebenhaller, S.; Kara, S.; Liese, A.; Syldatk, C.; Holtmann, D. Deep Eutectic Solvents as Efficient Solvents in Biocatalysis. Trends Biotechnol. 2019, 37, 943–959. [Google Scholar] [CrossRef]
- Xu, P.; Zheng, G.W.; Zong, M.H.; Li, N.; Lou, W.Y. Recent progress on deep eutectic solvents in biocatalysis. Bioresour. Bioprocess. 2017, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Hassani, F.; Amzazi, S.; Lavandera, I. The versatile applications of DES and their influence on oxidoreductase-mediated transformations. Molecules 2019, 24, 2190–2208. [Google Scholar] [CrossRef]
- Gotor-Fernández, V.; Paul, C. Deep eutectic solvents for redox biocatalysis. J. Biotechnol. 2019, 293, 24–35. [Google Scholar] [CrossRef]
- Panić, M.; Bubalo, M.C.; Redovniković, I.R. Designing a biocatalytic process involving deep eutectic solvents. J. Chem. Technol. Biotechnol. 2021, 96, 14–30. [Google Scholar] [CrossRef]
- Erol, Ö.; Hollmann, F. Natural deep eutectic solvents as performance additives for biocatalysis. Adv. Bot. Res. 2021, 97, 95–132. [Google Scholar] [CrossRef]
- Witayakran, S.; Ragauskas, A. Synthetic Applications of Laccase in Green Chemistry. Adv. Synth. Catal. 2009, 351, 1187–1209. [Google Scholar] [CrossRef]
- Bassanini, I.; Ferrandi, E.E.; Riva, S.; Monti, D. Biocatalysis with Laccases: An Updated Overview. Catalysts 2021, 11, 26. [Google Scholar] [CrossRef]
- Romero-Guido, C.; Baez, A.; Torres, E. Dioxygen Activation by Laccases: Green Chemistry for Fine Chemical Synthesis. Catalysts 2018, 8, 223. [Google Scholar] [CrossRef]
- Itoh, T.; Itoh, T. Laccase-catalyzed reactions in ionic liquids for green sustainable chemistry. ACS Sustain. Chem. Eng. 2021, 9, 1443–1458. [Google Scholar] [CrossRef]
- Solomon, E.; Sundaram, U.; Machonkin, T. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2605. [Google Scholar] [CrossRef]
- Morozova, O.V.; Shumakovich, G.P.; Gorbacheva, M.A.; Shleev, S.V.; Yaropolov, A.I. “Blue” Laccases. Biochemistry 2007, 72, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Walde, P.; Guo, Z. Enzyme-catalyzed chemical structure-controlling template polymerization. Soft Matter 2011, 7, 316–331. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I. Enzyme Initiated Radical Polymerizations. Polymers 2012, 4, 759–793. [Google Scholar] [CrossRef]
- Mogharabi, M.; Faramarzi, M. Laccase and Laccase-Mediated Systems in the Synthesis of Organic Compounds. Adv. Synth. Catal. 2014, 356, 897–927. [Google Scholar] [CrossRef]
- Morozova, O.V.; Shumakovich, G.P.; Shleev, S.V.; Yaropolov, Y.I. Laccase-mediator systems and their applications: A review. Appl. Biochem. Microbiol. 2007, 43, 523–535. [Google Scholar] [CrossRef]
- Khodaverdian, S.; Dabirmanesh, B.; Heydari, A.; Dashtban-moghadam, E.; Khajeh, K.; Ghazic, F. Activity, stability and structure of laccase in betaine based natural deep eutectic solvents. Int. J. Biol. Macromol. 2018, 107, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.; Pereira, M.; Freire, M.; Silva, J.; Coutinho, J.; Tavares, A. Laccase Activation in Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2019, 7, 11806–11814. [Google Scholar] [CrossRef]
- Delorme, A.E.; Andanson, J.M.; Verney, V. Improving laccase thermostability with aqueous natural deep eutectic solvents. Int. J. Biol. Macromol. 2020, 163, 919–926. [Google Scholar] [CrossRef]
- Ünlü, A.; Prasad, B.; Anavekar, K.; Bubenheim, P.; Liese, A. Investigation of a green process for the polymerization of catechin. Prep. Biochem. Biotechnol. 2017, 47, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Altundağ, A.; Ünlü, A.E.; Takaç, S. Deep eutectic solvent-assisted synthesis of polyaniline by laccase enzyme. J. Chem. Technol. Biotechnol. 2020, 96, 1107–1115. [Google Scholar] [CrossRef]
- Jankun, J.; Selman, S.; Swiercz, R.; Skrzypczak-Jankun, E. Why drinking green tea could prevent cancer. Nature 1997, 387, 561. [Google Scholar] [CrossRef]
