
Single Atomic Pt on SrTiO3 Catalyst in Reverse Water Gas Shift Reactions

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussions
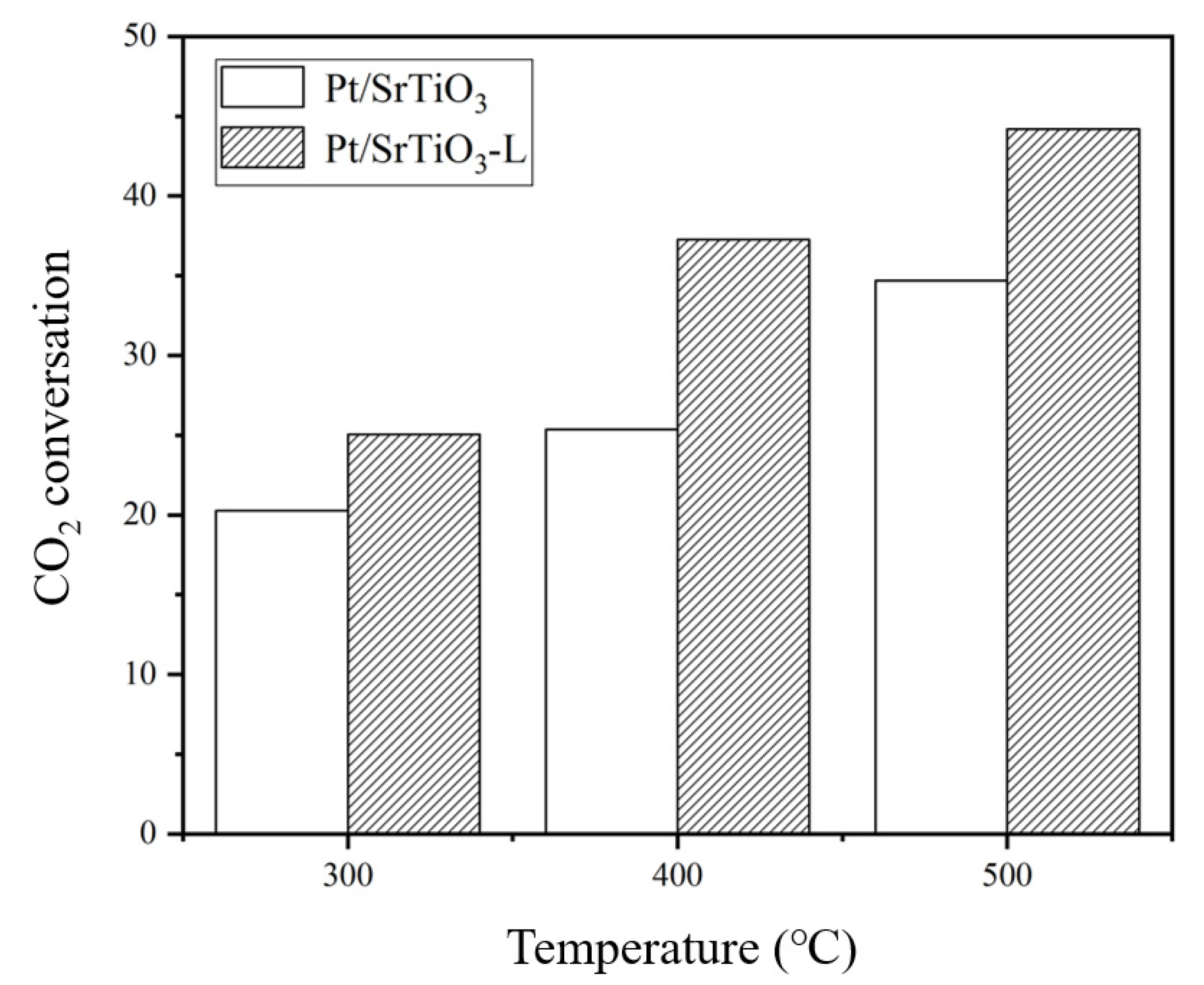
2.1. Catalytic Performances
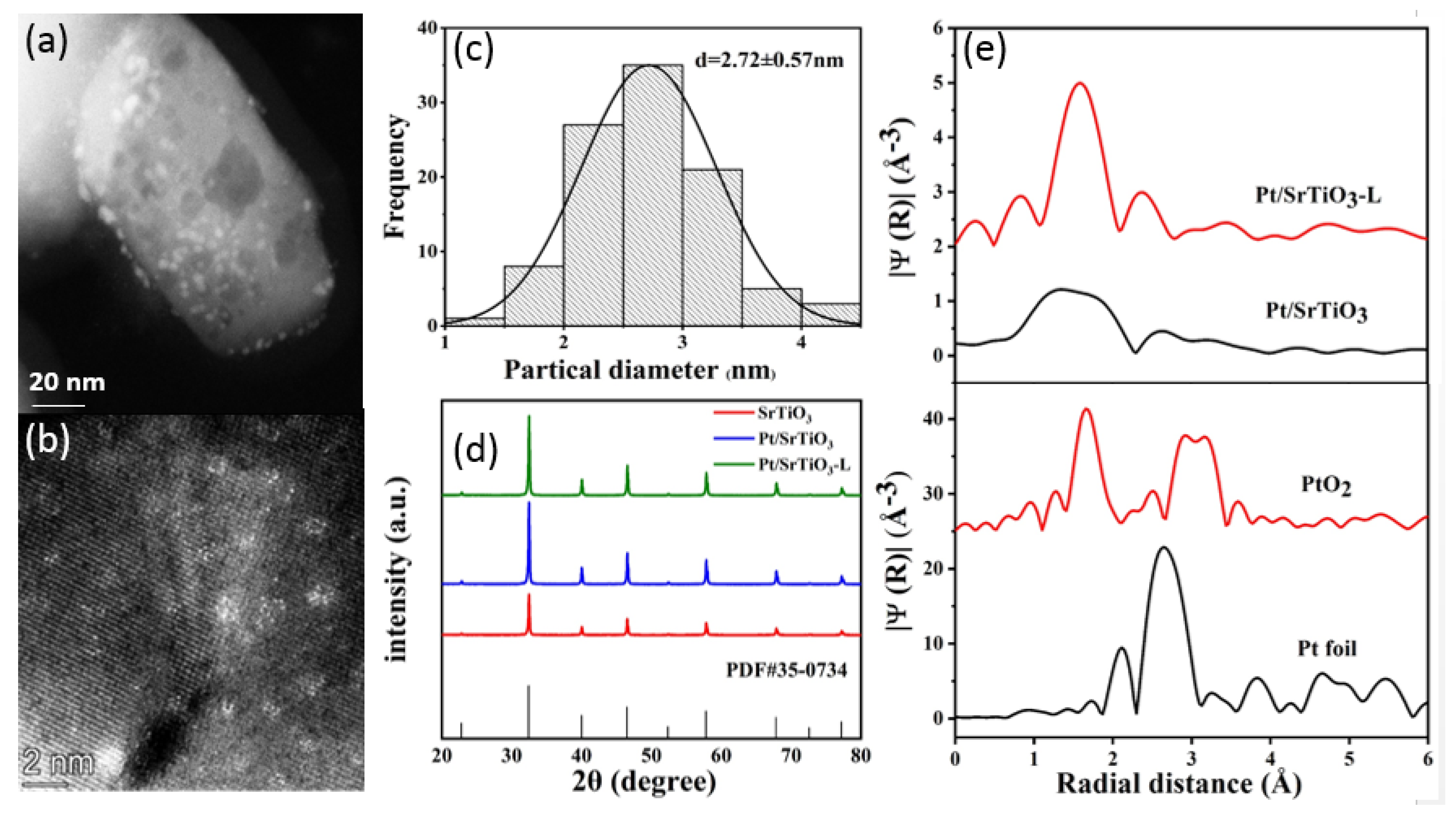
2.2. Textural Properties
2.3. H2-TPR
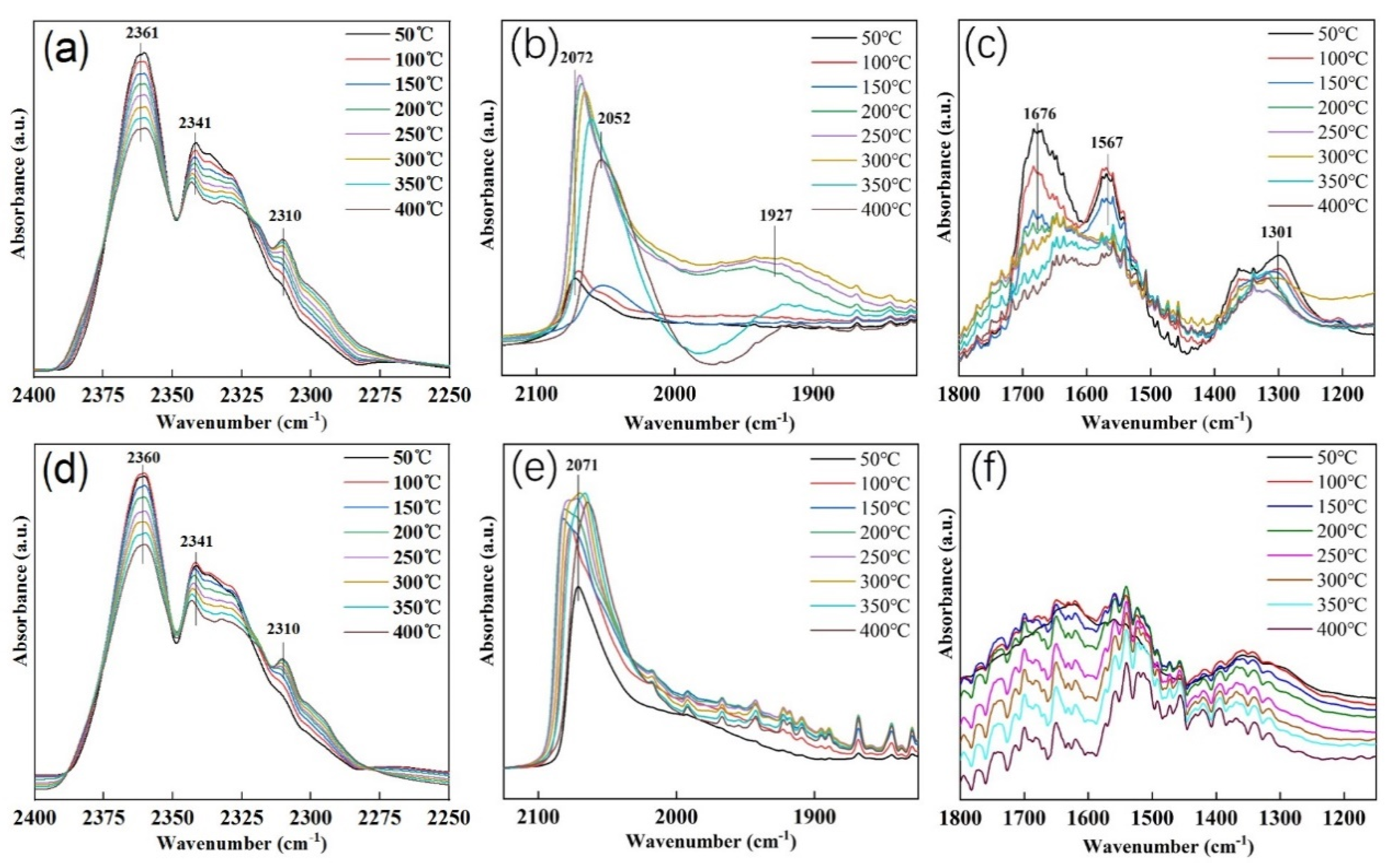
2.4. In Situ FTIR
2.4.1. CO2 and H2 Co-Adsorption on SrTiO3 Support
2.4.2. CO2 and H2 Adsorption on Pt/SrTiO3 and Pt/SrTiO3-L
- (1)
- CO2 pre-adsorption and purging by H2
- (2)
- CO2 and H2 co-adsorption
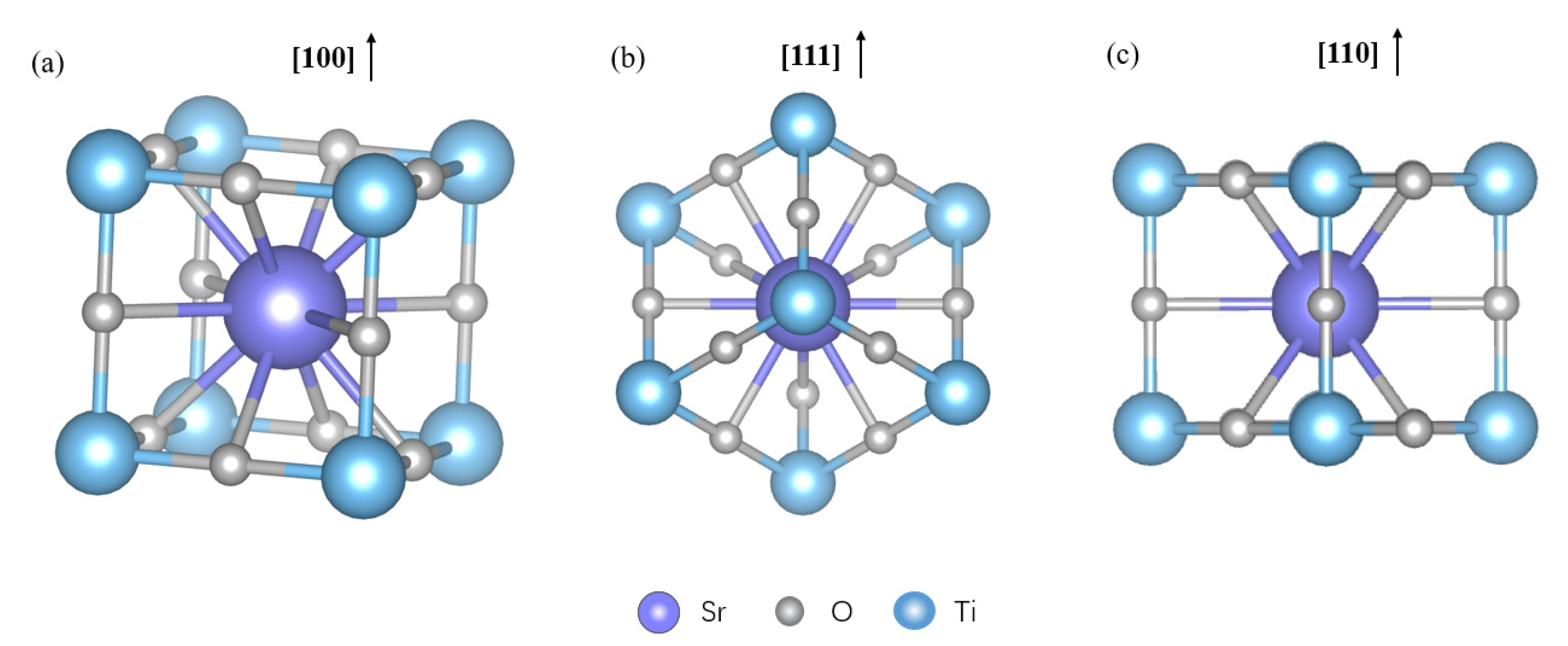
2.5. DFT Results and Reaction Mechanisms
3. Experimental Method
3.1. Catalyst Synthesis
3.2. Characterizations
3.2.1. Powder X-ray Diffraction (XRD)
3.2.2. BET and Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-OES)
3.2.3. H2-Temperature-Programmed Reduction (H2-TPR)
3.2.4. Scanning Transmission Electron Microscopy (STEM)
3.2.5. In Situ Fourier-Transformed Infrared Spectroscopy
3.2.6. Density Functional Theory (DFT) Calculation Details
3.2.7. Extended X-ray Absorption Fine Structure
3.3. Activity Tests
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Studt, F.; Sharafutdinov, I.; Abild-Pedersen, F.; Elkjær, C.F.; Hummelshøj, J.S.; Dahl, S.; Chorkendorff, I.; Nørskov, J.K. Discovery of a Ni-Ga Catalyst for Carbon Dioxide Reduction to Methanol. Nat. Chem. 2014, 6, 320–324. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Dai, Y.; Pu, Z.; Lao, Z.; Chen, Y.; Wang, M.; Zheng, X.; Zhu, J.; Zhang, W.; et al. Synergetic Interaction between Neighbouring Platinum Monomers in CO2 Hydrogenation. Nat. Nanotechnol. 