
Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene



Abstract
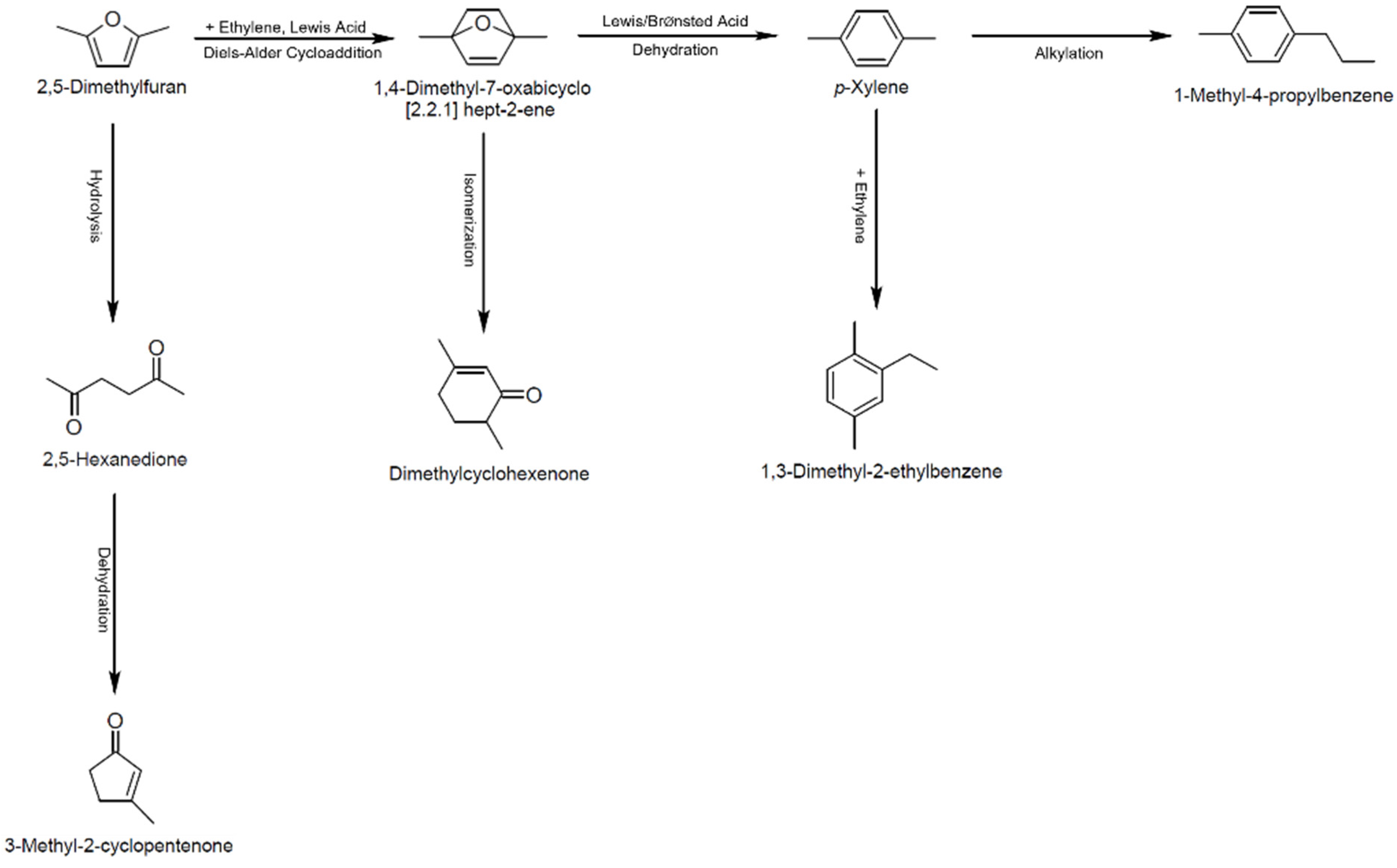
:1. Introduction
2. Results and Discussion
2.1. Catalyst Characterization
2.2. Catalytic Performance
3. Experimental Section
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalytic Test Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-C.; Green, S.K.; Williams, C.L.; Dauenhauer, P.J.; Fan, W. Ultra-selective cycloaddition of dimethylfuran for renewable p-Xylene with H-BEA. Green Chem. 2014, 16, 585–588. [Google Scholar] [CrossRef]
- Settle, A.E.; Berstis, L.; Rorrer, N.A.; Roman-Leshkóv, Y.; Beckham, G.T.; Richards, R.M.; Vardon, D.R. Heterogeneous Diels–Alder catalysis for biomass-derived aromatic compounds. Green Chem. 2017, 19, 3468–3492. [Google Scholar] [CrossRef]
- Brandvold, T.A. Carbohydrate Route to Para-Xylene and Terephthalic Acid. U.S. Patent 8314267B2, 20 November 2012. [Google Scholar]
- Román-Leshkov, Y.; Barrett, C.J.; Liu, Z.Y.; Dumesic, J.A. Production of dimethylfuran for liquid fuels from biomass-derived carbohydrates. Nature 2007, 447, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Osmundsen, C.M.; Taarning, E.; Dumesic, J.A. Selective production of aromatics from alkylfurans over solid acid catalysts. ChemCatChem 2013, 5, 2044–2050. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Kristianto, I.; Lee, H.; Jae, J. Production of renewable toluene from biomass-derived furans via Diels-Alder and dehydration reactions: A comparative study of Lewis acid catalysts. Fuel 2016, 182, 588–596. [Google Scholar] [CrossRef]
- Green, S.K.; Patet, R.E.; Nikbin, N.; Williams, C.L.; Chang, C.-C.; Yu, J.; Gorte, R.J.; Caratzoulas, S.; Fan, W.; Vlachos, D.G. Diels–Alder cycloaddition of 2-methylfuran and ethylene for renewable toluene. Appl. Catal. B Environ. 2016, 180, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.L.; Chang, C.-C.; Do, P.; Nikbin, N.; Caratzoulas, S.; Vlachos, D.G.; Lobo, R.F.; Fan, W.; Dauenhauer, P.J. Cycloaddition of biomass-derived furans for catalytic production of renewable p-Xylene. ACS Catal. 2012, 2, 935–939. [Google Scholar] [CrossRef]
- Nikbin, N.; Do, P.T.; Caratzoulas, S.; Lobo, R.F.; Dauenhauer, P.J.; Vlachos, D.G. A DFT study of the acid-catalyzed conversion of 2,5-dimethylfuran and ethylene to p-Xylene. J. Catal. 2013, 297, 35–43. [Google Scholar] [CrossRef]
- Feng, X.; Shen, C.; Tian, C.; Tan, T. Highly selective production of biobased p-Xylene from 2,5-dimethylfuran over SiO2–SO3H catalysts. Ind. Eng. Chem. Res. 2017, 56, 5852–5859. [Google Scholar] [CrossRef]
