Abstract
The valorisation of biomass-derived platform chemicals via catalytic hydrogenation is an eco-friendly tool which allows us to recover bio-based building blocks and produce fine chemicals with high industrial appeal. In the present study, a novel surfactant-type triazolyl-thioether ligand was prepared, showing excellent catalytic activity in the presence of bis(1,5-cyclooctadiene)diiridium(I) dichloride [Ir(COD)Cl]2 for the hydrogenation of furfural, cinnamaldehyde, levulinic acid, 5-hydroxymethylfurfural, vanillin, and citral. Easy recovery by liquid/liquid extraction allowed us to recover the catalyst, which could then be efficiently recycled up to 11 times for the hydrogenation of furfural. In-depth analysis revealed the formation of spherical structures with metal nanoparticles as big as 2–6 nm surrounded by the anionic ligand, preventing iridium nanoparticle degradation.
1. Introduction
Since the advent of green chemistry, the never-ending research on eco-friendly and sustainable approaches for the synthesis of valuable chemicals has generated an amazingly number of innovative procedures and protocols. Despite the massive body of literature available regarding efficient catalysts able to hydrogenate ketones or aldehydes [1,2,3,4], the transformation of biomass derived carbonyl compounds into high-value chemicals via environmentally safe solvents and mild reaction conditions are still actual, especially if including the principles of green chemistry [5,6,7,8]. In this context, homogenous catalysts generally offer high activity and selectivity with a wide substrate scope, but they are often scantly recyclable [9,10,11]. On the contrary, heterogenous catalysts have the advantage of being highly recyclable, but they generally lack in selectivity [12]. In fact, studies related to the application of heterogenous catalysts are generally limited to a single substrate since optimised conditions are not the same for different compounds. In the last decade, the use of ligands employed to stabilise transition metal nanoparticles (MNPs) generated from organometallic complexes emerged as a powerful tool combining the advantages derived from homogenous and heterogenous catalysis [13,14,15].
As demonstrated by pioneering studies in this field, MNPs can combine high activity and selectivity with efficient recycling of the catalyst as compared to molecular defined catalysts. Polymers, surfactants, and phosphorus-, nitrogen-, or sulphur-donor ligands or inorganic supports have been successfully employed to stabilise transition metals [16]. Several factors such as the shape, size, and distribution, deeply correlated to the preparation method, influence the reactivity of MNPs. Rare examples are present in the literature regarding the use of IrNPs as efficient catalysts for the hydrogenation of carbonyl compounds [16,17,18]. These reports deal with the use of pre-formed NPs usually synthetised in the presence of hydrogen atmosphere. However, the catalytic hydrogenation of carbonyl compounds still requires hydrogen pressure to be efficient. In contrast, the in situ formation of IrNPs is a well-known methodology that allows us to speed up the evaluation of new catalytic systems, thus avoiding multistep and costly catalyst preparation and isolation.
In this scenario, the design of NPs as stabilisers is crucial, and an ideal candidate should take advantage of cheap and commercially available reagents, benign solvents, and fast work-up procedures. Click chemistry, a term coined by K.B. Sharpless in 2001 to describe a novel concept of organic synthesis based on the use of a few simple high-yielding reactions [19,20,21], has widely been demonstrated to be a versatile tool for ligand design and catalyst synthesis.
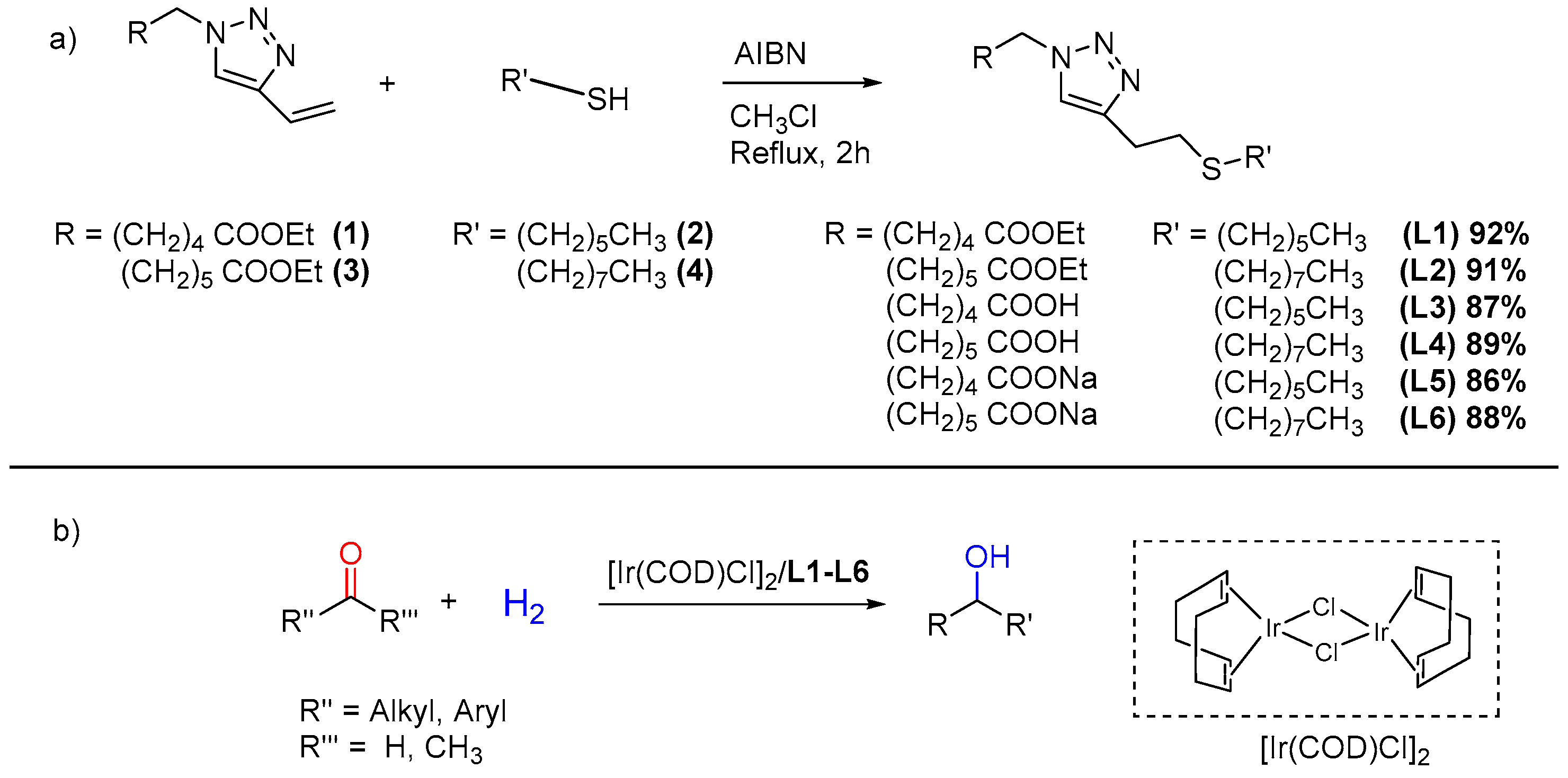
Our research group has long been involved in the study of innovative sustainable processes for fine chemistry and the manufacturing industry [22,23,24]. In this paper, the synthesis of a novel class of triazolyl-thioether surfactant-type ligands is reported, with a two-step strategy combining copper azide–alkyne cycloaddition (CuAAC) and thiol–ene click reactions (Scheme 1a). Further, these ligands (L1–L6) were employed in combination with bis(1,5-cyclooctadiene)diiridium(I) dichloride [Ir(COD)Cl]2 as a metal precursor for the in situ preparation of IrNPs, employed in the hydrogenation of carbonyl compounds (Scheme 1b). The recyclability of IrNPs was also assessed. To the best of our knowledge, there are no previous data in the literature regarding the use of recyclable non-supported IrNPs employed for the catalytic hydrogenation of biomass-derived substrates containing carbonyl and/or unsaturated functional groups.
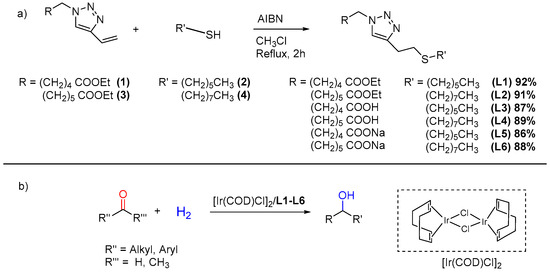
Scheme 1.
(a) Synthesis of ligands L1–L6 from vinyl triazoles with corresponding yield percentages; (b) Hydrogenation of carbonyl compounds as target reactions investigated in this work.
2. Results and Discussion
2.1. Ligand Synthesis
Copper azide–alkyne cycloaddition (CuAAC) is based on the reaction between an azide and alkyne in the presence of CuSO4·5H2O (or CuOAc) and sodium ascorbate as a reductant to give 1,4-disubstituted-1,2,3-triazoles. The modularity of the reaction allows us to easily tune the substituents while maintaining regioselectivity and high yields.