- Nakagawa, K.; Ninomiya, M.; Okubo, T.; Aoi, N.; Juneja, L.; Kim, M.; Yamanaka, K.; Miyazawa, T. Tea Catechin Supplementation Increases Antioxidant Capacity and Prevents Phospholipid Hydroperoxidation in Plasma of Humans. J. Agric. Food Chem. 1999, 47, 3967–3973. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, A.; Hrelia, S.; Angeloni, C.; Giordano, E.; Guarnieri, C.; Caldarera, C.; Biagi, P. Green tea protection of hypoxia/reoxygenation injury in cultured cardiac cells. J. Nutr. Biochem. 2002, 13, 103–111. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, J.; Chen, Y.; Agarwal, R. Anti-tumor-promoting activity of a polyphenolic fraction isolated from grape seeds in the mouse skin two-stage initiation-promotion protocol and identification of procyanidin B5-3′-gallate as the most effective antioxidant constituent. Carcinogenesis 1999, 20, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Nagarajan, R.; Kumar, J.; Salemme, A.; Togna, A.R.; Saso, L.; Bruno, F. Antioxidant Activity of Synthetic Polymers of Phenolic Compounds. Polymers 2020, 12, 1646. [Google Scholar] [CrossRef]
- Düweler, K.; Rohdewald, P. Urinary metabolites of French maritime pine bark extract in humans. Pharmazie 2000, 55, 364–368. Available online: https://pubmed.ncbi.nlm.nih.gov/11828617/ (accessed on 30 March 2021). [PubMed]
- Kurisawa, M.; Chung, J.; Uyama, H.; Kobayashi, S. Enzymatic synthesis and antioxidant properties of poly(rutin). Biomacromolecules 2003, 4, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Kurisawa, M.; Chung, J.; Uyama, H.; Kobayashi, S. Laccase-catalyzed synthesis and antioxidant property of poly(catechin). Macromol. Biosci. 2003, 3, 758–764. [Google Scholar] [CrossRef]
- Li, C.; Xie, B. Evaluation of the antioxidant and pro-oxidant effects of tea catechin oxypolymers. J. Agric. Food Chem. 2000, 48, 6362–6366. [Google Scholar] [CrossRef]
- Bruno, F.; Nagarajan, S.; Nagarajan, R.; Kumar, J.; Samuelson, L. Biocatalytic synthesis of water-soluble oligo(catechins). J. Macromol. Sci. A 2005, 42, 1547–1554. [Google Scholar] [CrossRef]
- Khlupova, M.E.; Morozova, O.V.; Vasil’eva, I.S.; Shumakovicha, G.P.; Zaitseva, E.A.; Pashintseva, N.V.; Kovalev, L.I.; Shishkin, S.S.; Chertkov, V.A.; Shestakova, A.K.; et al. Laccase catalyzed heterocoupling of dihydroquercetin and p-aminobenzoic acid: Effect of the reaction product on cultured cells. Biochemistry 2018, 83, 992–1001. [Google Scholar] [CrossRef]
- Weidmann, A. Dihydroquercetin: More than just an impurity? Eur. J. Pharmacol. 2012, 684, 19–26. [Google Scholar] [CrossRef]
- Pantouris, G.; Mowat, C. Antitumour agents as inhibitors of tryptophan 2,3-dioxygenase. Biochem. Biophys. Res. Comm. 2014, 443, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.; Morishima, C.; Lohmann, V.; Pal, S.; Lee, D.; Liu, Y.; Graf, T.; Oberlies, N. Identification of hepatoprotective flavonolignans from silymarin. Proc. Natl. Acad. Sci. USA 2010, 107, 5995–5999. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Murakami, K.; Uno, M.; Ikubo, H.; Nakagama, Y.; Katayama, S.; Akagi, K.; Irie, K. structure-activity relationship for (+)-taxifolin isolated from silymarin as an inhibitor of amyloid β aggregation. Biosci. Biotechnol. Biochem. 2013, 77, 1100–1103. [Google Scholar] [CrossRef]
- Makena, P.; Pierce, S.; Chung, K.T.; Sinclair, S. Comparative mutagenic effects of structurally similar flavonoids quercetin and taxifolin on tester strains Salmonella typhimurium TA102 and Escherichia coli WP-2 uvrA. Environ. Mol. Mutagen. 2009, 50, 451–459. [Google Scholar] [CrossRef]
- Ono, K.; Nakane, H.; Fukushima, M.; Chermann, J.C.; Barre-Sinoussi, F. Differential inhibitory effects of various flavonoids on the activities of reverse transcriptase and cellular DNA and RNA polymerases. Eur. J. Biochem. 1990, 190, 469–476. [Google Scholar] [CrossRef]
- Naki, A.; Varfolomeev, S.D. Mechanism of the inhibition of laccase activity from Polyporus versicolor by halide-ions. Biochemistry 1981, 46, 1344–1350. Available online: https://pubmed.ncbi.nlm.nih.gov/7295828/ (accessed on 30 March 2021).
