
Catalytic Liquefaction of Kraft Lignin with Solvothermal Approach

 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
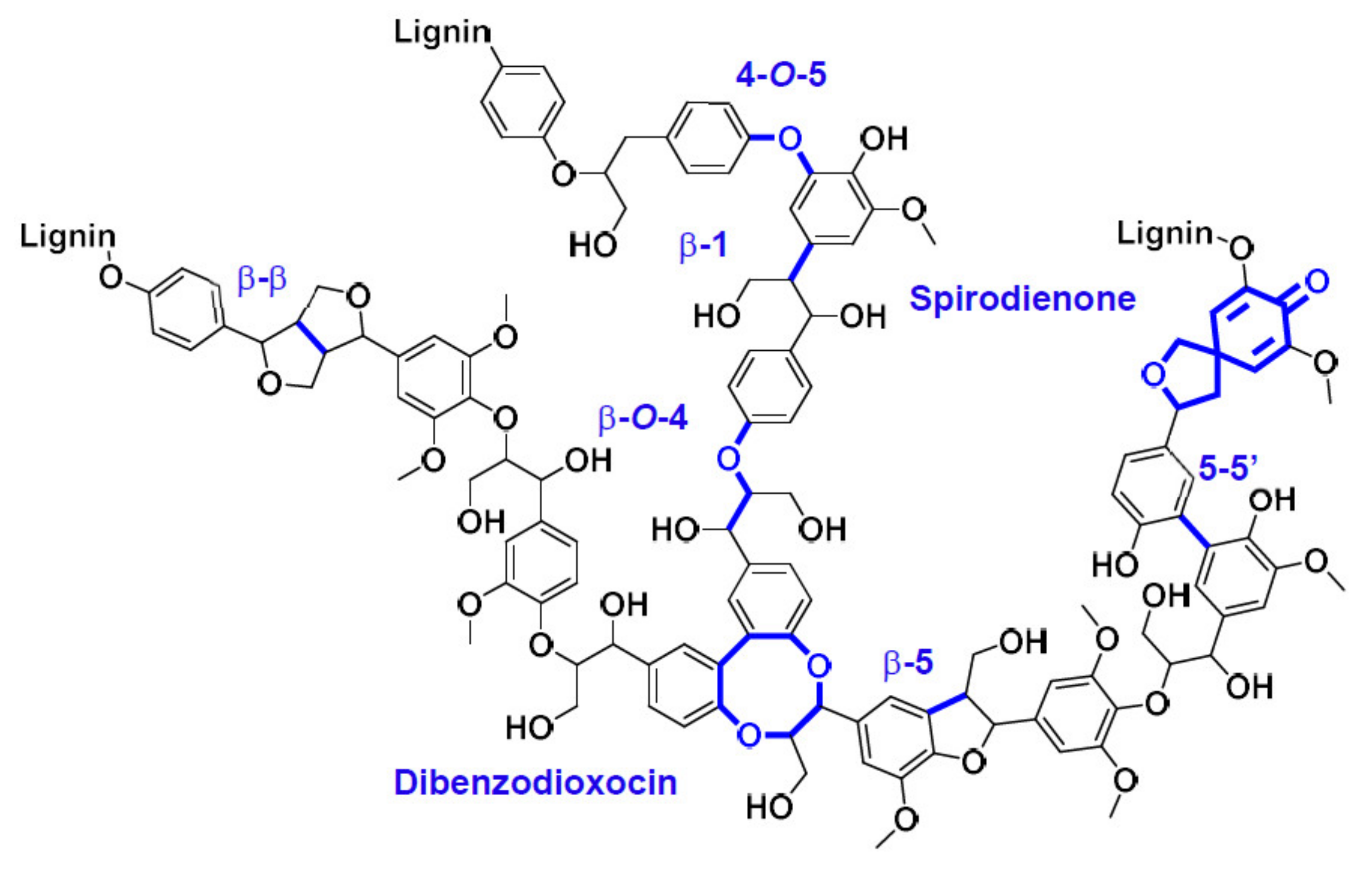
2.1. Characterization of Kraft Lignin
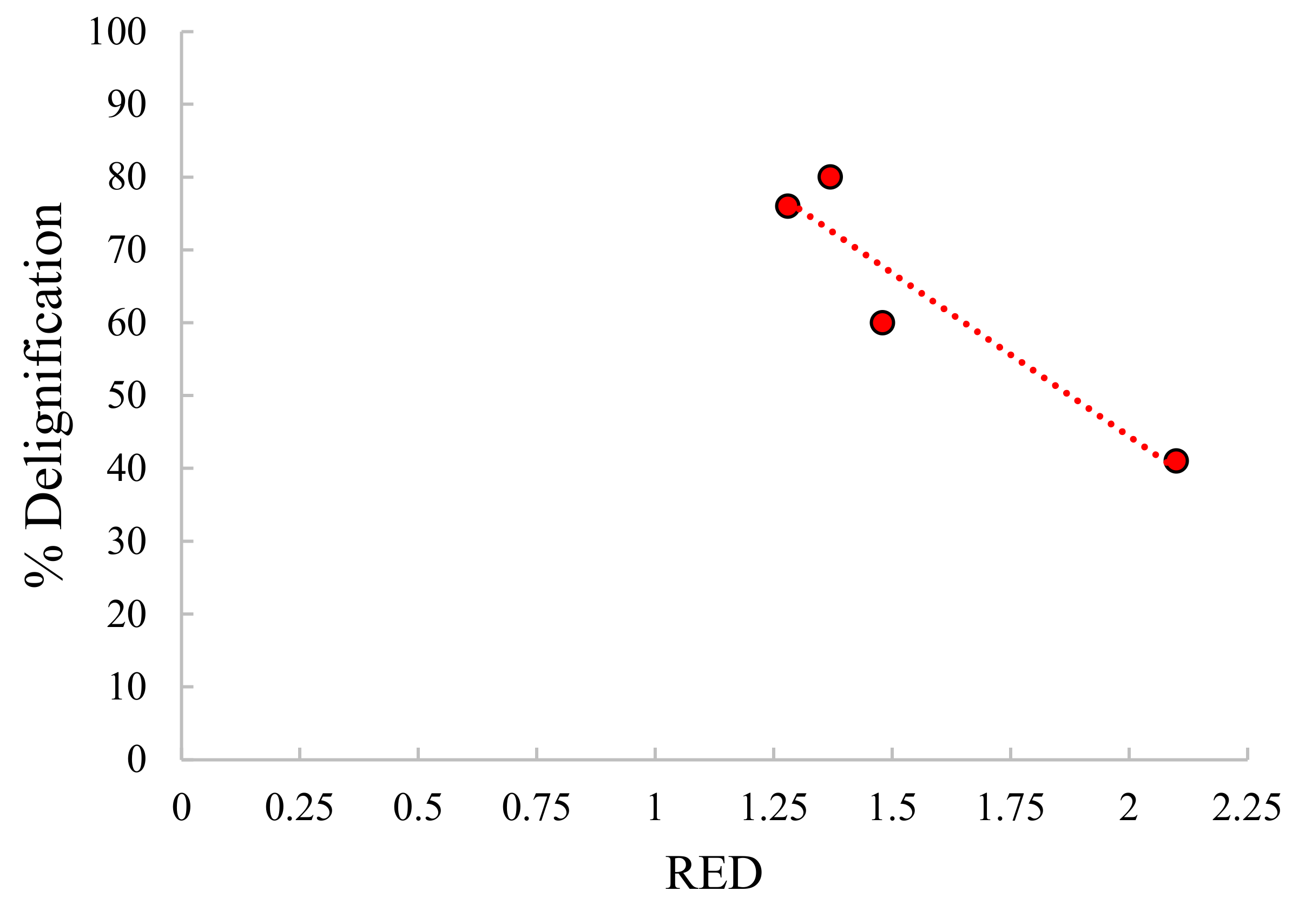
2.2. Hydrothermal Approach
2.2.1. Non-Catalytic Studies
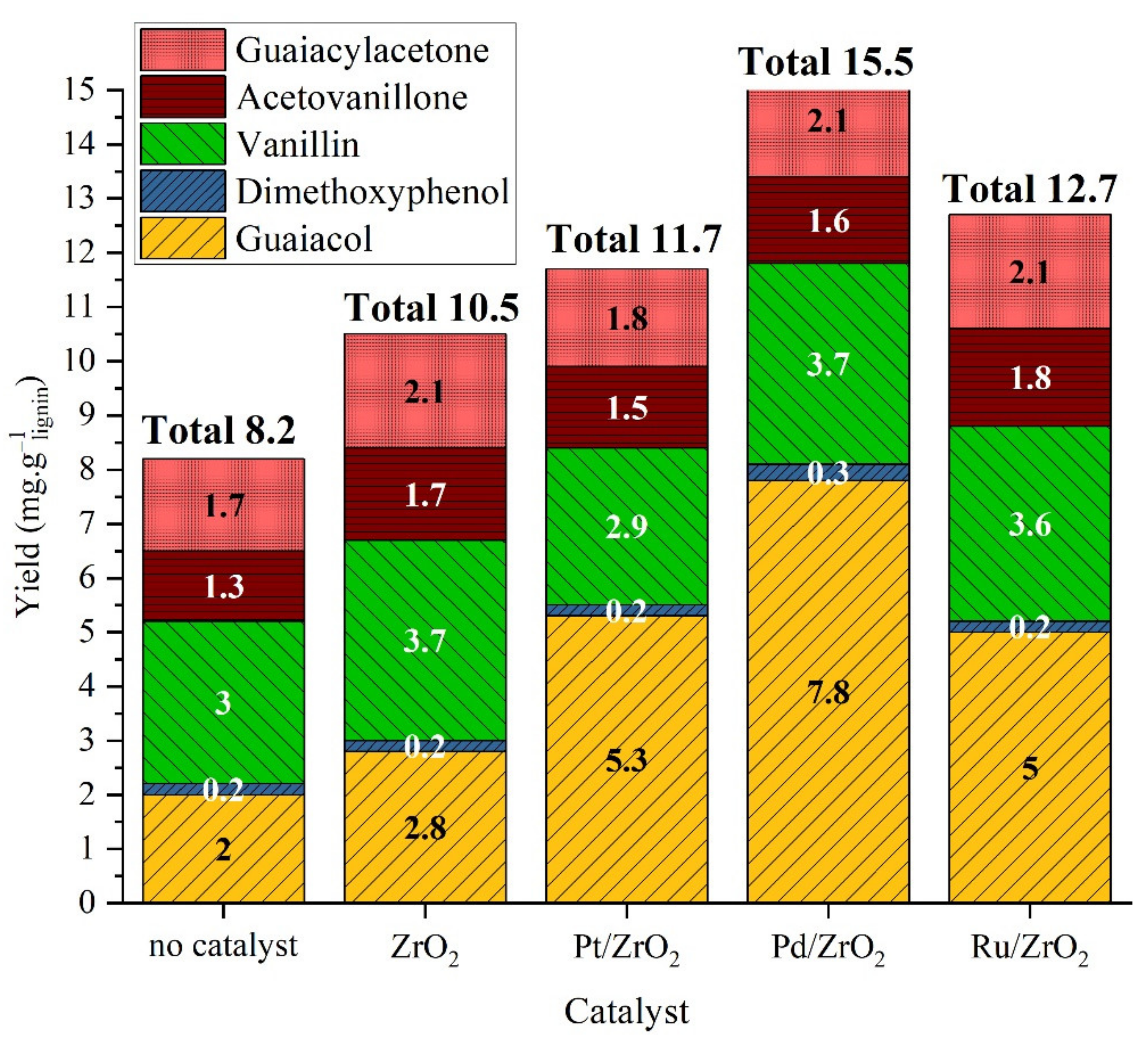
2.2.2. Catalytic Studies
2.3. Solvothermal Approach
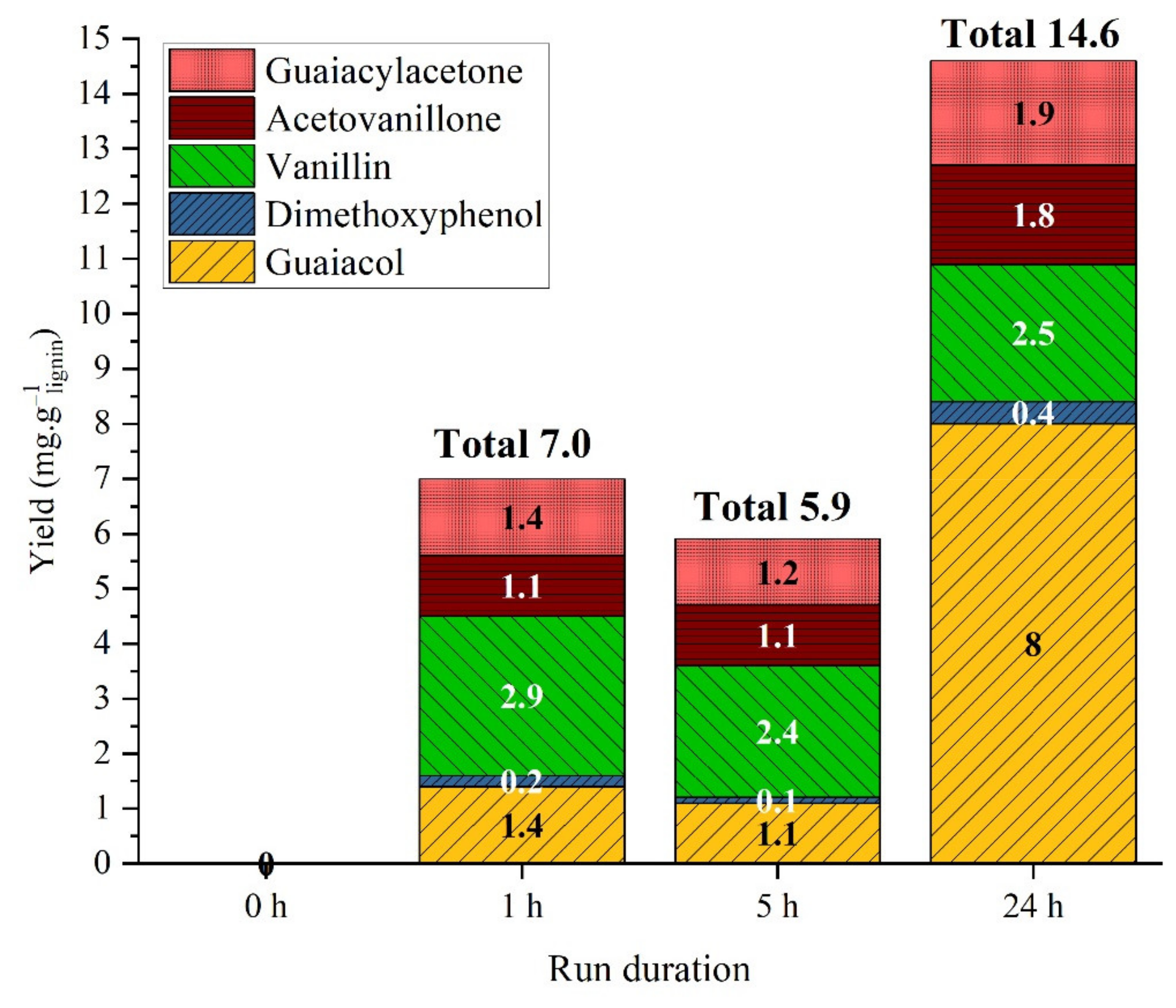
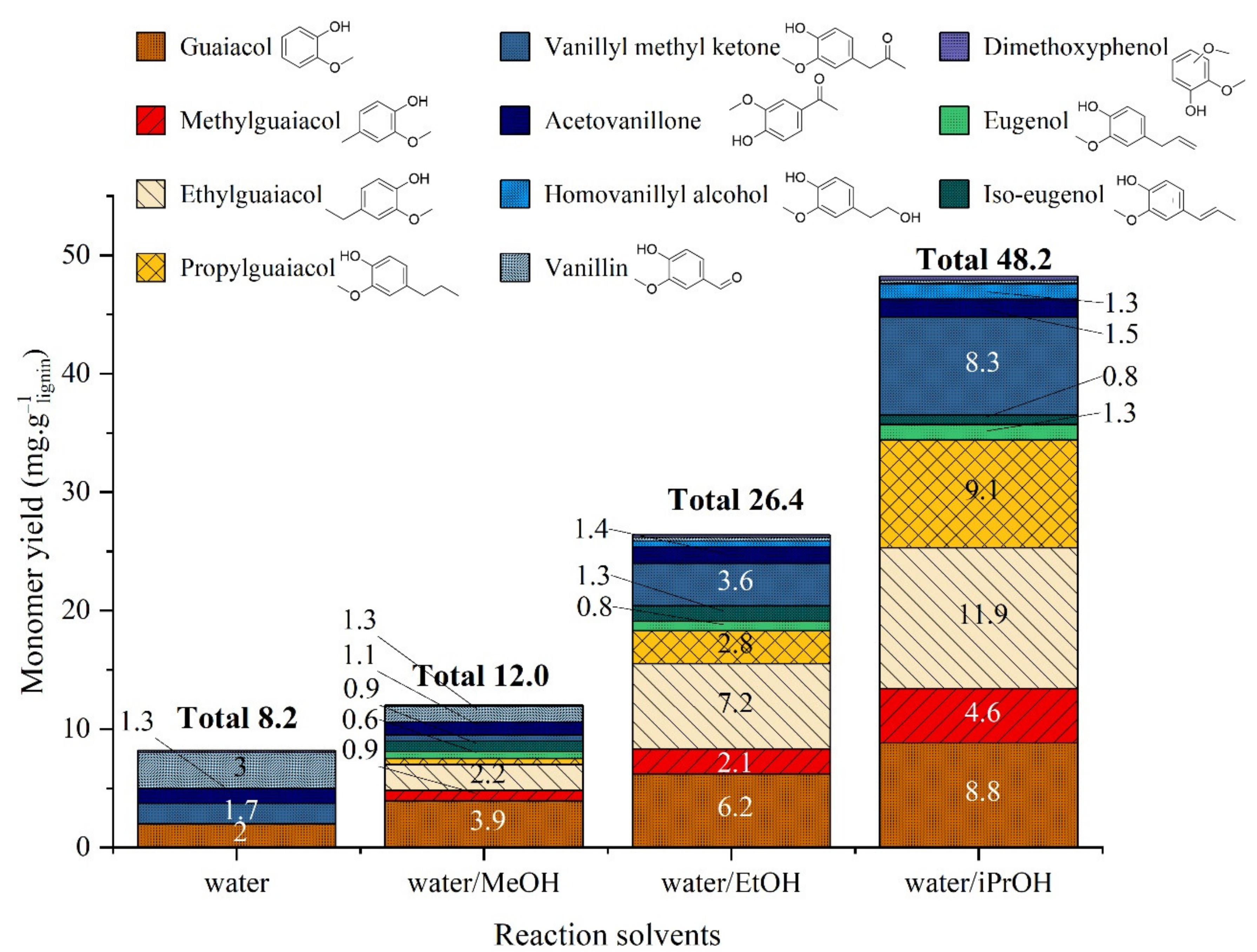
2.3.1. Non-Catalytic Studies
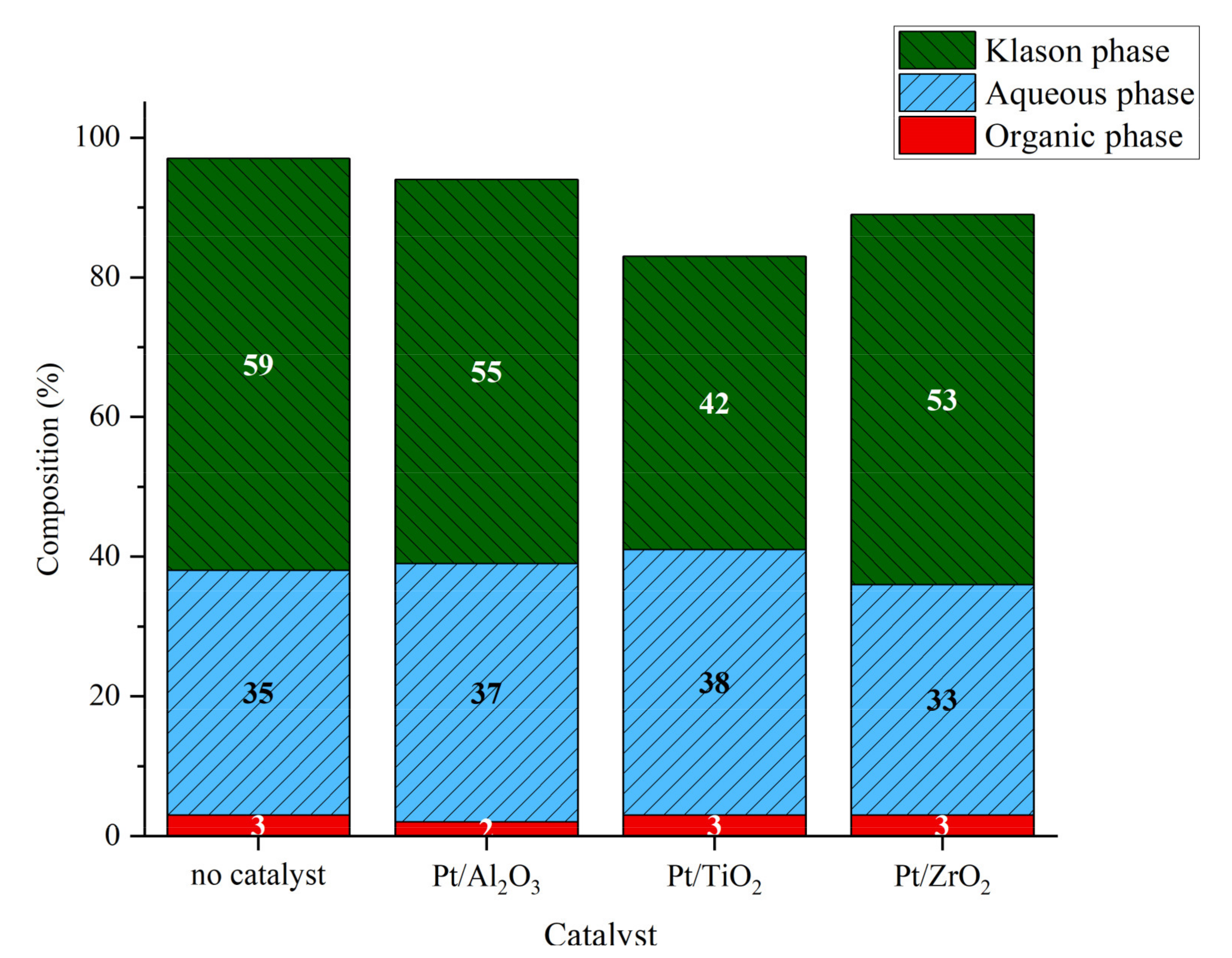