Similarly, the addition of a thiol to a C=C double bond, named thiol–ene addition, is an attracting click reaction mainly used for the polymerisation of monomers but also useful for the synthesis of small molecules [25,26,27,28].
With this in mind, a synthetic strategy was developed incorporating a double click strategy. While the synthesis of vinyl triazoles was accomplished as reported by Takizawa et al. (see Scheme S1 in the Supplementary Materials) [29], the method to synthetise the thioether moiety was specifically devised (Scheme 1a). A base-catalysed reaction between vinyltriazole (1) and hexanthiol (2), in the presence of triethylamine or sodium methoxide as a catalyst, did not allow us to recover the desired ligand (L1). This behaviour is probably due to the nature of the vinylic bond of compound (1), which is not sufficiently electron-poor to undergo nucleophilic attack by the thiolate. The most efficient procedure was found to be radical addition, in the presence of AIBN as a radical initiator, in chloroform under reflux for 2 h. With this methodology, two thiols with different aliphatic chain length were used to give ligands L1–L2 in high yields (>90%, Scheme 1). Basic hydrolysis was carried out by the addition of NaOH to obtain the corresponding acids L3–L4 or the sodium salts L5–L6 (Scheme 1). All syntheses were carried out without the need for inert atmosphere, and both intermediates and final ligands proved to be air stable. Characterisation by 1H, 13C NMR, ESI-MS spectrometry, and elemental analysis confirmed the structures of the ligands synthetised.
2.2. Hydrogenation of Furfural
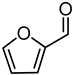
With a small library of ligands in hand, their catalytic potential in the presence of [Ir(COD)Cl]2 was evaluated for hydrogenation reactions. Given the wide commercial availability of furfural (FUR) and its appeal as a platform chemical [30,31], the hydrogenation of FUR to furfuryl alcohol (FA) was chosen as a model reaction for preliminary investigations into the catalytic activity, selectivity, and recyclability of the [Ir(COD)Cl]2/L1–L6 systems (Table 1).

Table 1.
Ligands and solvents screened for the hydrogenation of FUR to FA.
First, experiments were carried out according to reaction conditions similar to those generally adopted for the hydrogenation of carbonyl compounds [3,32,33]. Preliminarily, the catalytic activity of [Ir(COD)Cl]2, in the absence of any ligand, was tested in the hydrogenation of FUR. In fact, with a 0.1 mol% loading of [Ir(COD)Cl]2 in respect to FUR, at PH2 40 bar and 60 °C in a 1/1 vol/vol THF/H2O mixture, a mixture of FA/Tetrahydrofurfuryl alcohol (THFA) was obtained in a 4/1 ratio within 18 h. These data indicated that although [Ir(COD)Cl]2 had modest activity in the reaction conditions tested, low selectivity (Table 1, entry 1) was observed and no recycling was achieved (Table 1, entry 2) due to the formation of iridium black. Thus, a set of experiments was run to verify how the presence of the ligands would influence the catalytic activity of [Ir(COD)Cl]2 (Table 1, entries 3–9).
The experimental data revealed that ligands L1–L4 (Table 1, entries 3–6) did not significantly improve the catalytic activity of [Ir(COD)Cl]2, probably due to the low solubility of the ligands in THF/H2O; thus, different solvent mixtures were tested (Table S1), but with no significant improvement. Furthermore, the presence of ester moieties strongly inhibits the possibility for L1–L2 to promote the formation of surfactant-type structures able to preserve IrNPs. In fact, our research group previously reported an investigation of similar ligands and the effect on catalysis of the use of the Na salt [4,34]. As in this case, the L was scantly effective while the L-Na species were very soluble and, consequently, most active. As far as L3–L4 are concerned, possible interaction of the ligand via carboxylic group coordination to [Ir(COD)Cl]2 may also be a possible explanation for the lower activity in water. The performances of the catalytic systems formed by the combination of [Ir(COD)Cl]2 with L1–L4 were lower than the activity of [Ir(COD)Cl]2 when no ligand was used, suggesting that they were detrimental to the formation of IrNPs. In fact, TEM analysis of particles recovered after running the reaction with L1–L4 ligands revealed that iridium black particles were formed not surrounded by organic surfactants, similar to what is reported in Figure S1. On the other hand, the use of ligands as water-soluble salts (L5 and L6), prepared by addition to the reaction mixture of 10 eq. of NaOH in respect to the ligand, displayed good activity (Table 1, entries 7, 9), reaching yields in FA as high as 83% with L5 (Table 1, entry 7).
These data are in agreement with the literature, since anionic ligands are known as efficient water-soluble auxiliaries and may improve the water solubility of metallic species with respect to neutral or cationic ligands [35].
These preliminary experiments showed that ligand L5 was the best-performing ligand, even after separation and recycling of the catalyst phase (Table 1, entry 8). Me-THF (Table 1, entry 10), cyclopentyl methyl ether (CPME, entry 11), and water (Table 1, entry 12) were also tested, reaching lower yields in FA but with the advantage of higher sustainability of the solvents employed.
Importantly, selective formation of FA occurred in all cases when L1–L6 were added, while formation of the fully saturated THFA alcohol occurred (8%) only with [Ir(COD)Cl]2, probably due to the formation of iridium black nanoparticles (Table 1, entry 1). In fact, the reaction mixture recovered from this latter experiment was black, while under the best conditions (Table 1, entries 7 and 8), the reaction mixture was clear yellow. Upon standing at 4 °C for 24 h, however, some very fine and dark particles were also observed. This fact suggested that the formation of iridium nanoparticles may also occur in the presence of the ligand, and cooling down the temperature promoted the precipitation of the nanoparticles.
As pointed out by B. Chaudret et al. [36], it is not an easy matter to distinguish whether nanoparticles or well-defined molecular catalysts are involved in the reaction mechanism when solid or colloidal materials are detected after the reaction.
According to the literature, a key parameter which helps us to give a possible answer to the arcane is the behaviour of the catalytic system at variable precursor/ligand molar ratios [37]. It is not unusual that, unlike for molecular complexes, sub-stoichiometric amounts of ligand may have a beneficial effect on the stabilisation of nanoparticles, rather than stoichiometric or even higher quantities of ligand added [15,18,38]. Therefore, a study on the [Ir(COD)Cl]2/L5 ratio was performed, and the relevant results are summarised in Table 2. Furthermore, to optimise reaction conditions, several other parameters were screened, such as time, temperature, hydrogen pressure, catalyst loading, and amount of NaOH added.
Table 2.
Optimisation of the [Ir(COD)Cl]2/L5 system for the hydrogenation of FUR to FA.
When the [Ir(COD)Cl]2/L5 ratio was decreased by half at 60 °C and PH2 40 bar, yields as high as 87% in FA were achieved (compare entry 7, Table 1 with entry 1, Table 2).
Screening different [Ir(COD)Cl]2/L5 ratios at higher temperature (80 °C) confirmed that stochiometric or excess amounts of L5 were detrimental to the catalytic activity (compare entries 2–4, Table 2). On the contrary, the catalytic system performed better when the ligand was present in quantities of ≤0.5 equivalents (entries 5–6, Table 2), reaching highest yields in FA (95%) with a [Ir(COD)Cl]2/L5 molar ratio of 0.5. Interestingly, a reaction carried out under the best conditions but with 0.05 mol% [Ir(COD)Cl]2 loading gave equally good yields in FA: 95 and 84%, respectively (compare entries 5 and 7, Table 2).
The influence of the amount of NaOH in respect to L5 was then verified. Interestingly, it was found that 2 eq. of base was enough to promote catalyst activity (Table 2, entry 10), while large excess led to slightly lower efficiency (Table 2, entry 11).
The best reaction conditions for the hydrogenation of FUR to FA were found to be 80 °C with PH2 of 40 bar and a FUR/[Ir(COD)Cl]2/L5/NaOH molar ratio of 1.0/0.1/0.1/0.2, in a 1/1 vol/vol THF/H2O solvent mixture, reaching yields in FA of 95% in 18 h with total selectivity.
