
Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons


Abstract
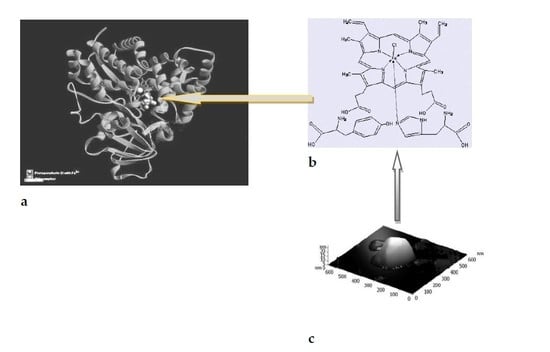
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Triple Systems Ni(acac)2+L2+PhOH That Are the Catalysts of Reaction of Selective Ethylbenzene Oxidation in α-Phenyl Ethyl Hydroperoxide
2.2. Possible Effect of Tyr Fragment
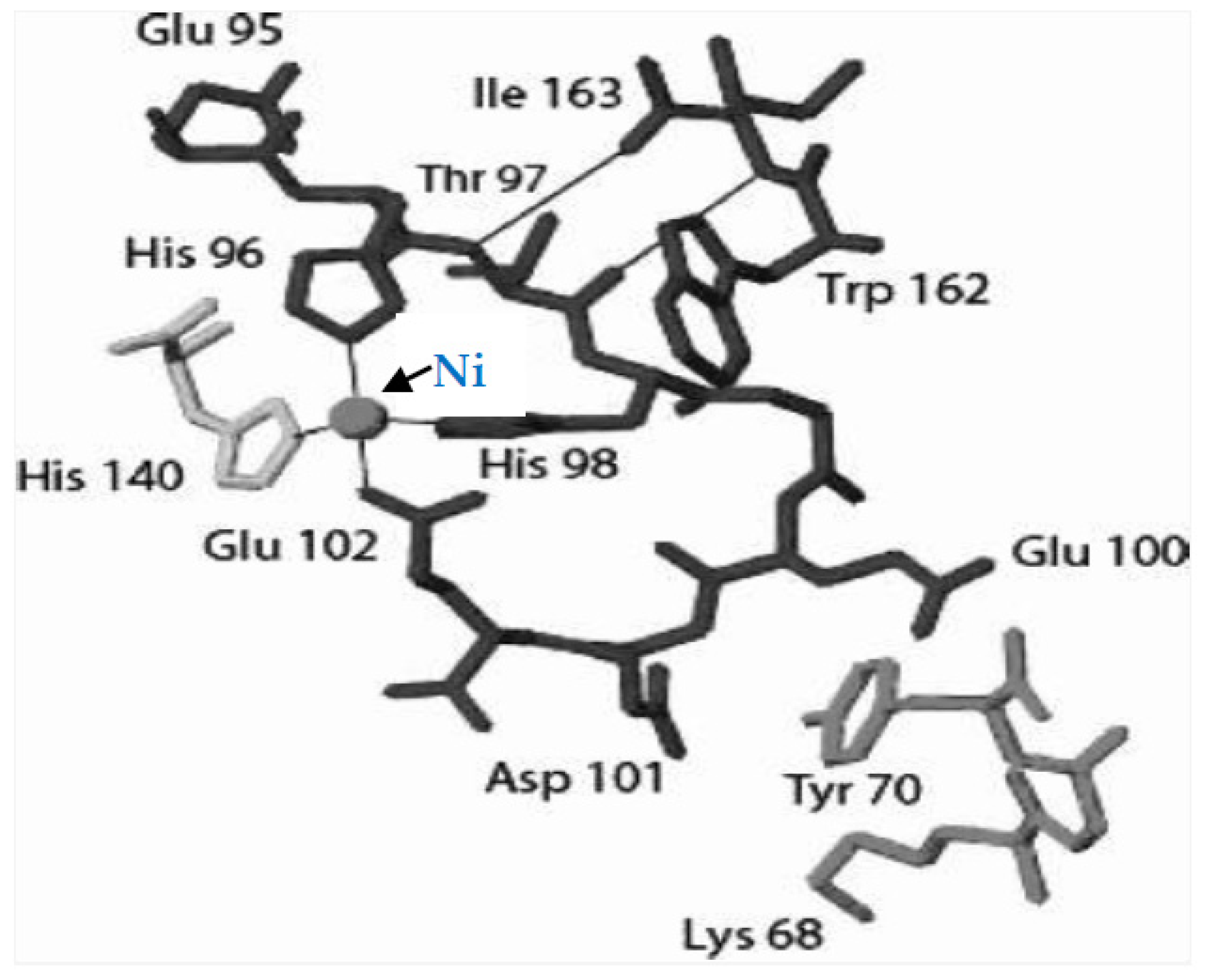
2.2.1. Catalysis with Acireductone Dioxygenases Ni(Fe)ARD
2.2.2. Catalysis with the Family of Cytochrome P450-Dependent Monooxygenases
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFM method | Atomic Force Microscopy method |
| (acac)− | Acetylacetonate ion |
| ARD | Acireductone ligand |
| 18C6 | 18-crown-6 |
| CO | Carbon monoxide |
| Dke1 | Fe-acetylacetone dioxygenase |
| HMPA | Hexamethylphosphorotriamide |
| Hacac | Acetylacetone |
| Hem | Hemin |
| His | L-histidine |
| L2 | Electron-donating mono-, or multidentate ligand |
| MSt | Stearates of Na, Li |
| NMP | N-methyl-2-pirrolidone |
| Ni(Fe)ARD | Ni(Fe) acireductone dioxygenases |
| (OAc)− | Acetate ion |
| PhOH | Phenol |
| Tyr | L-tyrosine |
| UV | Ultraviolet-spectroscopic data |
References
- Matienko, L.I.; Mosolova, L.A.; Zaikov, G.E. Selective Catalytic Hydrocarbons Oxidation. New Perspectives; Nova Science Publ. Inc.: New York, NY, USA, 2010; 150p. [Google Scholar]
- Matienko, L.I.; Mosolova, L.A.; Binyukov, V.I.; Mil, E.M.; Zaikov, G.E. The new approach to research of mechanism catalysis with nickel complexes in alkylarens oxidation. In Polymer Yearbook 2011; Nova Science Publishers: New York, NY, USA, 2012; pp. 221–230. [Google Scholar]
- Matienko, L.I.; Mosolova, L.A.; Binyukov, V.I.; Mil, E.M.; Zaikov, G.E. Supramolecular Nanostructures on the Basis of Catalytic Active Heteroligand Nickel Complexes and their Possible Roles in Chemical and Biological Systems. J. Biol. Res. 2012, 1, 37–44. [Google Scholar]