- Feng, X.; Cui, Z.; Ji, K.; Shen, C.; Tan, T. Ultra-selective p-Xylene production through cycloaddition and dehydration of 2,5-dimethylfuran and ethylene over tin phosphate. Appl. Catal. B Environ. 2019, 259, 118108. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Winoto, H.P.; Park, Y.-K.; Suh, D.J.; Lee, H.; Ha, J.-M.; Jae, J. Heteropolyacid catalysts for Diels-Alder cycloaddition of 2,5-dimethylfuran and ethylene to renewable p-Xylene. Catal. Today 2017, 293, 167–175. [Google Scholar] [CrossRef]
- Yin, J.; Shen, C.; Feng, X.; Ji, K.; Du, L. Highly selective production of p-Xylene from 2,5-dimethylfuran over hierarchical NbO x-based catalyst. ACS Sustain. Chem. Eng. 2018, 6, 1891–1899. [Google Scholar] [CrossRef]
- Zhao, R.; Zhao, Z.; Li, S.; Parvulescu, A.N.; Müller, U.; Zhang, W. Excellent Performances of Dealuminated H-Beta Zeolites from Organotemplate-Free Synthesis in Conversion of Biomass-derived 2,5-Dimethylfuran to Renewable p-Xylene. ChemSusChem 2018, 11, 3803–3811. [Google Scholar] [CrossRef] [PubMed]
- McGlone, J.; Priecel, P.; Da Vià, L.; Majdal, L.; Lopez-Sanchez, J.A. Desilicated ZSM-5 zeolites for the production of renewable p-Xylene via Diels–Alder cycloaddition of dimethylfuran and ethylene. Catalysts 2018, 8, 253. [Google Scholar] [CrossRef] [Green Version]
- Wijaya, Y.P.; Suh, D.J.; Jae, J. Production of renewable p-Xylene from 2,5-dimethylfuran via Diels–Alder cycloaddition and dehydrative aromatization reactions over silica−alumina aerogel catalysts. Catal. Commun. 2015, 70, 12–16. [Google Scholar] [CrossRef]
- Cho, H.J.; Ren, L.; Vattipalli, V.; Yeh, Y.-H.; Gould, N.; Xu, B.; Gorte, R.J.; Lobo, R.; Dauenhauer, P.J.; Tsapatsis, M. Renewable p-Xylene from 2,5-dimethylfuran and ethylene using phosphorus-containing zeolite catalysts. ChemCatChem 2017, 9, 398–402. [Google Scholar] [CrossRef]
- Gulbinski, J.; Ren, L.; Vattipalli, V.; Chen, H.; Delaney, J.; Bai, P.; Dauenhauer, P.; Tsapatsis, M.; Abdelrahman, O.A.; Fan, W. Role of silica support in phosphoric acid catalyzed production of p-Xylene from 2,5-dimethylfuran and ethylene. Ind. Eng. Chem. Res. 2020, 59, 22049–22056. [Google Scholar] [CrossRef]
- Zhao, P.; Boekfa, B.; Nishitoba, T.; Tsunoji, N.; Sano, T.; Yokoi, T.; Ogura, M.; Ehara, M. Theoretical study on 31P NMR chemical shifts of phosphorus-modified CHA zeolites. Microporous Mesoporous Mater. 2020, 294, 109908. [Google Scholar] [CrossRef]
- Jain, S.K.; Tabassum, T.; Li, L.; Ren, L.; Fan, W.; Tsapatsis, M.; Caratzoulas, S.; Han, S.; Scott, S.L. P-Site Structural Diversity and Evolution in a Zeosil Catalyst. J. Am. Chem. Soc. 2021, 143, 1968–1983. [Google Scholar] [CrossRef]
- Bortun, A.I.; Bortun, L.; Clearfield, A.; Villa-García, M.A.; García, J.R.; Rodríguez, J. Synthesis and characterization of a novel layered titanium phosphate. J. Mater. Res. 1996, 11, 2490–2498. [Google Scholar] [CrossRef]