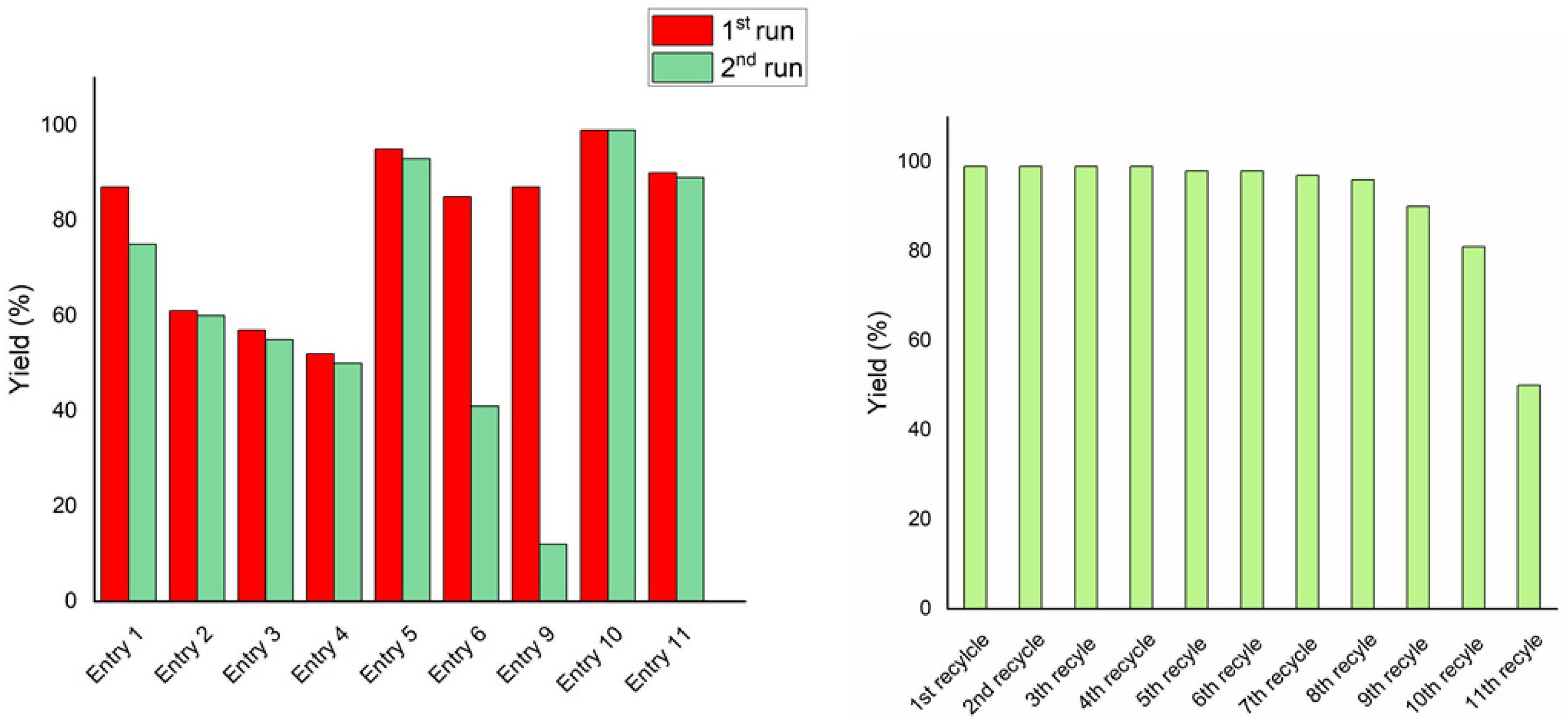
The recyclability of the catalytic system was further investigated. The catalytic mixtures of entries 1–6 and 9–11 recovered after a first catalytic cycle were recovered and recycled (Figure 1).
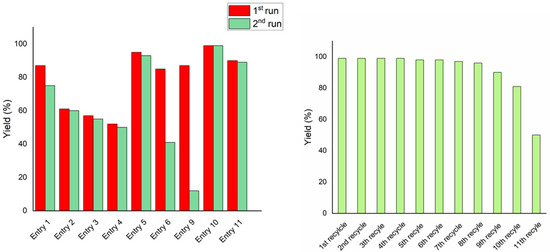
Figure 1.
(a) Yield of entries 1–6 and their first recycling. Entries refer to Table 2. (b) Recycling experiments of FUR hydrogenation in the presence of a FUR/[Ir(COD)Cl]2/L5/NaOH ratio of 1.0/0.1/0.1/0.2 mol/mol at 80 °C, PH2 40 bar, for 18 h; solvent: THF/H2O (3 mL/3 mL).
The data clearly show that both the base concentration and metal/ligand ratio affect the outcome of the recycling experiment. Excess base was undoubtedly beneficial in terms of recyclability (Figure 1a, entries 1–5 and 10–11). This is due to the role of the base, solubilising ligand L5 into the aqueous media. On the contrary, when only one equivalent of NaOH with respect to L5 was added, the recycled catalytic phase dropped at 12% of FA yield (Figure 1a, entry 9). Furthermore, when a metal-to-ligand ratio of <1:0.5 was used, lower activity and poorer recycling capability were observed (Figure 1a, entry 6). On the contrary, stoichiometric or excess L5 was demonstrated to preserve catalytic activity after recycling (Figure 1a).
Under optimised conditions, the catalyst proved to be very active, selective, and recyclable (see Figure 1b). In fact, recovery and reuse of the water phase containing the active catalyst showed no loss in activity for up to six recycles. Activity significantly dropped only after 10 recycling experiments.
When the active phase was recovered via liquid/liquid extraction, leaching of the catalyst in the organic phase was evaluated by ICP-MS analysis, revealing that leaching of the catalyst was limited to a maximum of 0.3%. This confirmed that the catalytic system was well confined into the aqueous phase, in accordance with its long-lasting recyclability.
2.3. Characterisation of IrNPs
The data reported above clearly highlight that the catalytic mechanism of [Ir(COD)Cl]2/L5 was not in line with classical homogeneous hydrogenation. The results achieved with FUR seem to indicate that the reactions may proceed via L5-stabilised MNPs.
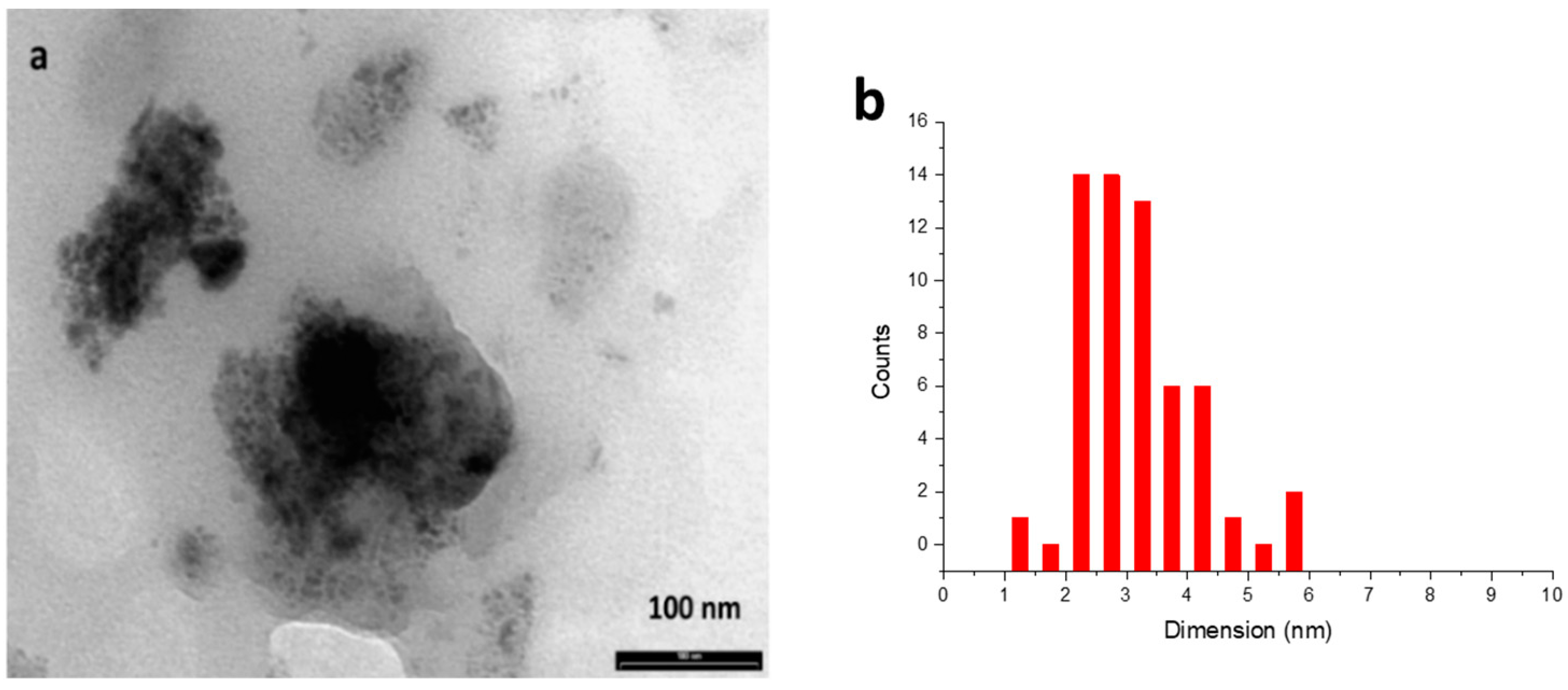
The most important parameters affecting the reactivity of MNPs are known to be their size, shape, and distribution. Hence, TEM analysis was performed to further gain insight into the catalyst’s nature. Iridium nanoparticles recovered after catalysis (Figure 2a) revealed sizes within the range of 2–6 nm.