2018, 13, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, W.; Zheng, X.; Chen, Y.; Wu, W.; Qiu, J.; Zhao, X.; Zhao, X.; Dai, Y.; Zeng, J. Incorporating Nitrogen Atoms into Cobalt Nanosheets as a Strategy to Boost Catalytic Activity toward CO2 Hydrogenation. Nat. Energy 2017, 2, 869–876. [Google Scholar] [CrossRef]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active Sites for CO2 Hydrogenation to Methanol on Cu/ZnO Catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barroso Quiroga, M.M.; Castro Luna, A.E. Kinetic Analysis of Rate Data for Dry Reforming of Methane. Ind. Eng. Chem. Res. 2007, 46, 5265–5270. [Google Scholar] [CrossRef]
- Baudouin, D.; Rodemerck, U.; Krumeich, F.; De Mallmann, A.; Szeto, K.C.; Ménard, H.; Veyre, L.; Candy, J.P.; Webb, P.B.; Thieuleux, C.; et al. Particle Size Effect in the Low Temperature Reforming of Methane by Carbon Dioxide on Silica-Supported Ni Nanoparticles. J. Catal. 2013, 297, 27–34. [Google Scholar] [CrossRef]
- Millet, M.-M.; Algara-Siller, G.; Wrabetz, S.; Mazheika, A.; Girgsdies, F.; Teschner, D.; Seitz, F.-D.; Tarasov, A.; Levchenko, S.V.; Schlögl, R.; et al. Ni Single Atom Catalysts for CO2 Activation. J. Am. Chem. Soc. 2019, 141, 2451−2461. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Chen, Y.; Wang, X.; Zhang, Z.; Wang, D.; Li, Y. Chemical Synthesis of Single Atomic Site Catalysts. Chem. Rev. 2020, 120, 11900–11955. [Google Scholar] [CrossRef]
- Du, Y.; Sheng, H.; Astruc, D.; Zhu, M. Atomically Precise Noble Metal Nanoclusters as Efficient Catalysts: A Bridge between Structure and Properties. Chem. Rev. 2020, 120, 526–622. [Google Scholar] [CrossRef]