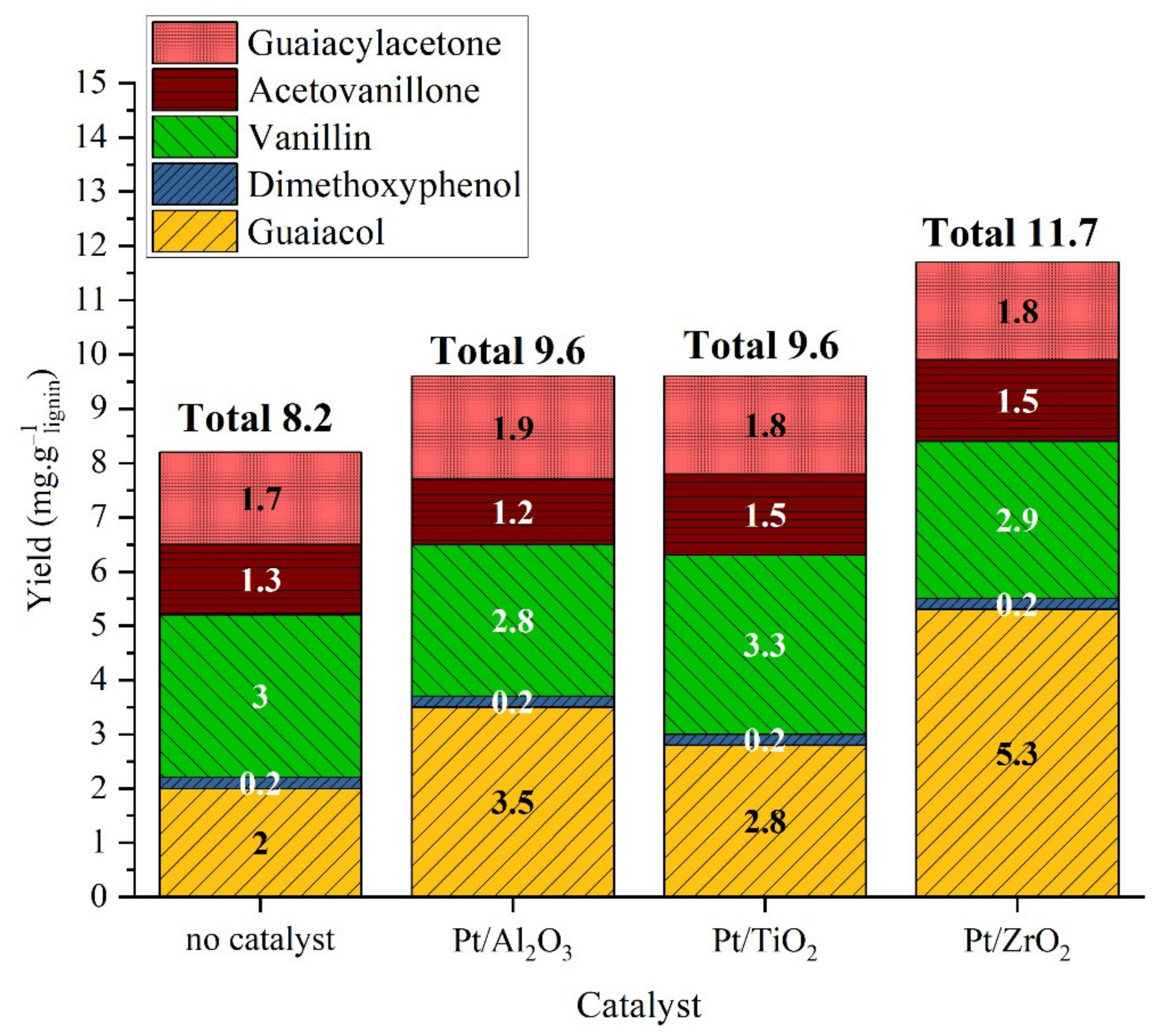
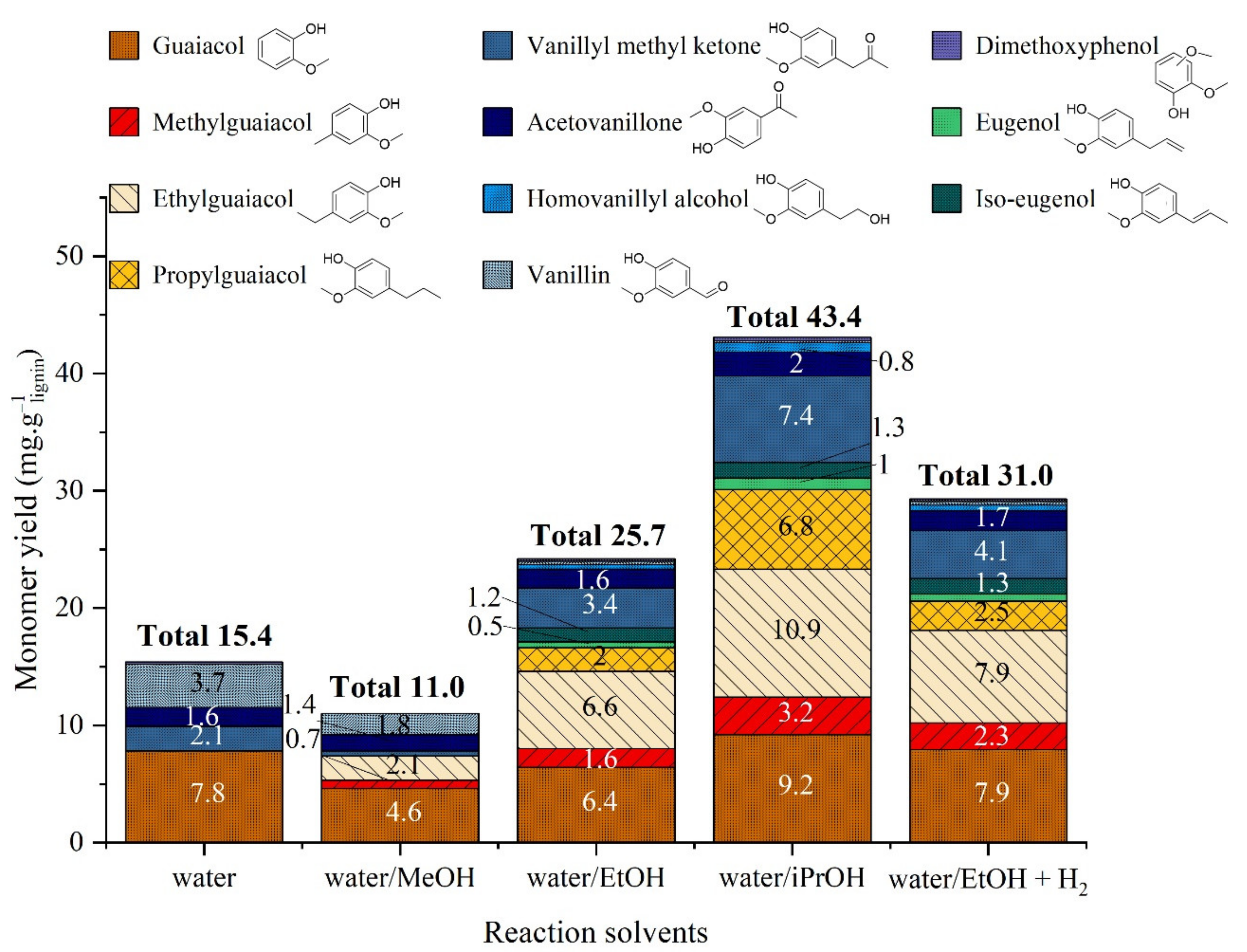
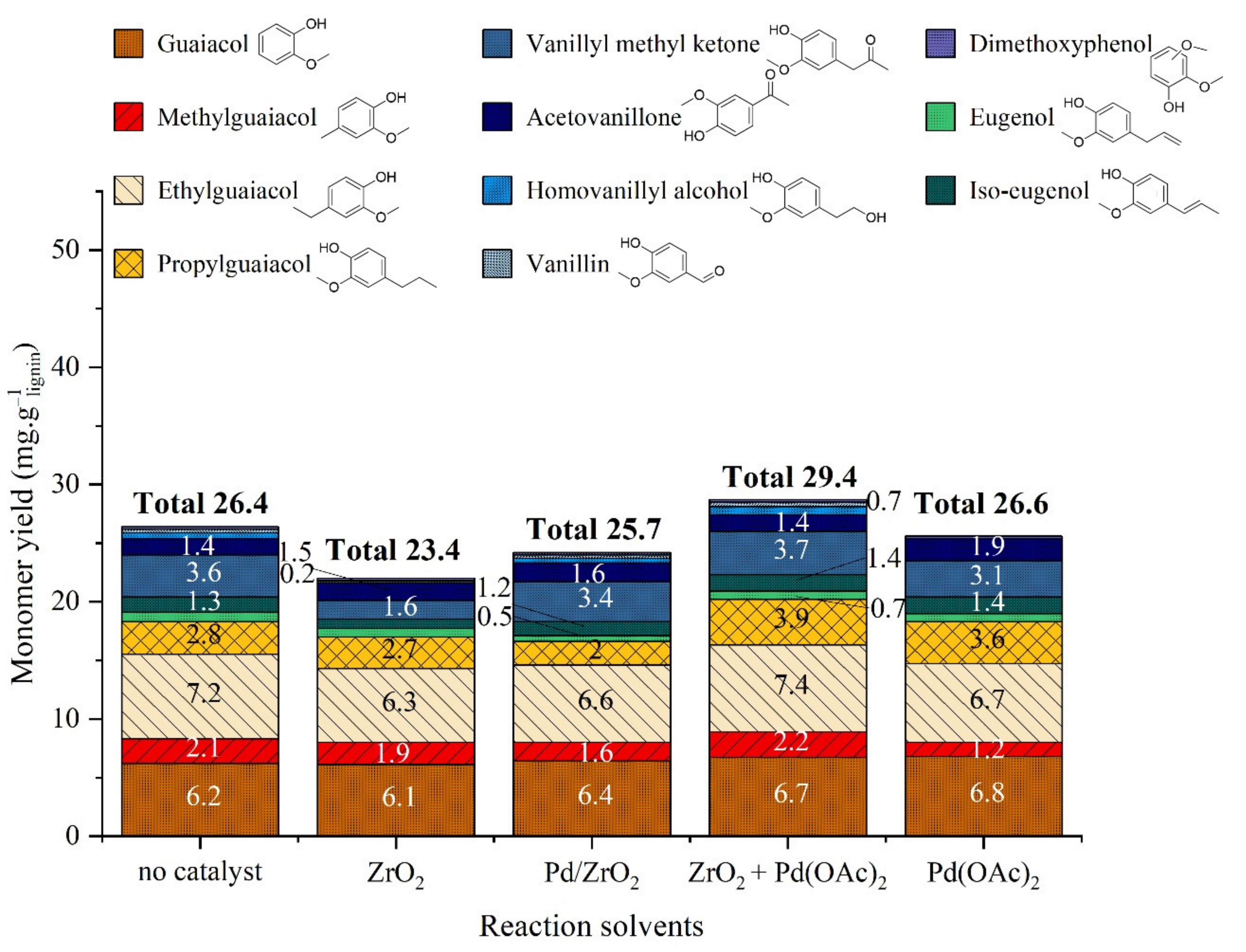
2.3.2. Catalytic Studies
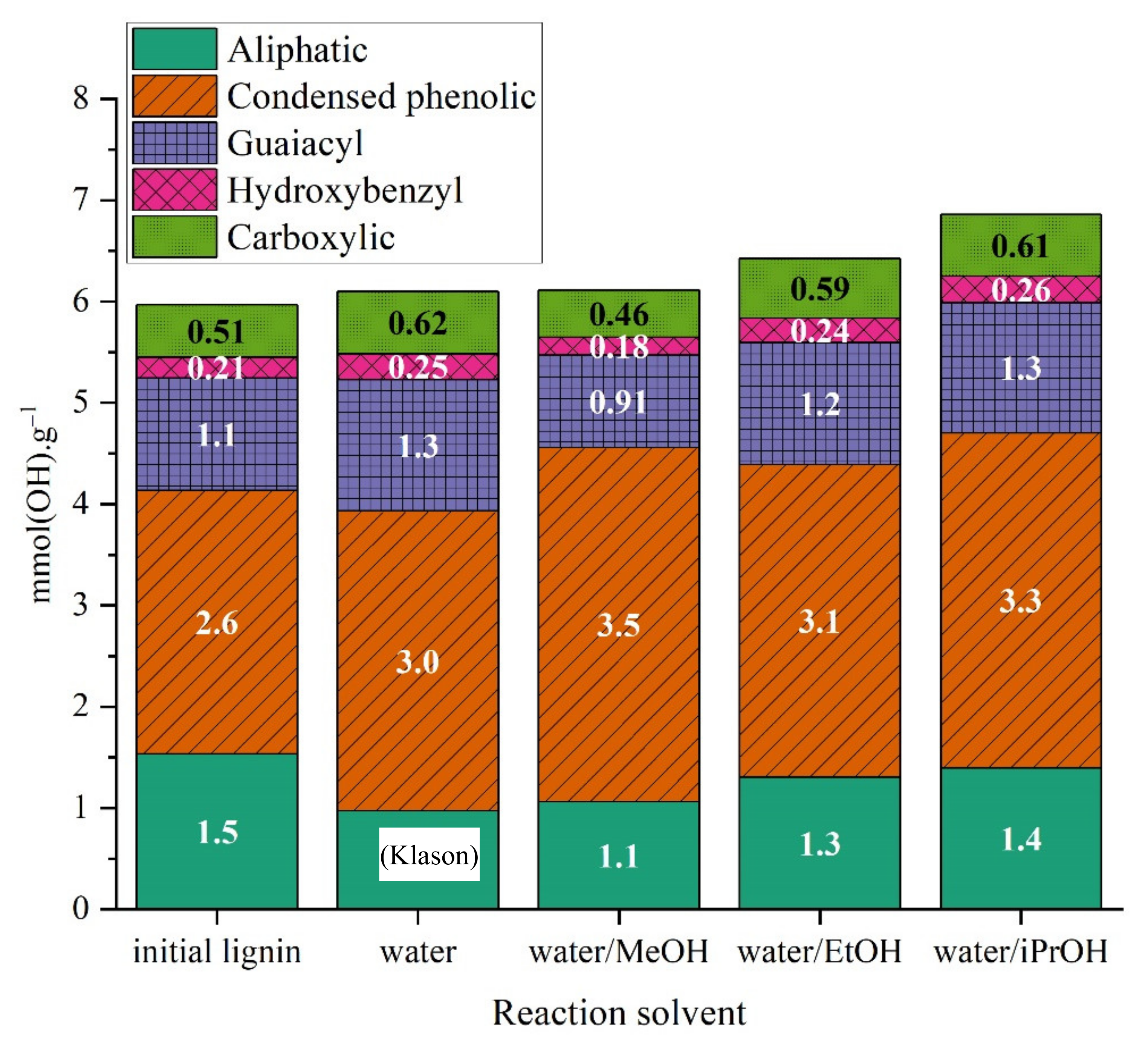
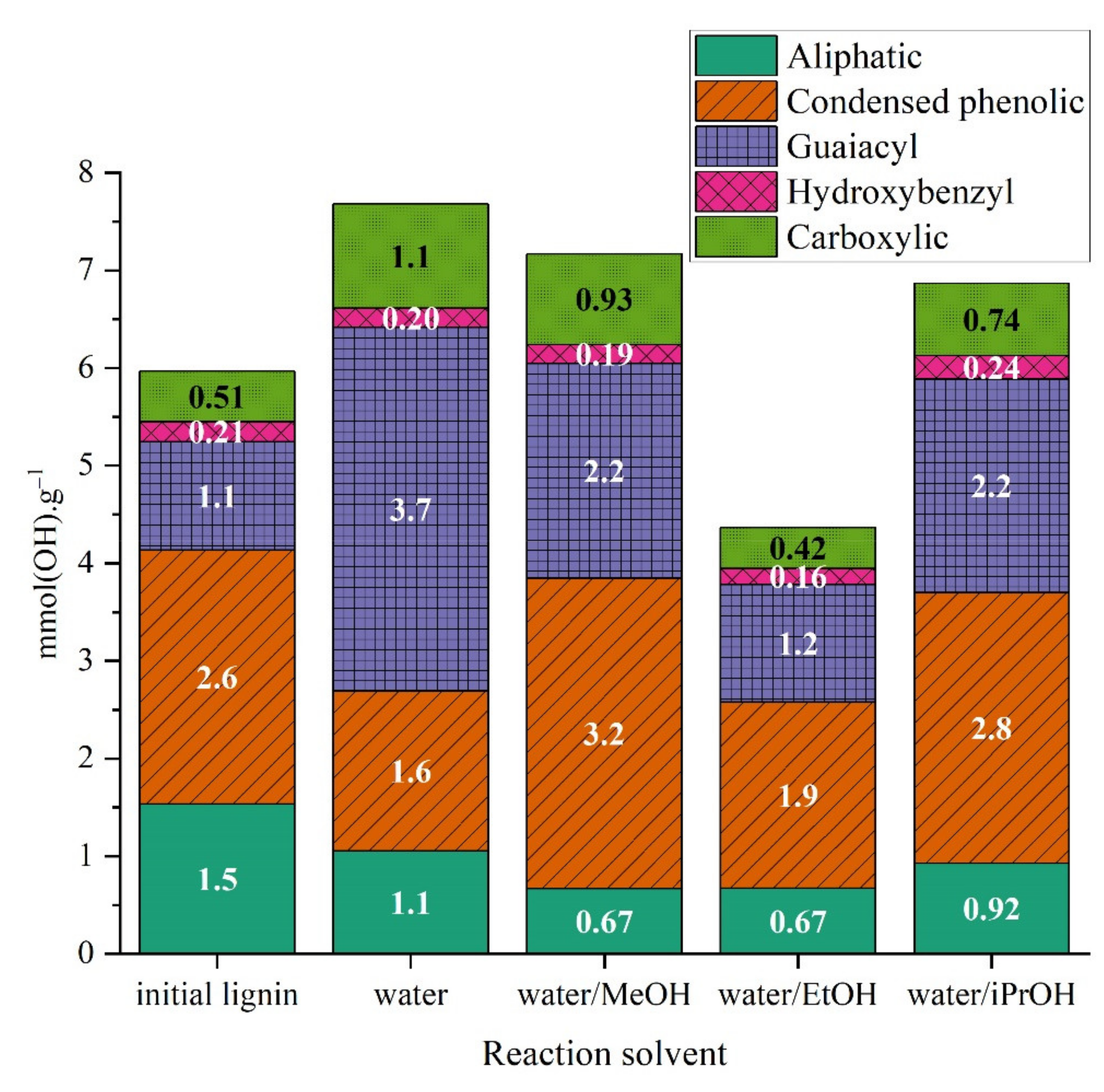
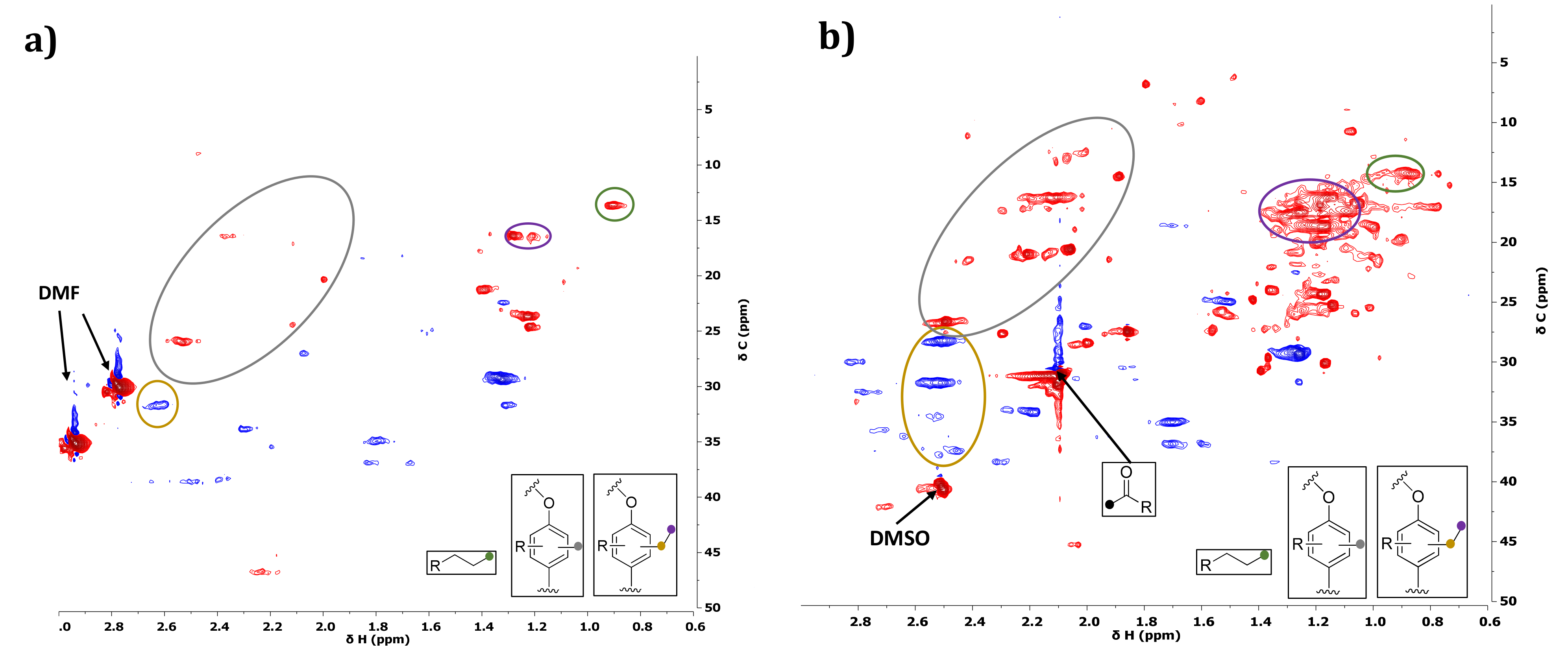
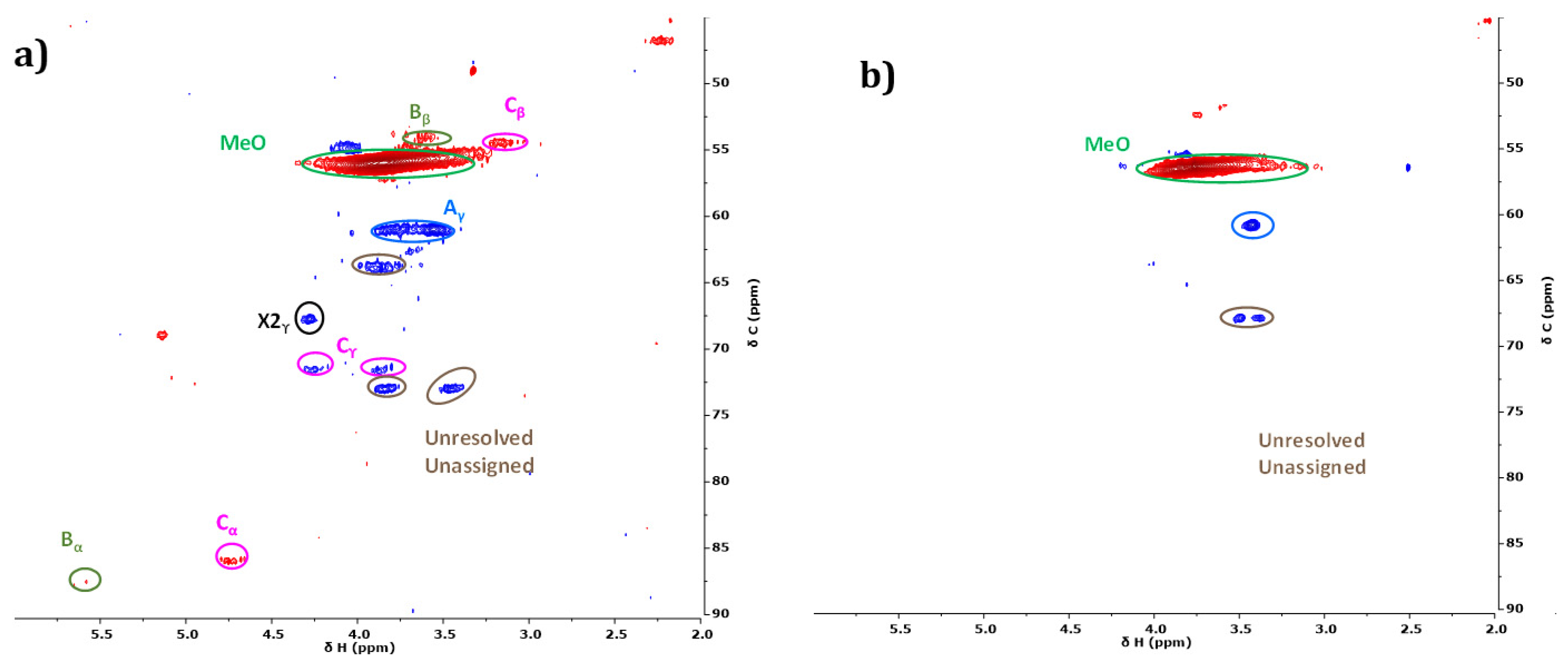
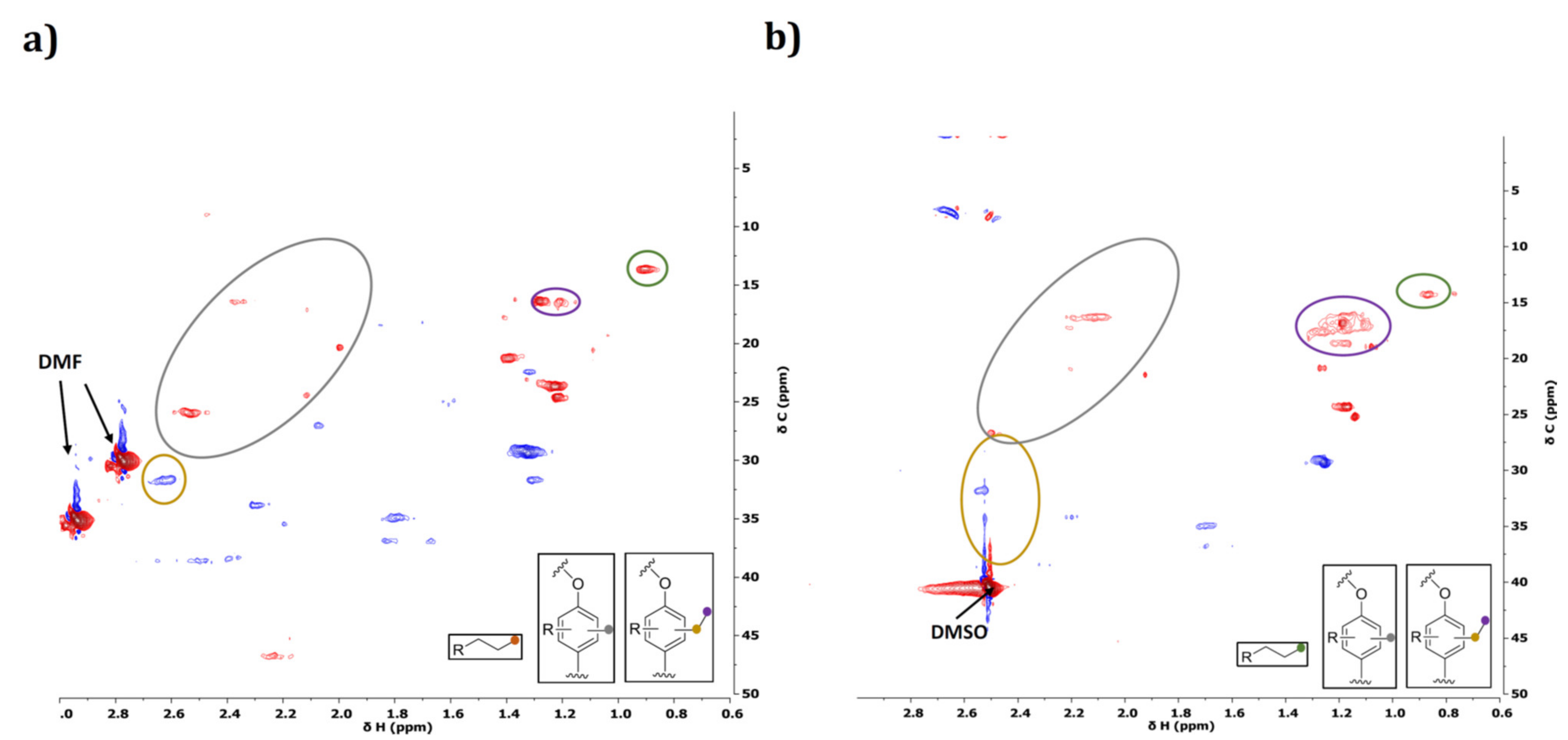
2.3.3. Characterization of Reaction Products
2.3.4. Investigation of Pd/ZrO2 Catalyst
- -
- In the presence of ZrO2 support alone, for comparison purpose (Run 1);
- -