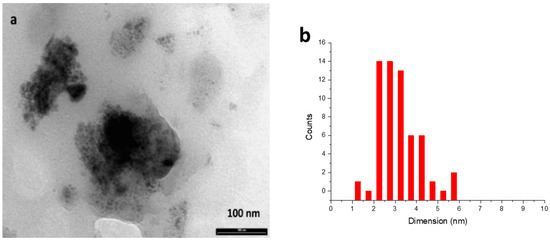
Figure 2.
(a) TEM analysis of iridium nanoparticles recovered after reaction of entry 10, Table 2; (b) Size distribution of nanoparticles.
Most of the particles showed a size distribution within the range of 2–3.5 nm, as reported in Figure 2b. The dimensions of the active particles are similar to those of other types of MNPs reported in the literature for catalytic hydrogenation reactions [17,35,38,39].
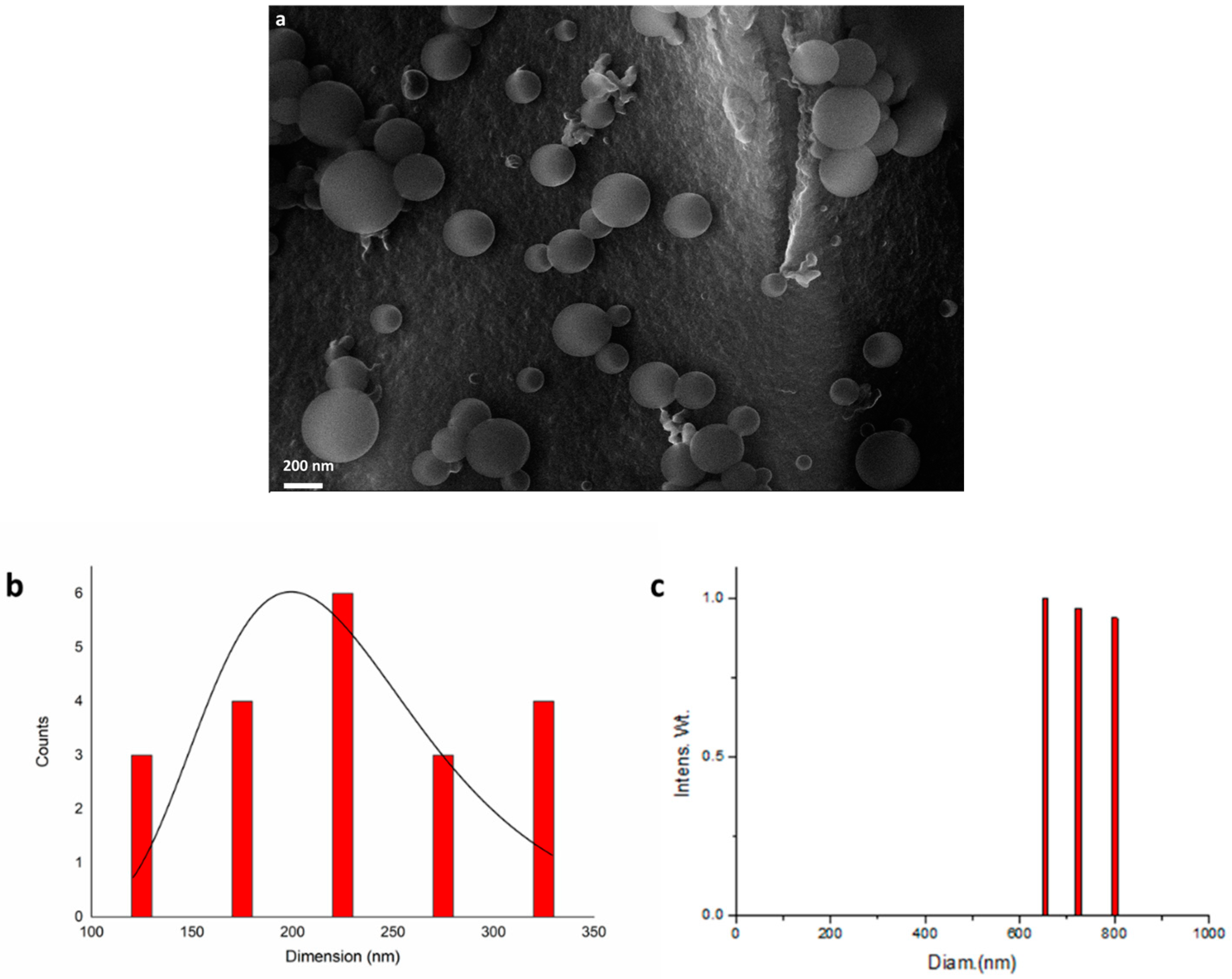
The MNPs shown by TEM analysis were clustered in spherical profiles, and an organic frame surrounding the metal species was visible. Driven by these indications, SEM analysis was performed, from which it was possible to observe spherical aggregates (Figure 3a) with dimensions within the range of 100 to 350 nm (Figure 3b).
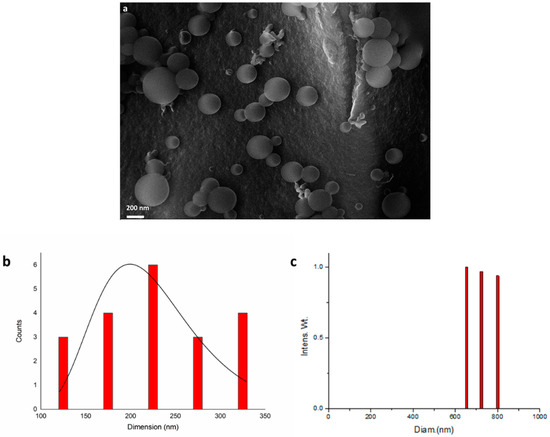
Figure 3.
(a) SEM analysis of iridium nanoparticles recovered after entry 11, Table 2; (b) Size distribution of micellar structures; (c) DLS analysis at 654 nm.
It is important to point out that all iridium nanoparticles were confined within the spherical surfactant structures, as observed by delay light scattering analysis (DLS), acquired at 654 nm, which showed only aggregates from 600 nm up to 800 nm and no traces of smaller aggregates (Figure 3c). Aggregates were found to be bigger with respect to SEM analysis because of the acquisition modality in solution, which is known to give higher values [40].
These data clearly confirmed that L5 works as a surfactant-type ligand. It should be mentioned that, despite L5 displaying the best catalytic performances in combination with [Ir(COD)Cl]2 when other ligands where employed, FA was in any case recovered but in variable, generally lower yields.
Analogous conclusions may be inferred for the ligand-to-metal molar ratio. Some representative TEM and SEM images acquired at different L/Ir ratios show that at L/Ir < 0.5, the formation of spherical structures surrounding the metal nanoparticles was not well defined (see Supplementary Materials, Figures S1–S2). On the contrary, at L/Ir > 0.5, the overabundant presence of the organic framework seemed to prevent metal nanoparticles from interacting with the substrate (see Supplementary Materials, Figures S3–S4).
Summing up, ligand L5 possesses the right properties in terms of chain lengths and hydrophobic/hydrophilic balance to form stable surfactant-type structures. This was confirmed by DLS, TEM, and SEM analysis performed on L6, which demonstrated the absence of surfactant-type structures (see Figure S5).