- Tang, Y.; Asokan, C.; Xu, M.; Graham, G.W.; Pan, X.; Christopher, P.; Li, J.; Sautet, P. Rh Single Atoms on TiO2 Dynamically Respond to Reaction Conditions by Adapting their Site. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Matsubu, J.C.; Yang, V.N.; Christopher, P. Isolated Metal Active Site Concentration and Stability Control Catalytic CO2 Reduction Selectivity. J. Am. Chem. Soc. 2015, 137, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Su, X.; Duan, H.; Liang, B.; Huang, Y.; Zhang, T. Catalytic Performance of the Pt/TiO2 Catalysts in Reverse Water Gas Shift Reaction: Controlled Product Selectivity and a Mechanism Study. Catal. Today 2017, 281, 312–318. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. The Effect of the Morphological Characteristics of TiO2 Supports on the Reverse Water–Gas Shift Reaction over Pt/TiO2 Catalysts. Appl. Catal. B Environ. 2012, 119–120, 100–108. [Google Scholar] [CrossRef]
- Matsubu, J.C.; Zhang, S.; DeRita, L.; Marinkovic, N.S.; Chen, J.G.; Graham, G.W.; Pan, X.; Christopher, P. Adsorbate-Mediated Strong Metal–Support Interactions in Oxide-Supported Rh Catalysts. Nat. Chem. 2017, 9, 120–127. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A Study on the Effect of Support’s Reducibility on the Reverse Water-Gas Shift Reaction Over Pt Catalysts. Appl. Catal. A Gen. 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Guo, Y.; Mei, S.; Yuan, K.; Wang, D.-J.; Liu, H.-C.; Yan, C.-H.; Zhang, Y.