- In the presence of Pd/ZrO2 and at the end of the reaction while the reactor is at 225 °C (hot filtration) sampling the reaction mixture (Run 2) for ICP analysis of the liquid phase;
- -
- In the presence of the support ZrO2 with the amount of palladium acetate (Pd(OAc)2) corresponding to leached palladium in Run 2. Pd(OAc)2 is entirely soluble in the reaction mixture at room temperature. This run was performed to determine the role of palladium and ZrO2 during the catalytic test;
- -
- In the presence of Pd(OAc)2 in concentration corresponding to leached palladium in Run 2. Pd(OAc)2 is entirely soluble in the reaction mixture at room temperature. This run was performed to determine the behavior of homogeneous palladium during the catalytic test (Table 6).
- -
- Heterogeneous 2%Pd/ZrO2 catalyst leads to global performances similar to homogeneous palladium 100 times less concentrated associated with ZrO2. Thus, the depolymerization of lignin to organic and aqueous phases is suspected to be catalyzed by homogeneous Pd species, coming either from Pd/ZrO2 leaching or Pd(OAc)2 in solution. Homogeneous Pd or Pd nanoparticles in solution are supposed to diffuse more easily in tridimensional structure of lignin.
- -
- The yield in monomers is higher in the absence of catalyst or in the presence of Pd(OAc)2 in combination with ZrO2. The degradation or recondensation of formed monomers could be catalyzed by Pd/ZrO2 catalyst only, explaining the lower amount of monomers in this case.
- -
- In the presence of H2, (see Section 2.3.2) the yield in monomers increases with Pd/ZrO2 with an evolution of products distribution. A higher rate of hydrogenolysis reactions catalyzed by Pd0 heterogeneous species is assumed in this case.
- -
- With the data collected, it is not possible to conclude on the type of active species when Pd(OAc)2 is used as a source of palladium. At room temperature, Pd complexes form in solution; at the end of the reaction, most of the palladium is in solid phase, probably forming nanoparticles. The exact form of palladium in reaction conditions is unknown.
- -
- The deposition of Pd on ZrO2 during reaction could happen but does not influence the catalytic results, given that Pd(OAc)2 leads to the same performances with and without ZrO2.