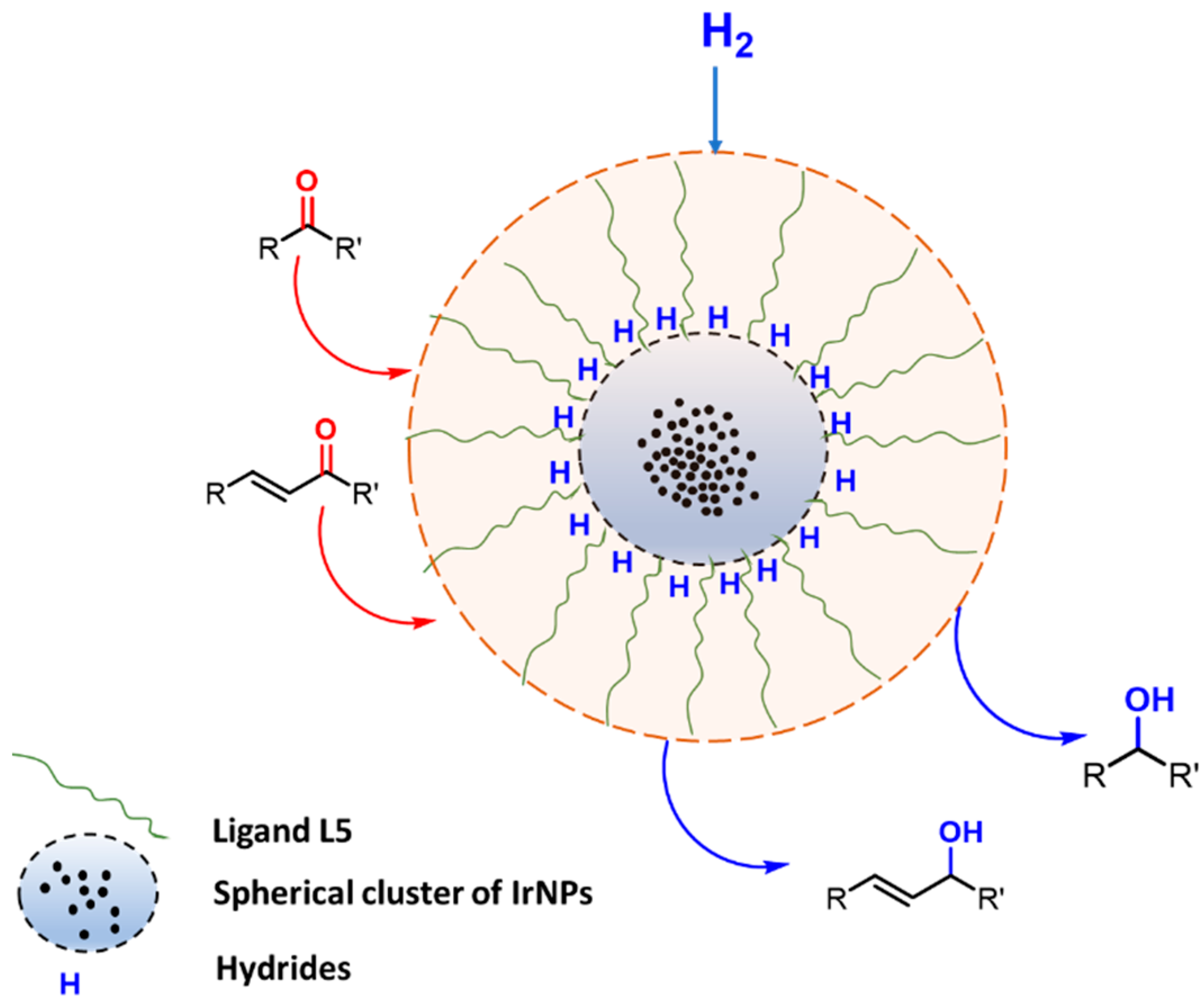
The mechanism by which the substrate interacts with the NPs is thought to be dependent on the presence of hydrides on the surface of iridium nanoparticles, as similarly demonstrated by B. Chaudret and co-workers by NMR experiments based on H/D exchange reactions [16,41]. A schematic representation of the possible catalytic system is presented in Figure 4.
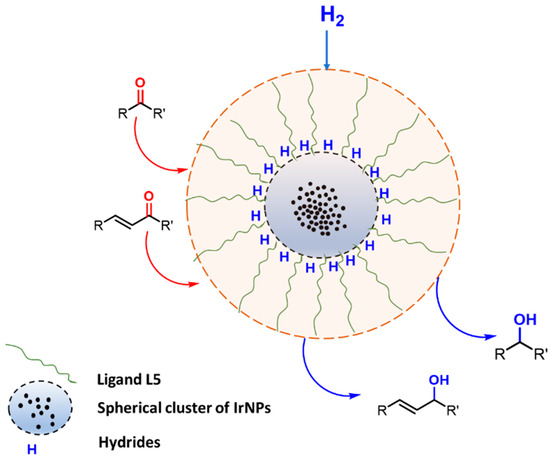
Figure 4.
A representation of the supposed mechanism for the hydrogenation of carbonyl compounds via L5-stabilised IrNPs.
However, it is worth mentioning that further studies are currently in progress to gain deeper insights into the IrNP stabilisation mechanism. In particular, more in-depth analyses supporting the formation of IrNPs and their identification as active species when combined with L5 are necessary and will be the object of future studies.
2.4. Hydrogenation of Biomass-Derived Platform Chenicals
A biomass-derived platform chemical is an intermediate molecule that is produced from biomass at a competitive cost and can be transformed into a number of valuable intermediates, preferably in a large-scale process [42,43].
Included in this class of compounds are cinnamaldehyde (CIN), levulinic acid (LA), 5-hydroxymethylfurfural (HMF), vanillin (VAN), and citral, which were taken into account herein to further explore the scope of the new catalytic system formed by [Ir(COD)Cl]2 and L5.
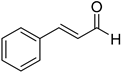
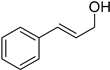
Notably, CIN was selectively converted to cinnamyl alcohol (CA) in relatively mild conditions, i.e., 40 °C and 40 bar (Table 3, entry 2). This high selectivity reached by metal nanoparticles is rare according to the literature [37]. Furthermore, this result confirmed that IrNPs performed better than other recently reported catalytic systems, as 10 times lower catalyst loading was required in our case (0.1 mol% vs. 1 mol%).
Table 3.
Hydrogenation of biomass-derived carbonyl compounds.
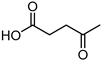
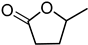
Optimised reaction conditions were also used for the conversion of LA to γ-valerolactone (GVL). At 80 °C and 40 bar, LA was fully reduced to GVL with almost complete selectivity (Table 3, entry 3). Recycling of the active catalyst led to satisfying activity of the aqueous phase with minimal activity retention.
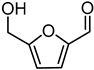
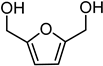
Reduction of 5-HMF leads to the production of the corresponding 2,5-bis(hydroxymethyl)furan diol (BHMF), used for the production of polymers, additives, bioactive compounds, and fuels [44,45]. Unfortunately, under the best reaction conditions optimised for FUR, yields in BHMF did not reach above 58% (Table 3, entry 4). This result is probably due to the formation of humins, since a brown precipitate was observed after the reaction. Humins are polymeric materials that are often formed when HMF is treated at a high temperature [46,47].
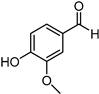
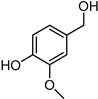
Hydrogenation via the [Ir(COD)Cl]2/L5 catalytic system allowed us to obtain VA with excellent yields (up to 88%) and recycle the aqueous phase with no loss in activity (Table 3, entry 5).
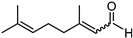
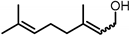
A further challenging substrate is citral, a mixture of geranial (trans isomer) and neral (cis isomer) that is mainly derived by distillation from lemongrass oil and commonly used as a flavouring agent. Due to the presence of a C=O and two C=C bonds, selectivity to the α,β–unsaturated product (a mixture of nerol and geraniol) is hard to achieve. In fact, most of the procedures reported in the literature make use of heterogenous catalysts, leading to poor yields of the unsaturated alcohol [48]. In our case, 75% yield of a 50/50 mixture of nerol and geraniol was achieved at 40 bar and 80 °C (Table 3, entry 6).
3. Materials and Methods
3.1. Materials
Commercial solvents (Aldrich, St. Louis, MO, USA) were purified as described in the literature [49]. Ethyl 5-bromovalerate, 3-butyn-1-ol, and 1-hexanthiol were purchased from Aldrich. Azobisisobutyronitrile (AIBN) was purchased from Acros organics (Geel, Belgium). Ethyl-5-azidopentanoate, ethyl-7-azidoeptanoate, and 5-(4-vinyl-1H-1,2,3-triazol-1-yl)pentanoate were prepared according to procedures reported in the literature [29].
3.2. Instrumentation
1H and 13C {1H} NMR spectra were recorded on a Bruker AVANCE 300 spectrometer (Billerica, MA, USA) operating at 300.21 and 75.44 MHz, respectively. The chemical shift values of the spectra are reported in δ units with reference to the residual solvent signal. The proton assignments were performed by standard chemical shift correlations, as well as by 1H 2D COSY experiments.
ESI-MS analyses were performed using a Finnigan LCQ-Duo ion-trap instrument (Thermo Fisher, Waltham, MA, USA), operating in positive ion mode (sheath gas N2, source voltage 4.0 KV, capillary voltage 21 V, capillary temperature 200 °C). All mass spectra were recorded on freshly prepared solutions.
Elemental analyses (C, H, N, S) were carried out using an Elementar Unicube microanalyser (Langenselbold, Germany). ICP-MS analyses were performed using an Agilent 7500a-Series instrument (Santa Clara, GA, USA).
The morphology was examined via Field Emission Electron Scanning Microscopy (FE-SEM) using a ZEISS LEO 1525 (Oberkochen, Germany). The samples were prepared by drying one drop of solution from the crude reaction mixture on silicon and observed by an inlens detector at 15 kV. The samples were observed without metallisation. TEM images were obtained using a Philips 208 Transmission Electron Microscope (Amsterdam, Netherlands). The samples were prepared by putting one drop of solution from the crude reaction mixture onto a copper grid pre-coated with a Formvar film and dried in air.
A Nicomp 380 ZLS photocorrelator (PSS, Santa Barbara, CA, USA) equipped with a 35 mW He/Ne laser (λ = 654nm) and an Avalanche photodiode detector was used to determine the size distribution. All data reported are the mean values achieved for a set of at least three experiments.