-W. Low-Temperature CO2 Methanation over CeO2-Supported Ru Single Atoms, Nanoclusters, and Nanoparticles Competitively Tuned by Strong Metal–Support Interactions and H-Spillover Effect. ACS Catal. 2018, 8, 6203–6215. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.C.; Tibiletti, D.; Breen, J.P.; Burch, R. Spectrokinetic Investigation of Reverse Water-Gas-Shift Reaction Intermediates over a Pt/CeO2 Catalyst. J. Phys. Chem. B 2004, 108, 20240–20246. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Khazaneh, M.T.; Widmann, D.; Behm, R. TAP Reactor Studies of the Oxidizing Capability of CO2 on a Au/CeO2 Catalyst—A First Step toward Identifying a Redox Mechanism in the Reverse Water–Gas Shift Reaction. J. Catal. 2013, 302, 20–30. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.; Breen, J.; Burch, R.; Petch, M.; Faurghenciu, A. Study of the Origin of the Deactivation of a Pt/CeO Catalyst During Reverse Water Gas Shift (RWGS) Reaction. J. Catal. 2004, 226, 382–392. [Google Scholar] [CrossRef]
- Fisher, I.A.; Bell, A.T. A Comparative Study of CO and CO2 Hydrogenation over Rh/SiO2. J. Catal. 1996, 162, 54–65. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Wang, X.; Shi, H.; Kwak, J.H.; Szanyi, J. Mechanism of CO2 Hydrogenation on Pd/Al2O3 Catalysts: Kinetics and Transient DRIFTS-MS Studies. ACS Catal. 2015, 5, 6337–6349. [Google Scholar] [CrossRef]