3. Materials and Methods
3.1. General Information
3.2. Catalysts
3.3. Standard Procedure for Lignin Conversion
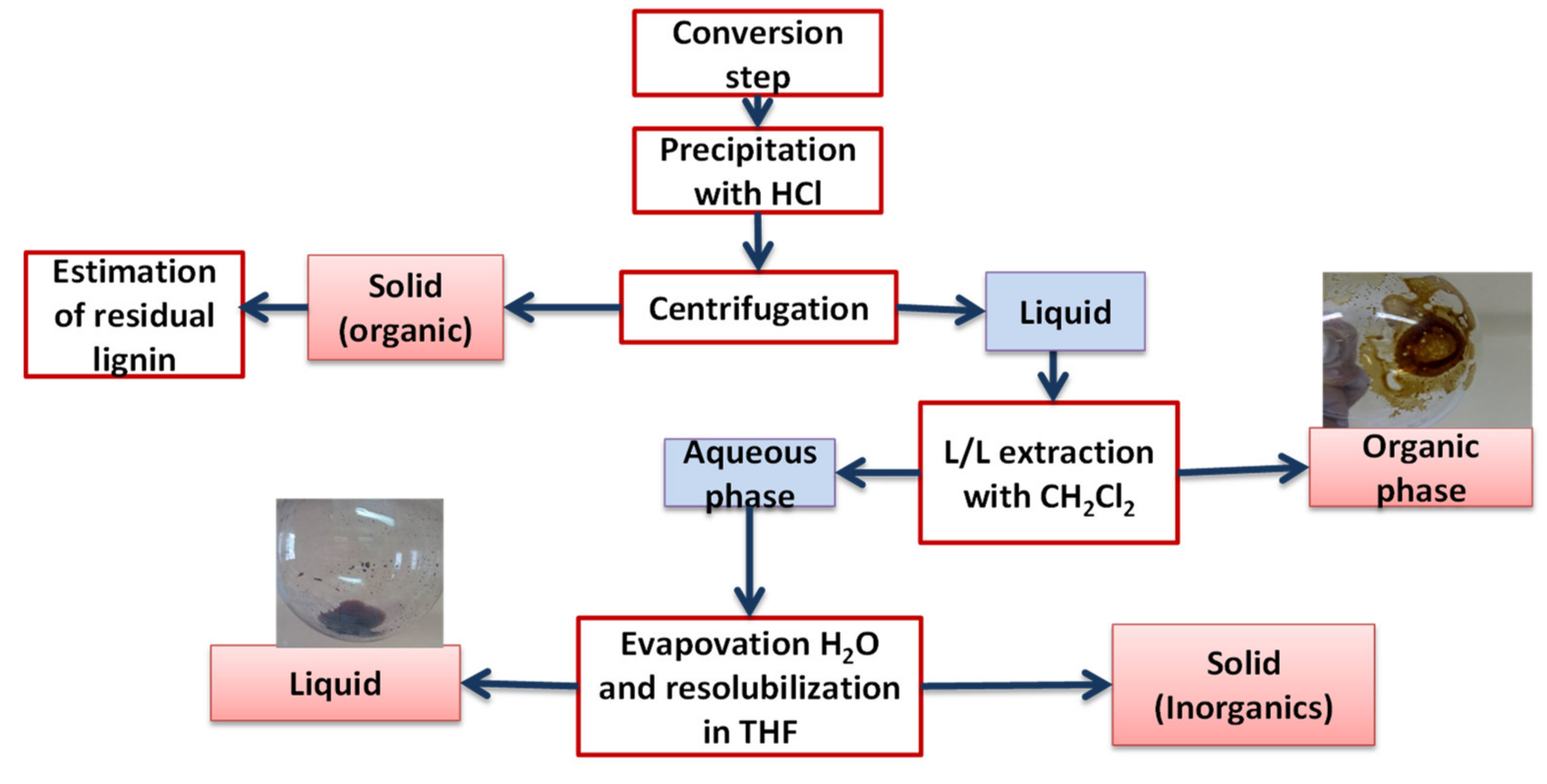
3.4. Products Recovery
3.5. Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vassilev, S.V.; Baxter, D.; Andersen, L.K.; Vassileva, C.G.; Morgan, T.J. An overview of the organic and inorganic phase composition of biomass. Fuel 2012, 94, 1–33. [Google Scholar] [CrossRef]
- Alén, R. Pulp mills and wood-based biorefineries. In Industrial Biorefineries & White Biotechnology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 91–126. ISBN 9780444634535. [Google Scholar]
- Sixta, H. Handbook of Pulp, 2nd ed.; Sixta, H., Ed.; Wiley: Lenzing, Austria, 2006; Volume 1–2, ISBN 9783527309993. [Google Scholar]
- Ragauskas, A.J.; Nagy, M.; Kim, D.H.; Eckert, C.A.; Hallett, J.P.; Liotta, C.L. From wood to fuels: Integrating biofuels and pulp production. Ind. Biotechnol. 2006, 2, 55–65. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Q.; Lee, D.J. Kraft lignin biorefinery: A perspective. Bioresour. Technol. 2018, 247, 1181–1183. [Google Scholar] [CrossRef] [PubMed]
- Katahira, R.; Elder, T.J.; Beckham, G.T. Chapter 1. A brief introduction to lignin structure. In RSC Energy and Environment Series; Royal Society of Chemistry: Cambridge, UK, 2018; Volume 2018, pp. 1–20. ISBN 9781782620426. [Google Scholar]
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crops Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Gierer, J. Chemical aspects of kraft pulping. Wood Sci. Technol. 1980, 14, 241–266. [Google Scholar] [CrossRef]
- Gellerstedt, G. Softwood kraft lignin: Raw material for the future. Ind. Crops Prod. 2015, 77, 845–854. [Google Scholar] [CrossRef]
- Poveda-Giraldo, J.A.; Solarte-Toro, J.C.; Cardona Alzate, C.A. The potential use of lignin as a platform product in biorefineries: A review. Renew. Sustain. Energy Rev. 2021, 138, 110688. [Google Scholar] [CrossRef]
- Cao, L.; Yu, I.K.M.; Liu, Y.; Ruan, X.; Tsang, D.C.W.; Hunt, A.J.; Ok, Y.S.; Song, H.; Zhang, S. Lignin valorization for the production of renewable chemicals: State-of-the-art review and future prospects. Bioresour. Technol. 2018, 269, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Gillet, S.; Aguedo, M.; Petitjean, L.; Morais, A.R.C.; da Costa Lopes, A.M.; Łukasik, R.M.; Anastas, P.T. Lignin transformations for high value applications: Towards targeted modifications using green chemistry. Green Chem. 2017, 19, 4200–4233. [Google Scholar] [CrossRef]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.-F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the way for lignin valorisation: Recent advances in bioengineering, biorefining and catalysis. Angew. Chem. Int. Ed. 2016, 55, 8164–8215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Zakzeski, J.; Weckhuysen, B.M. Lignin solubilization and aqueous phase reforming for the production of aromatic chemicals and hydrogen. ChemSusChem 2011, 4, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Vishtal, A.; Kraslawski, A. Challenges in industrial applications of technical lignins. BioResources 2011, 6, 3547–3568. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Romero, R.A.; Redondo, A.; Gnanakaran, S. Theoretical study of the remarkably diverse linkages in lignin. J. Phys. Chem. Lett. 2011, 2, 2660–2666. [Google Scholar] [CrossRef]
- Cheng, C.; Shen, D.; Gu, S.; Luo, K.H. State-of-the-art catalytic hydrogenolysis of lignin for the production of aromatic chemicals. Catal. Sci. Technol. 2018, 8, 6275–6296. [Google Scholar] [CrossRef]
- Garcia, A.C.; Cheng, S.; Cross, J.S. Solvolysis of kraft lignin to bio-oil: A critical review. Clean Technol. 2020, 2, 32. [Google Scholar] [CrossRef]
- Vermaas, J.V.; Crowley, M.F.; Beckham, G.T. Molecular lignin solubility and structure in organic solvents. ACS Sustain. Chem. Eng. 2020, 8, 17839–17850. [Google Scholar] [CrossRef]
- Margellou, A.; Triantafyllidis, K. Catalytic transfer hydrogenolysis reactions for lignin valorization to fuels and chemicals. Catalysts 2019, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, H.; Perras, F.A.; Naik, P.; Pruski, M.; Sadow, A.D.; Slowing, I.I. Two-step conversion of Kraft lignin to nylon precursors under mild conditions. Green Chem. 2020, 22, 4676–4682. [Google Scholar] [CrossRef]
- Jongerius, A.L.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Liquid-phase reforming and hydrodeoxygenation as a two-step route to aromatics from lignin. Green Chem. 2013, 15, 3049. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Ma, R.; Hao, W.; Chen, M.; Yan, F.; Cui, K.; Tian, Y.; Li, Y. Common pathways in ethanolysis of kraft lignin to platform chemicals over molybdenum-based catalysts. ACS Catal. 2015, 5, 4803–4813. [Google Scholar] [CrossRef]
- Cattelan, L.; Yuen, A.K.L.; Lui, M.Y.; Masters, A.F.; Selva, M.; Perosa, A.; Maschmeyer, T. Renewable aromatics from kraft lignin with molybdenum-based catalysts. ChemCatChem 2017, 9, 2717–2726. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Yang, C.; Zhu, Y.; Wang, J.; Wang, X.; Liu, C.; Liu, Y.; Lu, H.; Liang, B.; Li, Y. Synthesis-controlled α- and β-molybdenum carbide for base-promoted transfer hydrogenation of lignin to aromatic monomers in ethanol. Ind. Eng. Chem. Res. 2019, 58, 20270–20281. [Google Scholar] [CrossRef]
- Chen, M.; Ma, X.; Ma, R.; Wen, Z.; Yan, F.; Cui, K.; Chen, H.; Li, Y. Ethanolysis of kraft lignin over a reduction-modified MoO3 catalyst. Ind. Eng. Chem. Res. 2017, 56, 14025–14033. [Google Scholar] [CrossRef]
- Onwudili, J.A.; Williams, P.T. Catalytic depolymerization of alkali lignin in subcritical water: Influence of formic acid and Pd/C catalyst on the yields of liquid monomeric aromatic products. Green Chem. 2014, 16, 4740–4748. [Google Scholar] [CrossRef]
- Singh, S.K.; Ekhe, J.D. Cu–Mo doped zeolite ZSM-5 catalyzed conversion of lignin to alkyl phenols with high selectivity. Catal. Sci. Technol. 2015, 5, 2117–2124. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, L.; Gu, J.; Gou, L.; Xie, L.; Wang, Y.; Dai, L. Catalytic hydrotreatment of kraft lignin into aromatic alcohols over nickel-rhenium supported on niobium oxide catalyst. Bioresour. Technol. 2020, 299, 122582. [Google Scholar] [CrossRef]
- Yong, T.L.-K.; Matsumura, Y. Reaction kinetics of the lignin conversion in supercritical water. Ind. Eng. Chem. Res. 2012, 51, 11975–11988. [Google Scholar] [CrossRef]
- Gervasini, A.; Auroux, A. Acidity and basicity of metal oxide surfaces II. Determination by catalytic decomposition of isopropanol. J. Catal. 1991, 131, 190–198. [Google Scholar] [CrossRef]
- Huang, S.; Mahmood, N.; Zhang, Y.; Tymchyshyn, M.; Yuan, Z.; Xu, C. Reductive de-polymerization of kraft lignin with formic acid at low temperatures using inexpensive supported Ni-based catalysts. Fuel 2017, 209, 579–586. [Google Scholar] [CrossRef]
- Galkin, M.V.; Sawadjoon, S.; Rohde, V.; Dawange, M.; Samec, J.S.M. Mild heterogeneous palladium-catalyzed cleavage of β-o-4′-ether linkages of lignin model compounds and native lignin in air. ChemCatChem 2014, 6, 179–184. [Google Scholar] [CrossRef]