- Wu, B.-P.; Wen, Q.; Xu, H.; Yang, Z. Insights into the impact of deep eutectic solvents on horseradish peroxidase: Activity, stability and structure. J. Mol. Catal. B Enzym. 2014, 101, 101–107. [Google Scholar] [CrossRef]
- Dai, Y.; Spronsen, J.; Witkamp, G.J.; Verpoorte, R.; Choi, Y. Natural deep eutectic solvents as new potential media for green technology . Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hammond, O.S.; Bowron, D.T.; Edler, K.J. The effect of water upon deep eutectic solvent nanostructure: An unusual transition from ionic mixture to aqueous solution. Angew. Chem. Int. 2017, 56, 9782–9785. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Papale, F.; Bollino, F.; Piccolella, S.; Marciano, S.; Nocera, P.; Pacifico, S. Silica/quercetin sol-gel hybrids as antioxidant dental implant materials. Sci. Technol. Adv. Mater. 2015, 16, 035001. [Google Scholar] [CrossRef]
- Zu, S.; Yang, L.; Huang, J.; Ma, C.; Wang, W.; Zhao, C.; Zu, Y. Micronization of taxifolin by supercritical antisolvent process and evaluation of radical scavenging activity. Int. J. Mol. Sci. 2012, 13, 8869–8881. [Google Scholar] [CrossRef]
- Hasibi, F.; Nasirpour, A.; Varshosaz, J.; García-Manrique, P.; Blanco-López, M.; Gutiérrez, G.; Matos, M. Formulation and characterization of taxifolin-loaded lipid nanovesicles (liposomes, niosomes, and transfersomes) for beverage fortification. Eur. J. Lipid Sci. Technol. 2020, 122, 1900105–1900118. [Google Scholar] [CrossRef]
- Heneczkowski, M.; Kopacz, M.; Nowak, D.; Kużniar, A. Infrared spectrum analysis of some flavonoids. Acta. Pol. Pharm. 2001, 58, 415–420. [Google Scholar]
- Bovey, F.A.; Mireau, P. NMR of Polymers, 1st ed.; Academic Press: London, UK, 1996; p. 459. ISBN 978-012-11-9765-0. [Google Scholar]
- Kiehlmann, E.; Szczepina, M. Epimerization, transacylation and bromination of dihydroquercetin acetates; synthesis of 8-bromodihydroquercetin. Cent. Eur. J. Chem. 2011, 9, 492–498. [Google Scholar] [CrossRef]
- Kiehlmann, E.; Slade, P. Methylation of dihydroquercetin acetates: Synthesis of 5-o-methyldihydroquercetin. J. Nat. Prod. 2003, 66, 1562–1566. [Google Scholar] [CrossRef]
- Yamamura, S. Oxidation of phenols. In PATAI’S Chemistry of Functional Groups; Rapoport, Z., Ed.; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2003. [Google Scholar] [CrossRef]
- Jones, S.; Solomon, E.I. Electron transfer and reaction mechanism of laccases. E. Cell. Mol. Life Sci. 2015, 72, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007; 2357p, ISBN 978-047-17-2091-1. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, A.M.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Janesko, B.G.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Prescott, A.; Stamford, N.; Wheeler, G.; Firmin, J.L. In vitro properties of a recombinant flavonol synthase from Arabidopsis thaliana. Phytochemistry 2002, 60, 589–593. [Google Scholar] [CrossRef]