- Yang, X.; Su, X.; Chen, X.; Duan, H.; Liang, B.; Liu, Q.; Liu, X.; Ren, Y.; Huang, Y.; Zhang, T. Promotion Effects of Potassium on the Activity and Selectivity of Pt/Zeolite Catalysts for Reverse Water Gas Shift Reaction. Appl. Catal. B Environ. 2017, 216, 95–105. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.; Lin, L.; Yao, S.; Zhang, M.; Liu, X.; Wang, X.; Li, Y.-W.; Shi, C.; Ma, D. Highly Dispersed Copper over β-Mo2C as an Efficient and Stable Catalyst for the Reverse Water Gas Shift (RWGS) Reaction. ACS Catal. 2016, 7, 912–918. [Google Scholar] [CrossRef]
- Yan, B.; Wu, Q.; Cen, J.; Timoshenko, J.; Frenkel, A.; Su, D.; Chen, X.; Parise, J.B.; Stach, E.; Orlov, A.; et al. Highly Active Subnanometer Rh Clusters Derived from Rh-Doped SrTiO3 for CO2 Reduction. Appl. Catal. B Environ. 2018, 237, 1003–1011. [Google Scholar] [CrossRef]
- Parastaev, A.; Muravev, V.; Osta, E.H.; Van Hoof, A.J.F.; Kimpel, T.F.; Kosinov, N.; Hensen, E.J.M. Boosting CO2 Hydrogenation via Size-Dependent Metal–Support Interactions in Cobalt/Ceria-Based Catalysts. Nat. Catal. 2020, 3, 526–533. [Google Scholar] [CrossRef]
- Topalidis, D.E.; Petrakis, A.; Ladavos, L.; Loukatzikou, P.J.; Pomonis, A. A Kinetic Study of Methane and Carbon Dioxide in-Terconversion over 0.5%Pt/SrTiO3 Catalysts. Catal. Today 2007, 127, 238–245. [Google Scholar] [CrossRef]
- Saputra, L.; Sato, T.; Kojima, T.; Hara, T.; Ichikuni, N.; Shimazu, S. Preparation of a Highly Stable Pd-Perovskite Catalyst for Suzuki Couplings via a Low-Temperature Hydrothermal Treatment. ACS Omega 2018, 3, 17528–17531. [Google Scholar] [CrossRef] [Green Version]