- Belkheiri, T.; Andersson, S.-I.; Mattsson, C.; Olausson, L.; Theliander, H.; Vamling, L. Hydrothermal liquefaction of kraft lignin in subcritical water: Influence of phenol as capping agent. Energy Fuels 2018, 32, 5923–5932. [Google Scholar] [CrossRef]
- Kouris, P.D.; van Osch, D.J.G.P.; Cremers, G.J.W.; Boot, M.D.; Hensen, E.J.M. Mild thermolytic solvolysis of technical lignins in polar organic solvents to a crude lignin oil. Sustain. Energy Fuels 2020, 4, 6212–6226. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters; CRC Press: Boca Raton, FL, USA, 2007; ISBN 9780429127526. [Google Scholar]
- Muzart, J. Pd-Catalyzed hydrogen-transfer reactions from alcohols to C=C, C=O, and C=N bonds. Eur. J. Org. Chem. 2015, 2015, 5693–5707. [Google Scholar] [CrossRef]
- Cabaniss, S.E.; McVey, I.F. Aqueous infrared carboxylate absorbances: Aliphatic monocarboxylates. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1995, 51, 2385–2395. [Google Scholar] [CrossRef]
- Marton, J.; Marton, T.; Falkehag, S.I.; Adler, E. Alkali-catalyzed reactions of formaldehyde with lignins. In Lignin Structure and Reactions; Marton, J., Ed.; ACS Publications: New York, NY, USA, 1966; pp. 125–144. [Google Scholar]
- Horikawa, Y.; Uchino, Y.; Sako, T. Alkylation and acetal formation using supercritical alcohol without catalyst. Chem. Lett. 2003, 32, 232–233. [Google Scholar] [CrossRef]
- Huang, X.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Catalytic depolymerization of lignin in supercritical ethanol. ChemSusChem 2014, 7, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Sekiguchi, G.; Adschiri, T.; Arai, K. Control of reversible reactions in supercritical water: I. alkylations. AIChE J. 2004, 50, 665–672. [Google Scholar] [CrossRef]
- Albano, G.; Evangelisti, C.; Aronica, L.A. Hydrogenolysis of benzyl protected phenols and aniline promoted by supported palladium nanoparticles. ChemistrySelect 2017, 2, 384–388. [Google Scholar] [CrossRef]
- Lamblin, M.; Nassar-Hardy, L.; Hierso, J.-C.; Fouquet, E.; Felpin, F.-X. Recyclable heterogeneous palladium catalysts in pure water: Sustainable developments in Suzuki, Heck, Sonogashira and Tsuji-Trost reactions. Adv. Synth. Catal. 2010, 352, 33–79. [Google Scholar] [CrossRef]
- Conlon, D.A.; Pipik, B.; Ferdinand, S.; LeBlond, C.R.; Sowa, J.R.; Izzo, B.; Collins, P.; Ho, G.-J.; Williams, J.M.; Shi, Y.-J.; et al. Suzuki–Miyaura cross-coupling with quasi-heterogeneous palladium. Adv. Synth. Catal. 2003, 345, 931–935. [Google Scholar] [CrossRef]
- Yin, L.; Liebscher, J. Carbon−carbon coupling reactions catalyzed by heterogeneous palladium catalysts. Chem. Rev. 2007, 107, 133–173. [Google Scholar] [CrossRef] [PubMed]
- Ait Rass, H.; Essayem, N.; Besson, M. Selective aerobic oxidation of 5-HMF into 2,5-furandicarboxylic acid with Pt catalysts supported on TiO2-and ZrO2 -based supports. ChemSusChem 2015, 8, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Granata, A.; Argyropoulos, D.S. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a reagent for the accurate determination of the uncondensed and condensed phenolic moieties in lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]
- Popescu, C.-M.; Popescu, M.-C.; Singurel, G.; Vasile, C.; Argyropoulos, D.S.; Willfor, S. Spectral characterization of eucalyptus wood. Appl. Spectrosc. 2007, 61, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Cabral Almada, C.; Kazachenko, A.; Fongarland, P.; Da Silva Perez, D.; Kuznetsov, B.N.; Djakovitch, L. Oxidative depolymerization of lignins for producing aromatics: Variation of botanical origin and extraction methods. Biomass Convers. Biorefin. 2020. [Google Scholar] [CrossRef]
- Cabral Almada, C.; Kazachenko, A.; Fongarland, P.; Da Silva Perez, D.; Kuznetsov, B.N.; Djakovitch, L. Supported-metal catalysts in upgrading lignin to aromatics by oxidative depolymerization. Catalysts 2021, 11, 467. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Water Content (%wt) | 6 |
|---|---|
| Acid-soluble lignin (%wt) | 39.5 |
| Klason lignin (%wt) | 47.3 |
| Sugar content (%wt) | 1.4 |
| Ash content (%wt) | 20.0 |
| Organic matter composition (%wt) | C 48%; H 5%; O 34%; S 4% |
| Inorganic matter (ashes) composition (%wt, lignin basis) | Al 1.2%; K 1.2%; Na 1.2%; Mg 720 ppm; Ca 600 ppm; Fe 240 ppm; Li 240 ppm; V 50 ppm. |
| Monomeric Products | Yield (mg·glignin−1) | ||
|---|---|---|---|
| 225 °C | 250 °C | 275 °C | |
| Phenol | - | 0.5 | 0.4 |
| p-Cresol | - | - | 0.1 |
| Catechol | - | - | 3.6 |
| 4-Methylcatechol | - | - | 2.2 |
| Guaiacol | 4.4 | 15.2 | 1.2 |
| 3-Methylcatechol | - | - | 0.2 |
| m-Creosol | - | 0.4 | - |
| p-Creosol | - | 1.1 | 0.8 |
| 2,6-Dimethyl-hydroquinone | - | - | 0.1 |
| 4-Ethylcatechol | - | - | 0.2 |
| p-Ethylguaiacol | - | 1.1 | 1.0 |
| Vanillin | 2.5 | 2.7 | 0.2 |
| Total identified | 10.1 | 24.4 | 10.5 |
| Catalyst | Metal Loading (%wt) | Mole of Metal per Gram a (µmol) | Specific Surface Area BET b (m²·g−1) |
|---|---|---|---|
| Pt/TiO2 | 1.4 | 7.2 | 89 |
| Pt/Al2O3 | 2.3 | 7.2 | 302 |
| Pt/ZrO2 | 2.4 | 8.3 | 58 |
| Pd/ZrO2 | 2.0 | 8.5 | 61 |
| Ru/ZrO2 | 1.7 | 7.0 | 57 |
| δD | δP | δH | R0 | |
|---|---|---|---|---|
| Kraft Lignin | 21.7 | 14.2 | 16.9 | 13.5 |
| Water-MeOH (50/50) | 15.3 | 14.15 | 32.3 | - |
| Water-EtOH (50/50) | 15.65 | 12.4 | 30.85 | - |
| Water-iPrOH (50/50) | 15.65 | 11.05 | 28.9 | - |
| Initial Lignin | Water | Water/MeOH | Water/EtOH | Water/iPrOH | |||||
|---|---|---|---|---|---|---|---|---|---|
| OP | KP | OP | KP | OP | KP | OP | KP | ||
| Haliphatic | 69 | 113 | 51 | 316 | 104 | 259 | 91 | 294 | 77 |
| Hoxygenated aliphatic | 228 | 212 | 133 | 137 | 169 | 144 | 183 | 137 | 183 |
| Haromatic | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Entry | Catalyst | Mass of Pd Introduced in Reactor | Pd in Solid Phase before Reaction | Pd in Solid Phase after Reaction |
|---|---|---|---|---|
| Run 1 | ZrO2 | 0 | - | - |
| Run 2 | Pd/ZrO2 | 50.1 ppm | 2.0%wt | 1.9%wt a |
| Run 3 | ZrO2 + Pd(OAc)2 | 0.54 ppm | 210 ± 5 ppm b | 220 ± 5 ppm |
| Run 4 | Pd(OAc)2 | 0.50 ppm | n.d. | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebhat, W.; El Roz, A.; Fongarland, P.; Vilcocq, L.; Djakovitch, L. Catalytic Liquefaction of Kraft Lignin with Solvothermal Approach. Catalysts 2021, 11, 875. https://doi.org/10.3390/catal11080875
Sebhat W, El Roz A, Fongarland P, Vilcocq L, Djakovitch L. Catalytic Liquefaction of Kraft Lignin with Solvothermal Approach. Catalysts. 2021; 11(8):875. https://doi.org/10.3390/catal11080875
Chicago/Turabian StyleSebhat, Woldemichael, Ayman El Roz, Pascal Fongarland, Léa Vilcocq, and Laurent Djakovitch. 2021. "Catalytic Liquefaction of Kraft Lignin with Solvothermal Approach" Catalysts 11, no. 8: 875. https://doi.org/10.3390/catal11080875
APA StyleSebhat, W., El Roz, A., Fongarland, P., Vilcocq, L., & Djakovitch, L. (2021). Catalytic Liquefaction of Kraft Lignin with Solvothermal Approach. Catalysts, 11(8), 875. https://doi.org/10.3390/catal11080875