- Nifant’ev, E.; Mosyurov, S.; Kukhareva, T.; Vasyanina, L. Aminomethylation of Dihydroquercetin. Dokl. Chem. 2013, 448, 4–8. [Google Scholar] [CrossRef]
- Boursalian, B.; Ham, W.; Mazzotti, A.; Ritte, T. Charge transfer directed radical substitution enables para-selective C–H functionalization. Nat. Chem. 2016, 8, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Gorshina, E.S.; Rusinova, T.V.; Biryukov, V.V.; Morozova, O.V.; Shleev, S.V.; Yaropolov, A.I. The dynamics of oxidase activity during cultivation of basidiomycetes from the genus Trametes Fr. Appl. Biochem. Microbiol. 2006, 42, 558–563. [Google Scholar] [CrossRef]
- Ehresmann, B.; Imbault, P.; Well, J.H. Spectrophotometric determination of protein concentration in cell extracts containing tRNA’s and rRNA’s. Anal. Biochem. 1973, 54, 454–463. [Google Scholar] [CrossRef]
- Chertkov, V.; Davydov, D.; Shestakova, A. Regioselective N-arylation of nitroazoles. Determination of the structure of N-arylnitro-azoles on the basis of NMR spectroscopic data and quantum-chemical calculations. Chem. Heterocycl. Compd. 2011, 47, 45–54. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DES Components | Legend | HBA/HBD Molar Ratio | Temperature, °C | Aggregate State at Room Temperature | |
|---|---|---|---|---|---|
| HBA | HBD | ||||
| Betaine, B | Lactic acid, L | B-L (1:1) | 1:1 | 60 | Solid |
| B-L (1:2) | 1:2 | 40 | Clear liquid | ||
| B-L (1:3) | 1:3 | 60 | Clear liquid | ||
| Betaine, B | Glycerol, G | B-G (1:1) | 1:1 | 60 | Solid |
| B-G (1:2) | 1:2 | 60 | Highly viscous clear liquid | ||
| Betaine, B | Propionic acid, P | B-P (1:1) | 1:1 | 40 | Solid |
| B-P (1:2) | 1:2 | 40 | Clear liquid | ||
| B-P (1:3) | 1:3 | 40 | Clear liquid | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khlupova, M.; Vasil’eva, I.; Shumakovich, G.; Zaitseva, E.; Chertkov, V.; Shestakova, A.; Morozova, O.; Yaropolov, A. Enzymatic Polymerization of Dihydroquercetin (Taxifolin) in Betaine-Based Deep Eutectic Solvent and Product Characterization. Catalysts 2021, 11, 639. https://doi.org/10.3390/catal11050639
Khlupova M, Vasil’eva I, Shumakovich G, Zaitseva E, Chertkov V, Shestakova A, Morozova O, Yaropolov A. Enzymatic Polymerization of Dihydroquercetin (Taxifolin) in Betaine-Based Deep Eutectic Solvent and Product Characterization. Catalysts. 2021; 11(5):639. https://doi.org/10.3390/catal11050639
Chicago/Turabian StyleKhlupova, Maria, Irina Vasil’eva, Galina Shumakovich, Elena Zaitseva, Vyacheslav Chertkov, Alla Shestakova, Olga Morozova, and Alexander Yaropolov. 2021. "Enzymatic Polymerization of Dihydroquercetin (Taxifolin) in Betaine-Based Deep Eutectic Solvent and Product Characterization" Catalysts 11, no. 5: 639. https://doi.org/10.3390/catal11050639
APA StyleKhlupova, M., Vasil’eva, I., Shumakovich, G., Zaitseva, E., Chertkov, V., Shestakova, A., Morozova, O., & Yaropolov, A. (2021). Enzymatic Polymerization of Dihydroquercetin (Taxifolin) in Betaine-Based Deep Eutectic Solvent and Product Characterization. Catalysts, 11(5), 639. https://doi.org/10.3390/catal11050639