- Lyu, H.; Hisatomi, T.; Goto, Y.; Yoshida, M.; Higashi, T.; Katayama, M.; Takata, T.; Minegishi, T.; Nishiyama, H.; Yamada, T.; et al. An Al-Doped SrTiO3 Photocatalyst Maintaining Sunlight-Driven Overall Water Splitting Activity for over 1000 h of Constant Illumination. Chem. Sci. 2019, 10, 3196–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.-I.; Jeong, H.Y.; Lee, J.-S.; Kim, M.G.; Cho, J. A Bifunctional Perovskite Catalyst for Oxygen Reduction and Evolution. Angew. Chem. 2014, 126, 4670–4674. [Google Scholar] [CrossRef]
- Nishihata, Y.; Mizuki, J.I.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-Regeneration of a Pd-perovskite Catalyst for Automotive Emissions Control. Nat. Cell Biol. 2002, 418, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Guo, Y.; Lin, Q.-L.; Huang, L.; Ren, J.-T.; Liu, H.-C.; Yan, C.-H.; Zhang, Y.-W. Size Effect-Tuned Water Gas Shift Reaction Activity and Pathway on Ceria Supported Platinum Catalysts. J. Catal. 2021, 394, 121–130. [Google Scholar] [CrossRef]
- Chen, X.; Su, X.; Liang, B.; Yang, X.; Ren, X.; Duan, H.; Huang, Y.; Zhang, T. Identification of Relevant Active Sites and a Mechanism Study for Reverse Water Gas Shift Reaction over Pt/CeO2 Catalysts. J. Energy Chem. 2016, 25, 1051–1057. [Google Scholar] [CrossRef]
- Jeong, H.; Shin, D.; Kim, B.S.; Bae, J.; Shin, S.; Choe, C.; Han, J.W.; Lee, H. Controlling the Oxidation State of Pt Single Atoms for Maximizing Catalytic Activity. Angew. Chemi. 2020, 59, 20691–20696. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, J.-X.; Allard, L.F.; Lee, S.; Liu, J.; Li, H.; Wang, J.; Wang, J.; Oh, S.H.; Li, W.; et al. Surpassing the Single-Atom Catalytic Activity Limit through Paired Pt-O-Pt Ensemble Built from Isolated Pt1 Atoms. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Puengjinda, P.; Muroyama, H.; Matsui, T.; Eguchi, K. Stability of Solid Oxide Fuel Cell Anodes Based on YST–SDC Composite with Ni Catalyst. J. Power Sources 2012, 216, 409–416. [Google Scholar] [CrossRef]
- Peng, R.; Sun, X.; Li, S.; Chen, L.; Fu, M.; Wu, J.; Ye, D. Shape Effect of Pt/CeO 2 Catalysts on the Catalytic Oxidation of Toluene. Chem. Eng. J. 2016, 306, 1234–1246. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; Lykhach, Y.; Migani, A.; Staudt, T.; Petrova, G.P.; Tsud, N.; Skála, T.; Bruix, A.; Illas, F.; Prince, K.C.; et al. Support Nanostructure Boosts Oxygen Transfer to Catalytically Active Platinum Nanoparticles. Nat. Mater. 2011, 10, 310–315. [Google Scholar] [CrossRef]
- Sola, A.C.; De La Piscina, P.R.; Homs, N. Behaviour of Pt/TiO2 Catalysts with Different Morphological and Structural Characteristics in the Photocatalytic Conversion of Ethanol Aqueous Solutions. Catal. Today 2020, 341, 13–20. [Google Scholar] [CrossRef]
- Toda, Y.; Hirayama, H.; Kuganathan, N.; Torrisi, A.; Sushko, P.; Hosono, H. Activation and Splitting of Carbon Dioxide on the Surface of an Inorganic Electride Material. Nat. Commun. 2013, 4, 2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, G.; Davis, B.H. Reverse Water-Gas Shift Reaction: Steady State Isotope Switching Study of the Reverse Water-Gas Shift Reaction using in Situ DRIFTS and a Pt/Ceria Catalyst. Appl. Catal. A Gen. 2005, 284, 31–38. [Google Scholar] [CrossRef]