- Matienko, L.I.; Mosolova, L.A.; Binyukov, V.I.; Mil, E.M.; Zaikov, G.E. Triple systems, based on Ni(acac)2, introduced ligands- modifiers HMPA, N-methylpirrolidone-2, PhOH, or Tyrosine, as effective catalysts in selective ethylbenzene oxidation with dioxygen, as models of Ni-ARD Dioxygenase. Oxid. Commun. 2017, 40, 569–580. [Google Scholar]
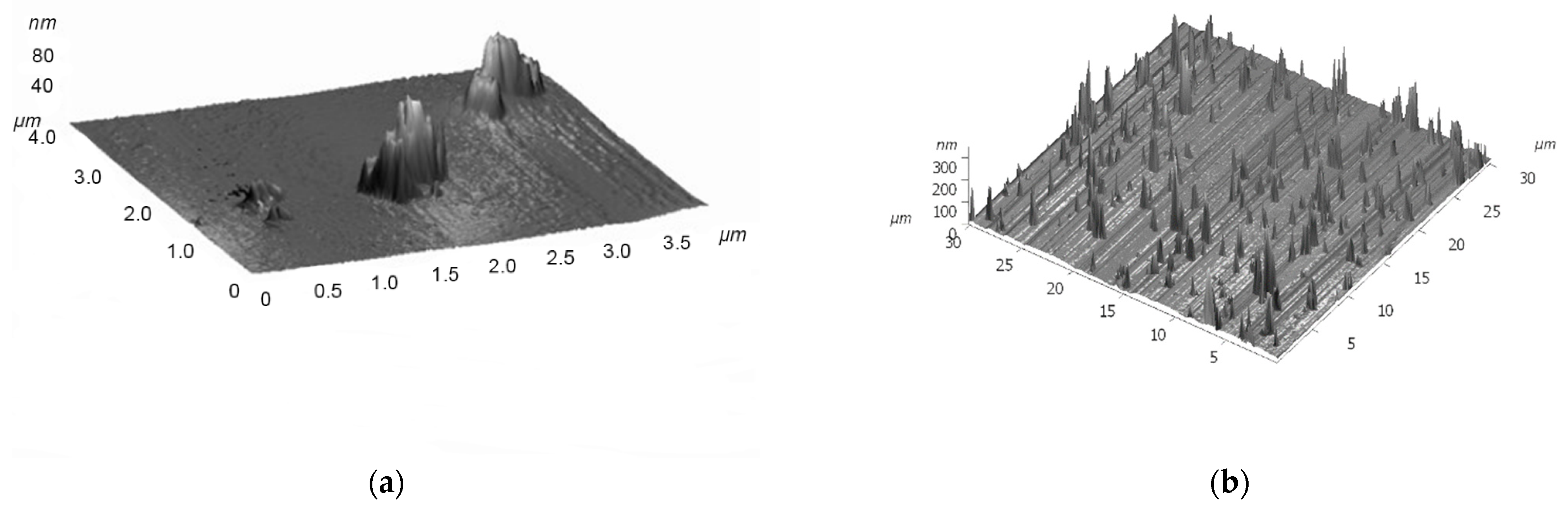
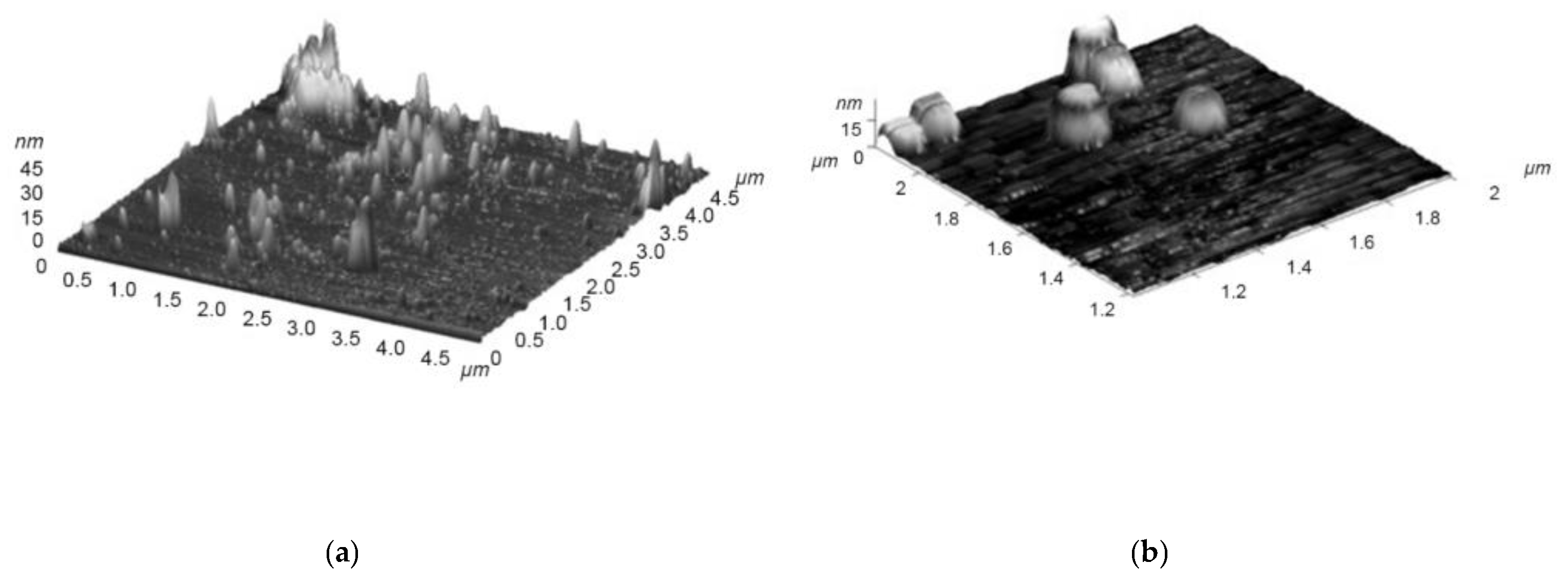
- Matienko, L.I.; Mil, E.M.; Binyukov, V.I. AFM Research of Supramolecular Structures. Russ. J. Phys. Chem. B 2020, 14, 559–563. [Google Scholar] [CrossRef]
- Ma, J.C.; Dougherty, D.A. The Cation−π Interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.D.; Buytendyk, A.M.; Wang, D.; Bowen, K.H.; Collins, K.D. Strong, Low-Barrier Hydrogen Bonds May Be Available to Enzymes. Biochemistry 2014, 53, 344–349. [Google Scholar] [CrossRef]
- Radi, R. Protein Tyrosine Nitration; Biochemical Mechanisms and Structure Basis of Functional Effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, S.; Sidhu, S.S. The importance of being tyrosine: Lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem. Biol. 2009, 4, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, V.V.; Roth, J.P. Tyrosine oxidation in heme oxygenase: Examination of long-range proton-coupled electron transfer. J. Biol. Inorg. Chem. 2014, 19, 1137–1148. [Google Scholar] [CrossRef]