3.3. Generic Procedure for Catalytic Hydrogenation
Hydrogenation reactions were performed in a 50 mL glass reactor equipped with a magnetic stirring bar and a septum purged with N2. [Ir(η4-COD)Cl]2, the ligand, solvents, dodecane, and reagents were added in this order to the glass reactor under a stream of N2 and stirred for 10 min until a yellow homogenous solution was obtained. Ligands L5–L6 were previously mixed with an aqueous solution of NaOH at known concentration before being added to the reaction mixture. The glass reactor was then transferred into a pre-purged 150 mL stainless steel autoclave, which was then sealed. The autoclave was purged with 3 cycles of N2 followed by 3 cycles of H2, pressurised with hydrogen to the desired pressure, and then heated to the desired temperature. After the required time, the autoclave was cooled to room temperature and depressurised, and the glass reactor was carefully removed. Afterwards, the extracting solvent was added, and the organic phase was separated, dried over anhydrous MgSO4, and filtered. A sample was taken and analysed by GC. The water phase was briefly concentrated under vacuo to remove residual THF and then reused for recycling experiments.
3.4. Ligand Synthesis
3.4.1. Synthesis of Ethyl 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)pentanoate (L1)
Ethyl 4-(4-vinyl-1H-1,2,3-triazol-1-yl)butanoate (1) (1.82 g, 8.14 mmol),1-hexanthiol (1.38 mL, 9.76 mmol), and AIBN (0.67 g, 4.07 mmol) were dissolved in chloroform (25 mL) and refluxed. The reaction was monitored by 1H NMR. After 2 h, complete conversion of (1) was observed and the reaction was stopped. The crude mixture was then purified by flash chromatography (hexane/EtOAc 7:3) to yield L1 as a yellow oil (92% yield).
EA calcd for C17H31O2N3S: C(59.79) H(9.15) N(12.30) S(9.39); found: C(59.21) H(9.54) N(12.66) S(9.57).
1H NMR (300 MHz, CDCl3) δ (ppm) = 7.40 (s, 1H), 4.36 (t, J = 7.1 Hz, 2H), 4.15 (q, J = 7.1 Hz, 2H), 3.02 (t, J = 7.4 Hz, 2H), 2.86 (t, J = 7.4 Hz, 2H), 2.55 (t, J = 7.4 Hz, 2H), 2.36 (t, J = 7.2 Hz, 2H), 1.96 (m, 2H), 1.77–1.50 (m, 4H), 1.48–1.05 (m, 11H), 0.91 (t, J = 6.6 Hz, 3H).
13C NMR (300 MHz, CDCl3) δ (ppm) = 172.9, 146.4, 121.1, 60.5, 49.8, 33.4, 32.2, 31.7, 31.4, 29.6, 28.6, 26.3, 22.5, 22.5, 21.8, 14.2, 14.0.
HRMS (ESI) calcd for C17H31O2N3S [M+H]+: 341.21371 found: 341.21354.
3.4.2. Synthesis of Ethyl 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)hexanoate (L2)
L2 was synthetised via the same procedure as above and obtained as a pale yellow oil (91% yield).
EA calcd for C19H35O2N3S: C(61.75) H(9.55) N(11.37) S(8.67); found: C(60.82) H(9.04) N(12.46) S(9.01).
1H NMR (300 MHz, CDCl3) δ (ppm) = 7.37 (s, 1H), 4.44 (t, J = 7.1 Hz, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.00 (t, J = 7.4 Hz, 2H), 2.83 (t, J = 7.4 Hz, 2H), 2.52 (t, J = 7.4 Hz, 2H), 2.34 (t, J = 7.2 Hz, 2H), 1.95 (m, 2H), 1.65 (m, 4H), 1.25 (m, 13H), 0.88 (t, J = 6.6 Hz, 3H).
13C NMR (300 MHz, CDCl3) δ (ppm) = 172.8, 145.9, 120.3, 60.3, 50.8, 41.9, 32.4, 31.2, 30.9, 30.4, 29.4, 28.3, 26.7, 23.3, 22.9, 21.7, 14.6, 14.3.
HRMS (ESI) calcd for C19H35O2N3S [M+H]+: 369.2451 found: 369.2342.
3.4.3. Synthesis of 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)pentanoic Acid (L3)
L1 (0.90 g, 2.67 mmol) was dissolved in EtOH (6 mL), a solution of NaOH (0.20 g, 5 mmol) and H2O (4 mL) was added, and the mixture was gently stirred for 16 h at room temperature. At the end of this period, the EtOH was removed under vacuo and the aqueous layer was diluted with HCl 1 M (25 mL) and extracted with CH2Cl2 (15 mL × 3). The organic fractions were dried over MgSO4, filtered, and concentrated to yield L3 as a white solid (87% overall yield).
EA calcd for C15H26O2N3S: C(57.66) H(8.39) N(13.45) S(10.26); found: C(57.11) H(8.54) N(13.54) S(10.56).
1H NMR (300 MHz, CDCl3) δ (ppm) 10.51 (bs, 1H), 7.43 (s, 1H), 4.34 (t, J = 7.0 Hz, 2H), 2.99 (t, J = 7.2 Hz, 2H), 2.81 (s, 2H), 2.50 (t, J = 7.4 Hz, 2H), 2.38 (t, J = 7.2 Hz, 2H), 2.05–1.87 (m, 2H), 1.60 (m, 4H), 1.43–1.18 (m, 9H), 0.85 (t, J = 6.7 Hz, 3H).
13C NMR (300 MHz, CDCl3) δ (ppm) = 178.4, 130.7, 121.9, 52.1, 33.4, 32.2, 31.7, 31.4, 29.6, 28.6, 26.3, 22.5, 22.5, 21.8, 14.2.
HRMS (ESI) calcd for C15H26O2N3S [M+H]+: 312.17458 found: 312.17323.
3.4.4. Synthesis of 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)hexanoicanoic Acid (L4)
L4 was synthetised via the same procedure as above and obtained as a white solid (89% yield).
EA calcd for C18H33O2N3S: C(60.81) H(9.36) N(11.82) S(9.02); found: C(60.17) H(9.01) N(12.14) S(9.86).
1H NMR (300 MHz, CDCl3) δ (ppm) 10.01 (bs, 1H), 7.37 (s, 1H), 4.44 (t, J = 7.0 Hz, 2H), 3.00 (t, J = 7.2 Hz, 2H), 2.83 (s, 2H), 2.52 (t, J = 7.4 Hz, 2H), 2.34 (t, J = 7.2 Hz, 2H), 1.95 (m, 2H), 1.65 (m, 4H), 1.25 (m, 13H), 0.88 (t, J = 6.7 Hz, 3H).
13C NMR (300 MHz, CDCl3) δ (ppm) = 173.4, 146.9, 121.5, 60.9, 50.4, 33.9, 32.7, 32.1, 30.1, 29.6, 29.3, 26.8, 26.3, 23.1, 22.3, 14.7.8, 14.5.
HRMS (ESI) calcd for C18H33O2N3S [M+H]+: 355.2293 found: 355.2343.
3.4.5. Synthesis of Sodium 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)pentanoate (L5)
An aqueous solution of NaOH 1M (10 mL) was added to a round-bottom flask containing carboxyl ligand L3 (L3/NaOH = 1:1) The heterogenous mixture was vigorously stirred for 2 h until a homogenous colourless solution was obtained. Evaporating the water under vacuo allowed us to then obtain L5 as a white solid (86% overall yield).
EA calcd for C15H25O2N3SNa: C(53.87) H(7.53) N(12.56) S(9.59); found: C(54.11) H(7.34) N(13.12) S(9.78).
(300 MHz, CDCl3) δ (ppm) 10.51 (bs, 1H), 7.43 (s, 1H), 4.34 (t, J = 7.0 Hz, 2H), 2.99 (t, J = 7.2 Hz, 2H), 2.81 (s, 2H), 2.50 (t, J = 7.4 Hz, 2H), 2.38 (t, J = 7.2 Hz, 2H), 2.05–1.87 (m, 2H), 1.60 (m, 4H), 1.43–1.18 (m, 9H), 0.85 (t, J = 6.7 Hz, 3H).
HRMS (ESI) calcd for C15H26O2N3S [M+H]+: 312.17458 found: 312.17323.
3.4.6. Synthesis of Sodium 5-(4-(2-(hexylthio)ethyl)-1H-1,2,3-triazol-1-yl)pentanoate (L6)
L6 was synthetised via the same procedure as above and obtained as a colourless liquid (88% overall yield).
EA calcd for C18H32O2N3SNa: C(60.81) H(9.36) N(11.82) S(9.02); found: C(60.17) H(9.01) N(12.14) S(9.86).
1H NMR (300 MHz, D2O) δ (ppm) 7.33 (s, 1H), 4.26 (t, J = 7.0 Hz, 2H), 3.24 (t, J = 7.2 Hz, 2H), 2.87 (s, 2H), 2.51 (t, J = 7.4 Hz, 2H), 2.33 (t, J = 7.2 Hz, 2H), 1.97 (m, 2H), 1.61 (m, 4H), 1.24 (m, 13H), 0.4 (t, J = 6.7 Hz, 3H).