- Derita, L.; Dai, S.; Lopez-Zepeda, K.; Pham, N.; Graham, G.W.; Pan, X.; Christopher, P. Catalyst Architecture for Stable Single Atom Dispersion Enables Site-Specific Spectroscopic and Reactivity Measurements of CO Adsorbed to Pt Atoms, Oxidized Pt Clusters, and Metallic Pt Clusters on TiO2. J. Am. Chem. Soc. 2017, 139, 14150–14165. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Lian, C.; Hu, S.; Deng, Z.; Gong, J.; Li, M.; Liu, H.; Xing, M.; Zhang, J. Size-Dependent Activity and Selectivity of Carbon Dioxide Photocatalytic Reduction over Platinum Nanoparticles. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöchl, P.E. Projector Augmented-Wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 300 | 400 | 500 | |
|---|---|---|---|
| TOF (s−1) | 0.154 | 0.455 | 0.643 |
| Sample | SrTiO3 | Pt/SrTiO3 | Pt/SrTiO3-L |
|---|---|---|---|
| BET(/g) | 17 | 11 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, Y.; Ouyang, M.; Zhang, L.; Yang, M.; Wu, X.; Ran, R.; Weng, D.; Kang, F.; Si, Z. Single Atomic Pt on SrTiO3 Catalyst in Reverse Water Gas Shift Reactions. Catalysts 2021, 11, 738. https://doi.org/10.3390/catal11060738
Xing Y, Ouyang M, Zhang L, Yang M, Wu X, Ran R, Weng D, Kang F, Si Z. Single Atomic Pt on SrTiO3 Catalyst in Reverse Water Gas Shift Reactions. Catalysts. 2021; 11(6):738. https://doi.org/10.3390/catal11060738
Chicago/Turabian StyleXing, Yimeng, Mengyao Ouyang, Lingling Zhang, Ming Yang, Xiaodong Wu, Rui Ran, Duan Weng, Feiyu Kang, and Zhichun Si. 2021. "Single Atomic Pt on SrTiO3 Catalyst in Reverse Water Gas Shift Reactions" Catalysts 11, no. 6: 738. https://doi.org/10.3390/catal11060738