- Mbughuni, M.M.; Meier, K.K.; Münck, E.; Lipscomb, J.D. Substrate-Mediated Oxygen Activation by Homoprotocatechuate 2,3-Dioxygenase: Intermediates Formed by a Tyrosine 257 Variant. Biochemistry 2012, 51, 8743–8754. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Klinman, J.P. Enzymatic Methyl Transfer: Role of an Active Site Residue in Generating Active Site Compaction That Correlates with Catalytic Efficiency. J. Am. Chem. Soc. 2011, 133, 17134–17137. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, S.; Dirk, L.M.A.; Yesselman, J.D.; Nimtz, J.S.; Adhikari, U.; Mehl, R.A.; Scheiner, S.; Houtz, R.L.; Al-Hashimi, H.M.; Trievel, R.C. Conservation and Functional Importance of Carbon–Oxygen Hydrogen Bonding in AdoMet-Dependent Methyltransferases. J. Am. Chem. Soc. 2013, 135, 15536–15548. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.S.; Beynon, R.J. The Astacin Family of Metalloendpeptidases. Protein Sci. 1995, 4, 1247–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, J.; Eser Bekir, E.; Mannikko, D.; Krzyaniak Matthew, D.; Fitzpatrick Paul, F. HYSCORE Analysis of the Effects of Substrates on Coordination of Water to the Active Site Iron in Tyrosine Hydroxylase. Biochemistry 2015, 54, 3759–3771. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Hole Hopping Through Tyrosine/Tryptophan Chains Protects Proteins from Oxidative Damage. Proc. Natl. Acad. Sci. USA 2015, 112, 10920−10925. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Groves John, T. Oxygen Activation and Radical Transformations in Heme Protein and Metalloporphyrins. Chem. Rev. 2018, 118, 2491−2553. [Google Scholar] [CrossRef] [Green Version]
- Chai, S.C.; Dang, T.; Ju, M.; Goldsmith, R.B.; Maroney, M.J.; Pochapsky, T.C. Characterization of Metal Binding in the Active Sites of Acireductone Dioxygenase Isoforms from Klebsiella ATCC 8724. Biochemistry 2008, 47, 2428–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Garber, A.; Ryan, J.; Deshpande, A.; Ringe, D.; Pochapsky, T.C. A Model