HRMS (ESI) calcd for C18H33O2N3SNa [M+H]+: 377.2113; found: 377.2578.
3.5. Product Characterisation
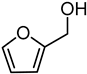
Furfuryl alcohol [44]. Pale yellow oil. 1H-NMR (300 MHz, CDCl3) δ (ppm) = 7.36 (m, 1H), 6.31 (m, 1H), 6.24 (d, J = 3.6 Hz, 1H), 4.52 (s, 2H), 3.07 (bs, 1H). 13C-NMR (400 MHz, CDCl3) δ (ppm) = 154.1, 142.4, 110.3, 107.7, 57.1.
Cinnamyl alcohol [32]. Pale yellow oil. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 7.41–7.22 (m, 5H), 6.62 (d, 1H), 6.37 (m, 1H), 4.33 (m, 2H), 1.49 ppm (br s, 1H); 13C-NMR (75 MHz, CDCl3): δ (ppm) = 136.7, 131.1, 128.6, 128.6, 127.7, 126.5, 63.7.
γ-valerolactone [50]. Colourless oil. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 4.64 (m, 1H), 2.53 (m, 2H), 2.36 (m, 1H), 1.81 (m, 1H), 1.47 (m, 3H). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 177.2, 77.6, 29.8, 28.9, 20.9.
2,5-Di(hydroxymethyl)furan [44]. White solid. 1H-NMR (300 MHz, DMSO-d6) δ (ppm) = 6.19 (s, 2H), 5.19 (bs, 2H), 4.36 (s, 1H). 13C-NMR (300 MHz, DMSO-d6) δ (ppm) = 154.7, 107.5, 55.8.
Vanillyl alcohol [51]. White solid. 1H-NMR (300 MHz, DMSO-d6) δ (ppm) = 6.86 (br d, J = 1.3 Hz, 1H), 6.73–6.65 (m, 2H), 4.42 (s, 2H), 3.75 ppm (s, 3H). 13C-NMR (300 MHz, DMSO-d6) δ (ppm) = 148.0, 146.5, 133.1, 119.6, 115.4, 111.5, 63.3, 56.0 ppm.
Nerol [52]. Colourless oil. 1H-NMR (300 MHz, CDCl3) δ (ppm) = 5.44 (t, J = 7 Hz, 2H), 5.05 (m, 1H), 4.09 (dd, J = 0.8, 7.1 Hz, 2H), 1.71 (s, 3H), 1.70 (s, 3H), 1.64 (s, 3H). 13C-NMR (300 MHz, CDCl3) δ (ppm) = 139.9, 132.4, 124.7, 123.8, 58.9, 31.9, 26.5, 25.6, 23.4, 17.6.
Geraniol [53]. Colourless oil. 1H-NMR (300 MHz, CDCl3) δ (ppm) = 5.45–5.50 (m, 1 H), 5.05–5.15 (m, 1 H), 4.07–4.20 (m, 2 H), 2.00–2.15 (m, 4 H), 1.61–1.69 (m, 9 H). 13C-NMR (300 MHz, CDCl3) δ (ppm) = 139.6, 131.5, 124.5, 123.8, 58.9, 39.5, 26.3, 25.7, 17.9, 16.3.
4. Conclusions
In summary, a robust and innovative catalytic system was successfully employed for the hydrogenation of biomass-derived carbonyl compounds. The small library of novel ligands synthesised by means of click chemistry was revealed to be versatile, easy to recover, and highly efficient in stabilising iridium nanoparticles. In fact, in-depth SEM, TEM, and DLS analyses confirmed that L5 was able to preserve nanoparticles via a surfactant-type assembly in aqueous media as large as 100–300 nm. It is noteworthy that the nanoparticles were found to be very active towards the hydrogenation of carbonyl compounds, with a focus on platform chemicals derived from biomass. Furthermore, the active species were stable enough to be recycled via an easy liquid/liquid extraction, thus maintaining catalytic activity for several runs. The new method developed herein provided alcohols with high yields and selectivity, representing a promising alternative for the valorisation of biomass-derived platform chemicals. Further studies are currently ongoing to gain deeper insights regarding the identification and characterisation of active species by more in-depth analysis.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/catal11080914/s1, Figure S1: TEM (left) and SEM (right) of [Ir(COD)Cl]2 with no surfactant ligand added, Figure S2: TEM (left) and SEM (right) of [Ir(COD)Cl]2/L5 1:0.25, Figure S3: TEM (left) and SEM (right) of [Ir(COD)Cl]2/L5 1:0.75, Figure S4: TEM (left) and SEM (right) of [Ir(COD)Cl]2/L5 1:1, Figure S5: TEM (left) and SEM (right) of [Ir(COD)Cl]2/L6 1:0.5, Table S1: Ligands and solvent screening for the hydrogenation of FUR to FA.
Author Contributions
All authors: conceptualisation, methodology, writing—original draft; R.S., C.B., N.B., A.D.M. and V.B.: investigation, data curation; R.S., S.P. and V.B.: supervision, project administration; All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the institutional funding of Ca’ Foscari University Venice (ADIR 2018 and 2019).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cornils, B.; Herrmann, W.A.; Beller, M.; Paciello, R. (Eds.) Applied Homogeneous Catalysis with Organometallic Compounds; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; ISBN 9788578110796. [Google Scholar]
- De Vries, J.G.; Elsevier, C.J. (Eds.) The Handbook of Homogeneous Hydrogenation; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; ISBN 9783527311613. [Google Scholar]
- Li, A.Y.; Moores, A. Carbonyl Reduction and Biomass: A Case Study of Sustainable Catalysis. ACS Sustain. Chem. Eng. 2019, 7, 10182–10197. [Google Scholar] [CrossRef]
- Paganelli, S.; Alam, M.M.; Beghetto, V.; Scrivanti, A.; Amadio, E.; Bertoldini, M.; Matteoli, U. A pyridyl-triazole ligand for ruthenium and iridium catalyzed C=C and C=O hydrogenations in water/organic solvent biphasic systems. Appl. Catal. A Gen. 2015, 503, 20–25. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, R. Catalytic Hydrogenation for Biomass Valorization; Rinaldi, R., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; ISBN 9781782620099. [Google Scholar]
- Sole, R.; Taddei, L.; Franceschi, C.; Beghetto, V. Efficient Chemo-Enzymatic Transformation of Animal Biomass Waste for Eco-Friendly Leather Production. Molecules 2019, 24, 2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornils, B.; Herrmann, W.A.; Horvat, J.; Leitner, W.; Mecking, S.; Olivier-Bourbigou, H.; Vogt, D. (Eds.) Multiphase Homogenous Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; ISBN 9783527307210. [Google Scholar]
- Scrivanti, A.; Bortoluzzi, M.; Sole, R.; Beghetto, V. Synthesis and characterization of yttrium, europium, terbium and dysprosium complexes containing a novel type of triazolyl–oxazoline ligand. Chem. Pap. 2018, 72, 799–808. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A. Aqueous-Phase Organometallic Catalysis; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2004; ISBN 3527307125. [Google Scholar]
- Beghetto, V.; Scrivanti, A.; Bertoldini, M.; Aversa, M.; Zancanaro, A.; Matteoli, U. A practical, enantioselective synthesis of the fragrances canthoxal and silvial®, and evaluation of their olfactory activity. Synthesis 2015, 47, 272–278. [Google Scholar] [CrossRef]
- Kliewer, C.J.; Bieri, M.; Somorjai, G.A. Hydrogenation of the α,β-unsaturated aldehydes acrolein, crotonaldehyde, and prenal over Pt single crystals: A kinetic and sum-frequency generation vibrational spectroscopy study. J. Am. Chem. Soc. 2009, 131, 9958–9966. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D. (Ed.) Nanoparticles and Catalysis; Wiley-VCH Verlag: Weinheim, Germany, 2008; ISBN 9783527305070. [Google Scholar]
- Serp, P.; Philippot, K. (Eds.) Nanomaterials in Catalysis: First Edition; Wiley-VCH Verlag: Weinheim, Germany, 2012; ISBN 9783527331246. [Google Scholar]
- Martínez-Prieto, L.M.; Chaudret, B. Organometallic Ruthenium Nanoparticles: Synthesis, Surface Chemistry, and Insights into Ligand Coordination. Acc. Chem. Res 2018, 51, 376–384. [Google Scholar] [CrossRef]
- Ruiz-Varilla, A.M.; Baquero, E.A.; Chaudret, B.; de Jesús, E.; Gonzalez-Arellano, C.; Flores, J.C. Water-soluble NHC-stabilized platinum nanoparticles as recoverable catalysts for hydrogenation in water. Catal. Sci. Technol. 2020, 10, 2874–2881. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Chen, P.; Zeng, M.; Jiang, J.; Jin, Z. Thermoregulated phase-transfer iridium nanoparticle catalyst: Highly selective hydrogenation of the C=O bond for α,β-unsaturated aldehydes and the C=C bond for α,β-unsaturated ketones. Catal. Sci. Technol. 2016, 6, 7386–7390. [Google Scholar] [CrossRef]