- Djafer, L.; Ayral, A.; Boury, B.; Laine, R.M. Surface modification of titania powder P25 with phosphate and phosphonic acids–Effect on thermal stability and photocatalytic activity. J. Colloid Interface Sci. 2013, 393, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Radwan, N.R.; Hagar, M.; Afifi, T.H.; Al-wadaani, F.; Okasha, R.M. Catalytic Activity of Sulfated and Phosphated Catalysts towards the Synthesis of Substituted Coumarin. Catalysts 2018, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Ghoreishi, K.; Asim, N.; Yarmo, M.; Samsudin, M. Mesoporous phosphated and sulphated silica as solid acid catalysts for glycerol acetylation. Chem. Pap. 2014, 68, 1194–1204. [Google Scholar] [CrossRef]
- Haihong, X.; Daishi, G.; Qizhong, J.; Zifeng, M.; Wanjun, L.; Zheng, W. Catalytic performance of sulfated silica MCM-41 for the cyclization of pseudoionone to ionones. Chin. J. Catal. 2006, 27, 1080–1086. [Google Scholar] [CrossRef]
- Hosseini-Sarvari, M.; Sodagar, E.; Doroodmand, M.M. Nano sulfated titania as solid acid catalyst in direct synthesis of fatty acid amides. J. Org. Chem. 2011, 76, 2853–2859. [Google Scholar] [CrossRef]
- Bai, X.; Pan, L.; Zhao, P.; Fan, D.; Li, W. A new solid acid SO42−/TiO2 catalyst modified with tin to synthesize 1, 6-hexanediol diacrylate. Chin. J. Catal. 2016, 37, 1469–1476. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET (m2/g) | Vtotal (cm3/g) | Vmicro (cm3/g) | Total NH3 Desorbed (mmol/g) |
|---|---|---|---|---|
| SiO2 | 175 | 1.45 | 0.27 | - |
| SiP 300 | 128 | 0.70 | 0.13 | 0.676 |
| SiP 500 | 75 | 0.53 | 0.16 | 0.424 |
| SiS 500 | 44 | 0.46 | 0.19 | - |
| TiO2 | 36 | 0.58 | 0.38 | - |
| TiP 300 | 31 | 0.56 | 0.18 | 0.432 |
| TiP 500 | 27 | 0.45 | 0.11 | 0.934 |
| TiS 500 | 47 | 0.54 | 0.21 | - |
| Catalyst | Mass Fraction (%) | |||
|---|---|---|---|---|
| C | H | N | S | |
| SiS 300 | 0.17 | 0.23 | 0.03 | 0.02 |
| SiS 500 | 0.14 | 0 | 0.07 | 0 |
| TiS 300 | 0.18 | 0.19 | 0 | 0.53 |
| TiS 500 | 0.19 | 0.19 | 0 | 0.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Jae, J. Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene. Catalysts 2021, 11, 1074. https://doi.org/10.3390/catal11091074
Kim H, Jae J. Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene. Catalysts. 2021; 11(9):1074. https://doi.org/10.3390/catal11091074
Chicago/Turabian StyleKim, Hanbyeol, and Jungho Jae. 2021. "Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene" Catalysts 11, no. 9: 1074. https://doi.org/10.3390/catal11091074
APA StyleKim, H., & Jae, J. (2021). Diels–Alder Cycloaddition of Biomass-Derived 2,5-Dimethylfuran and Ethylene over Sulfated and Phosphated Metal Oxides for Renewable p-Xylene. Catalysts, 11(9), 1074. https://doi.org/10.3390/catal11091074