for the Solution Structure of Human Fe(II)-Bound Acireductone Dioxygenase and Interactions with the Regulatory Domain of Matrix Metalloproteinase I (MMP-I). Biochemistry 2020, 59, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Matienko, L.I.; Binykov, V.I.; Mil, E.M.; Zaikov, G.E. Role of Supramolecular Structures in Mechanisms of Catalytic Oxidation, and Action of Ni(Fe)ARD Dioxygenases on Model Systems. Chem. Chem. Tech. 2020, 14, 304–311. [Google Scholar] [CrossRef]
- Leitgeb, S.; Straganz, G.D.; Nidetzky, B. Functional characterization of an orphan cupin protein from Burkholderia xenovorans reveals a mononuclear nonheme Fe2+-dependent oxygenase that cleaves β-diketones. FEBS J. 2009, 276, 5983–5997. [Google Scholar] [CrossRef]
- Cook Sarah, A.; Hill Ethan, A.; Borovik, A.S. Lessons from Nature: A Bio-Inspired Approach to Molecular Design. Biochemistry 2015, 54, 4167−4180. [Google Scholar]
- Savino, C.; Montemiglio, L.C.; Giuliano, S.; Miele Adriana, E.; Kendrew Jemth Per Steven, G.; Gianni, S. Investigating the Structural Plasticity of a Cytochrome P450. Three-dimensional structures of P450 EryK and binding to its physiological substrate. J. Biol. Chem. 2009, 284, 29170–29179. [Google Scholar] [CrossRef] [Green Version]
- Beletskaya, I.; Tyurin, V.S.; Tsivadze, A.Y.; Guilard, R.R.; Stem, C. Supramolecular Chemistry of Metalloporphyrins. Chem. Rev. 2009, 109, 1659–1713. [Google Scholar] [CrossRef] [PubMed]
- Murry, D.T.; Tysko, R. Side Chain Hydrogen-Bonding Interactions within Amiloid-like Fibrils Formed by the Low-Complexity Domain of FUS: Evidence from Solid State Nuclear Magnetic Resonance Spectroscopy. Biochemistry 2020, 59, 304–378. [Google Scholar]