- Cano, I.; Tschan, M.J.L.; Martínez-Prieto, L.M.; Philippot, K.; Chaudret, B.; Van Leeuwen, P.W.N.M. Enantioselective hydrogenation of ketones by iridium nanoparticles ligated with chiral secondary phosphine oxides. Catal. Sci. Technol. 2016, 6, 3758–3766. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Beghetto, V.; Gatto, V.; Conca, S.; Bardella, N.; Buranello, C.; Gasparetto, G.; Sole, R. Development of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride cross-linked carboxymethyl cellulose films. Carbohydr. Polym. 2020, 249, 116810. [Google Scholar] [CrossRef]
- Sole, R.; Agostinis, L.; Conca, S.; Gatto, V.; Bardella, N.; Morandini, A.; Buranello, C.; Beghetto, V. Synthesis of Amidation Agents and Their Reactivity in Condensation Reactions. Synthesis 2021, 53, 1672–1682. [Google Scholar] [CrossRef]
- Sole, R.; Gatto, V.; Conca, S.; Bardella, N.; Morandini, A.; Beghetto, V. Sustainable Triazine-Based Dehydro-Condensation Agents for Amide Synthesis. Molecules 2021, 26, 191. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Lowe, A.B.; Bowman, C.N. Thiol-click chemistry: A multifaceted toolbox for small molecule and polymer synthesis. Chem. Soc. Rev. 2010, 39, 1355–1387. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Polymer Chemistry Thiol—Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Huang, S.; Sinha, J.; Podgo, M.; Zhang, X.; Claudino, M.; Bowman, C.N. Mechanistic Modeling of the Thiol−Michael Addition Polymerization Kinetics: Structural Effects of the Thiol and Vinyl Monomers. Macromolecules 2018, 51, 5979–5988. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.P.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2013, 26, 724–744. [Google Scholar] [CrossRef]
- Takizawa, K.; Nulwala, H.; Thibault, R.J.; Lowenhielm, P.; Yoshinaga, K.; Wooley, K.L.; Hawker, C.J. Facile Syntheses of 4-Vinyl-1,2,3-triazole Monomers by Click Azide/Acetylene Coupling. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 2897–2912. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; López Granados, M. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Deuss, P.J.; Barta, K.; De Vries, J.G. Homogeneous catalysis for the conversion of biomass and biomass-derived platform chemicals. Catal. Sci. Technol. 2014, 4, 1174–1196. [Google Scholar] [CrossRef] [Green Version]
- Puylaert, P.; van Heck, R.; Fan, Y.; Spannenberg, A.; Baumann, W.; Beller, M.; Medlock, J.; Bonrath, W.; Lefort, L.; Hinze, S.; et al. Selective Hydrogenation of α,β-Unsaturated Aldehydes and Ketones by Air-Stable Ruthenium NNS Complexes. Chemistry 2017, 23, 8473–8481. [Google Scholar] [CrossRef]
- Werkmeister, S.; Neumann, J.; Junge, K.; Beller, M. Pincer-Type Complexes for Catalytic (De)Hydrogenation and Transfer (De)Hydrogenation Reactions: Recent Progress. Chemistry 2015, 21, 12226–12250. [Google Scholar] [CrossRef] [PubMed]
- Scrivanti, A.; Beghetto, V.; Alam, M.M.; Paganelli, S.; Canton, P.; Bertoldini, M.; Amadio, E. Biphase hydroformylation catalyzed by rhodium in combination with a water-soluble pyridyl-triazole ligand. Inorg. Chim. Acta 2017, 455, 613–617. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Ma, Y.; Wu, J.; Wang, F.; Xiang, J.; Zhu, J.; Wang, Q.; Deng, J. Surfactant-accelerated asymmetric transfer hydrogenation with recyclable water-soluble catalyst in. RSC Adv. 2013, 3, 1825–1834. [Google Scholar] [CrossRef]
- Jansat, S.; Picurelli, D.; Pelzer, K.; Philippot, K.; Gómez, M.; Muller, G.; Lecante, P.; Chaudret, B. Synthesis, characterization and catalytic reactivity of ruthenium nanoparticles stabilized by chiral N-donor ligands. New J. Chem. 2006, 30, 115–122. [Google Scholar] [CrossRef]
- An, K.; Somorjai, G.A. Size and Shape Control of Metal Nanoparticles for Reaction Selectivity in Catalysis. ChemCatChem 2012, 4, 1512–1524. [Google Scholar] [CrossRef]
- Li, J.; Tang, Y.; Wang, Q.; Li, X.; Cun, L.; Zhang, X.; Zhu, J.; Li, L.; Deng, J. Chiral Surfactant-Type Catalyst for Asymmetric Reduction of Aliphatic Ketones in Water. J. Am. Chem. Soc. 2012, 134, 18522–18525. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Lipshutz, B.H.; Isley, N.A.; Fennewald, J.C.; Slack, E.D. On the Way Towards Greener Transition-Metal- Catalyzed Processes as Quantified by E Factors. Angew. Chem. Int. Ed. 2013, 52, 10952–10958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Sorella, G.; Strukul, G.; Scarso, A. Recent advances in catalysis in micellar media. Green Chem. 2015, 17, 644. [Google Scholar] [CrossRef]
- Pery, T.; Pelzer, K.; Buntkowsky, G.; Philippot, K.; Limbach, H.-H.; Chaudret, B. Direct NMR Evidence for the Presence of Mobile Surface Hydrides on Ruthenium Nanoparticles. ChemPhysChem 2005, 6, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Program, B.; Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass Volume I-Results of Screening for Potential Candidates from Sugars and Synthesis Gas; Pacific Northwest National Laboratory (PNNL): Richland, WA, USA; National Renewable Energy Laboratory (NREL) Office of Biomass: Golden, CO, USA, 2004. [Google Scholar]
- Sun, Z.; Fridrich, B.; De Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Xu, J.; Zhou, S.; He, A.; Tang, X.; Lin, L.; Xu, J.; Zhao, Y. Catalytic Advances in the Production and Application of Biomass-Derived 2,5-Dihydroxymethylfuran. ACS Catal. 2018, 8, 2959–2980. [Google Scholar] [CrossRef]
- Sole, R.; Bortoluzzi, M.; Spannenberg, A.; Tin, S.; Beghetto, V.; de Vries, J.G. Synthesis, characterization and catalytic activity of novel ruthenium complexes bearing NNN click based ligands. Dalt. Trans. 2019, 48, 13580–13588. [Google Scholar] [CrossRef]
- Van Putten, R.J.; Van Der Waal, J.C.; De Jong, E.; Rasrendra, C.B.; Heeres, H.J.; De Vries, J.G. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef] [PubMed]
- Stolle, A.; Gallert, T.; Schmöger, C.; Ondruschka, B. Hydrogenation of citral: A wide-spread model reaction for selective reduction of α,β-unsaturated aldehydes. RSC Adv. 2013, 3, 2112–2153. [Google Scholar] [CrossRef]
- Armarego, W.L.F. (Ed.) Purification of Laboratory Chemicals, 8th ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netehrlands, 2017. [Google Scholar]
- Bellè, A.; Tabanelli, T.; Fiorani, G.; Perosa, A.; Cavani, F.; Selva, M. A Multiphase Protocol for Selective Hydrogenation and Reductive Amination of Levulinic Acid with Integrated Catalyst Recovery. ChemSusChem 2019, 12, 3343–3354. [Google Scholar] [CrossRef] [PubMed]
- Figliolia, R.; Cavigli, P.; Comuzzi, C.; Del Zotto, A.; Lovison, D.; Strazzolini, P.; Susmel, S.; Zuccaccia, D.; Ballico, M.; Baratta, W. CNN pincer ruthenium complexes for efficient transfer hydrogenation of biomass-derived carbonyl compounds. Dalt. Trans. 2020, 49, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Wajs-Bonikowska, A.; Malarz, J.; Stojakowska, A. Composition of Essential Oils from Roots and Aerial Parts of Carpesium divaricatum, a Traditional Herbal Medicine and Wild Edible Plant from South-East Asia, Grown in Poland. Molecules 2019, 24, 4418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, M.; Liu, Q.Z.; Liu, Z.L. Identification of insecticidal constituents from the essential oil from the aerial parts Stachys riederi var. Japonica. Molecules 2018, 23, 1200. [Google Scholar] [CrossRef] [Green Version]
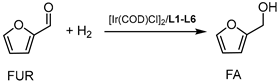
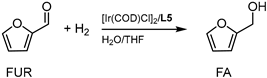
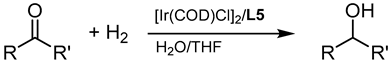
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).