- Matienko, L.I.; Binyukov, V.I.; Mil, E.M.; Mosolova, L.A.; Zaikov, G.E. Application of the AFM method to studying the role of Supramolecular structures and Tyr-fragment in the mechanism of Ni(Fe)ARD action on model systems. Oxid. Commun. 2018, 41, 429–440. [Google Scholar]
- Biedermann, F.; Schneider, H.-J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef]
- Basom, E.J.; Bryce, A.M.; Thielges, M.C. Conformational Heterogeneity and the Affinity of Substrate Molecular Recognition by Cytochrome P450cam. Biochemistry 2017, 56, 3248–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, R. Molecular level structural studies of metalloproteins/metalloenzymes by scanning tunneling microscopy; Scopes and promises. Curr. Sci. 2003, 84, 1202–1210. [Google Scholar]
- Sarkar, R.; Xie, T.-Z.; Endres, K.J.; Wang, Z.; Moorefield, C.N.; Saunders, M.J.; Ghorai, S.; Patri, A.K.; Wesdemiotis, C.; Dobrynin, A.V.; et al. Sierpinski Pyramids by Molecular Entanglement. J. Am. Chem. Soc. 2020, 142, 5528–5530. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matienko, L.; Binyukov, V.; Mil, E.; Goloshchapov, A. Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons. Catalysts 2021, 11, 1032. https://doi.org/10.3390/catal11091032
Matienko L, Binyukov V, Mil E, Goloshchapov A. Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons. Catalysts. 2021; 11(9):1032. https://doi.org/10.3390/catal11091032
Chicago/Turabian StyleMatienko, Ludmila, Vladimir Binyukov, Elena Mil, and Alexander Goloshchapov. 2021. "Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons" Catalysts 11, no. 9: 1032. https://doi.org/10.3390/catal11091032
APA StyleMatienko, L., Binyukov, V., Mil, E., & Goloshchapov, A. (2021). Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons. Catalysts, 11(9), 1032. https://doi.org/10.3390/catal11091032