1. Introduction
The natural diastereomeric mixture of silybin A and B (approximately in the ratio 4:5) is isolated from the crude extract of the fruits (achenes, bot. cypselae) of
Silybum marianum L. (Asteraceae). This extract, which contains silybin and other silybin congeners, such as isosilybin, silychristin, and silydianin, as well as some other flavonoids and a polymeric fraction, is called silymarin. Silybin and its congeners belong to a subclass of flavonoids named flavonolignans [
1]. They are characterized by a phenylpropanoid moiety fused to the flavan skeleton, often taxifolin. The biosynthesis of flavonolignans in
S. marianum is not stereospecific; flavonolignans are typically diastereomeric mixtures (except silydianin). For a detailed review on silybin and other flavonolignans, see ref. [
2,
3,
4].
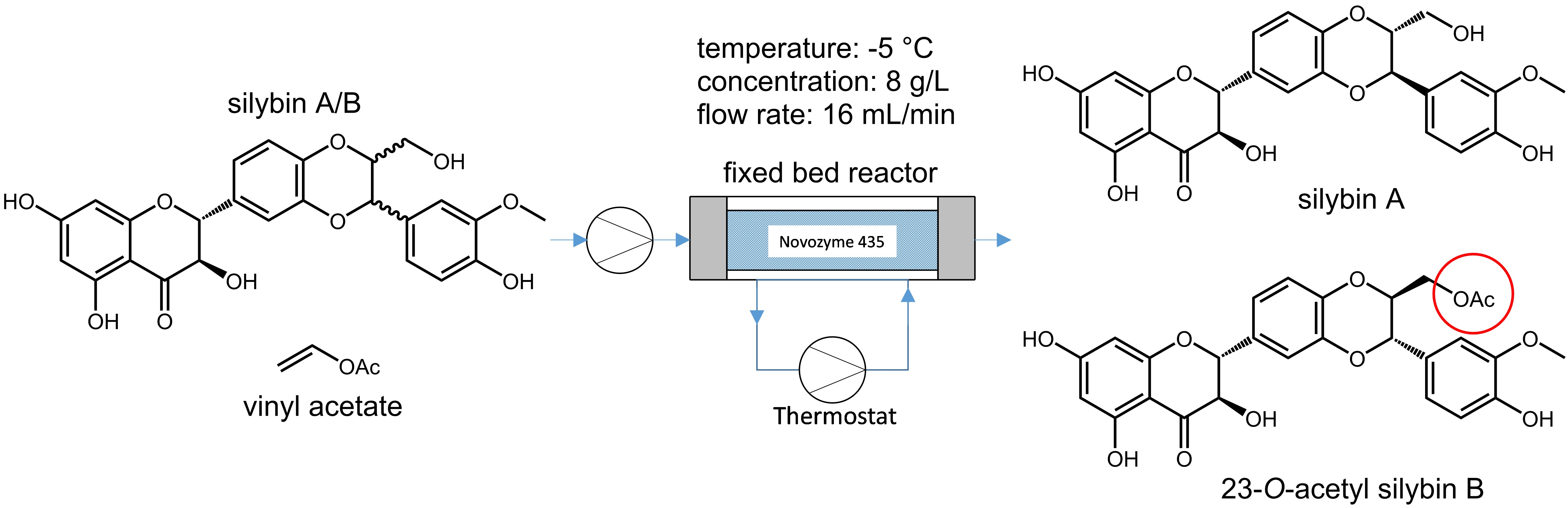
The diastereomeric pair of silybin A and silybin B (
Figure 1) behave like enantiomers in most cases, making their separation challenging. Because of this obstacle, even in modern studies to determine biological activities, a natural mixture of silybins (or even the crude extract—silymarin) is sometimes used [
5,
6], which is methodologically incorrect. Biological structures, such as typical 3D-systems, require stereochemically defined ligands and, therefore, results generated from mixtures have no—or very limited—validity. Indeed, there are significant differences between silybin diastereomers in terms of various biological activities [
3], as well as in bioavailability and metabolism [
3,
7]. Therefore, a single compound is required for biological studies instead of the natural mixture, especially in cases where the composition varies significantly [
8,
9,
10].
In fact, silybin was considered as a single compound from its first isolation in 1959 [
11] until 1979 [
12], when a separation into two peaks was observed using their HPLC method (C18, water/MeOH 60/40 with 5% of acetic acid). This observation was followed by the final assignment of the absolute configuration of the two silybin diastereomers in 2003 [
13]. The same paper [
13] describes what is believed to be the first semi-preparatory separation of milligram quantities of silybin A from silybin B by repeated preparative HPLC chromatography. More authors used this same laborious and cumbersome approach [
14,
15], which later allowed about 20 mg of each diastereomer to be obtained in 80 HPLC injections [
9] and probably reached its limits when 154 injections were used to obtain about 5 g of each diastereomer [
16].
In 2010, a chemo-enzymatic approach using lipases for the separation of silybin diastereomers was investigated [
17]. Lipases, as chiral catalysts, are capable of catalyzing the hydrolysis (solvolysis) of esters and also the formation of new esters by the reverse process. In nature, lipases are digestive enzymes that catalyze the hydrolysis of lipids into fatty acids and glycerol at the surface of lipid micelles and therefore can operate in hydrophobic environments, which distinguishes them from esterases. They belong to one of the few classes of enzymes that can tolerate high concentrations of organic solvents [
18]. The chiral property of the enzyme can recognize chirality and discriminate between enantiomers, or in this case, diastereomers. The different stereomers react at different rates, resulting in optically enriched mixtures of compounds that can then be separated as distinct compounds.
Monti et al. tested a panel of commercial lipases for the acylation or solvolysis of silybin A/B [
17]. Diastereomeric discrimination was observed with enantiomeric ratio (E) values ranging from 2 to 20. In subsequent work, the two best enzymes [
19] (
Candida rugosa lipase and
C. antarctica lipase B, CALB) were studied, the latter being chosen because it is cheaply available immobilized on acrylate (as Novozym
® 435) and because
C. rugosa lipase did not give reproducible results in bulk. CALB showed a preference for silybin B, especially during acetylation. The 23-
O-acetyl-silybin B formed can be easily separated from silybin A by simple flash column chromatography and then quantitatively hydrolyzed [
19]. This work [
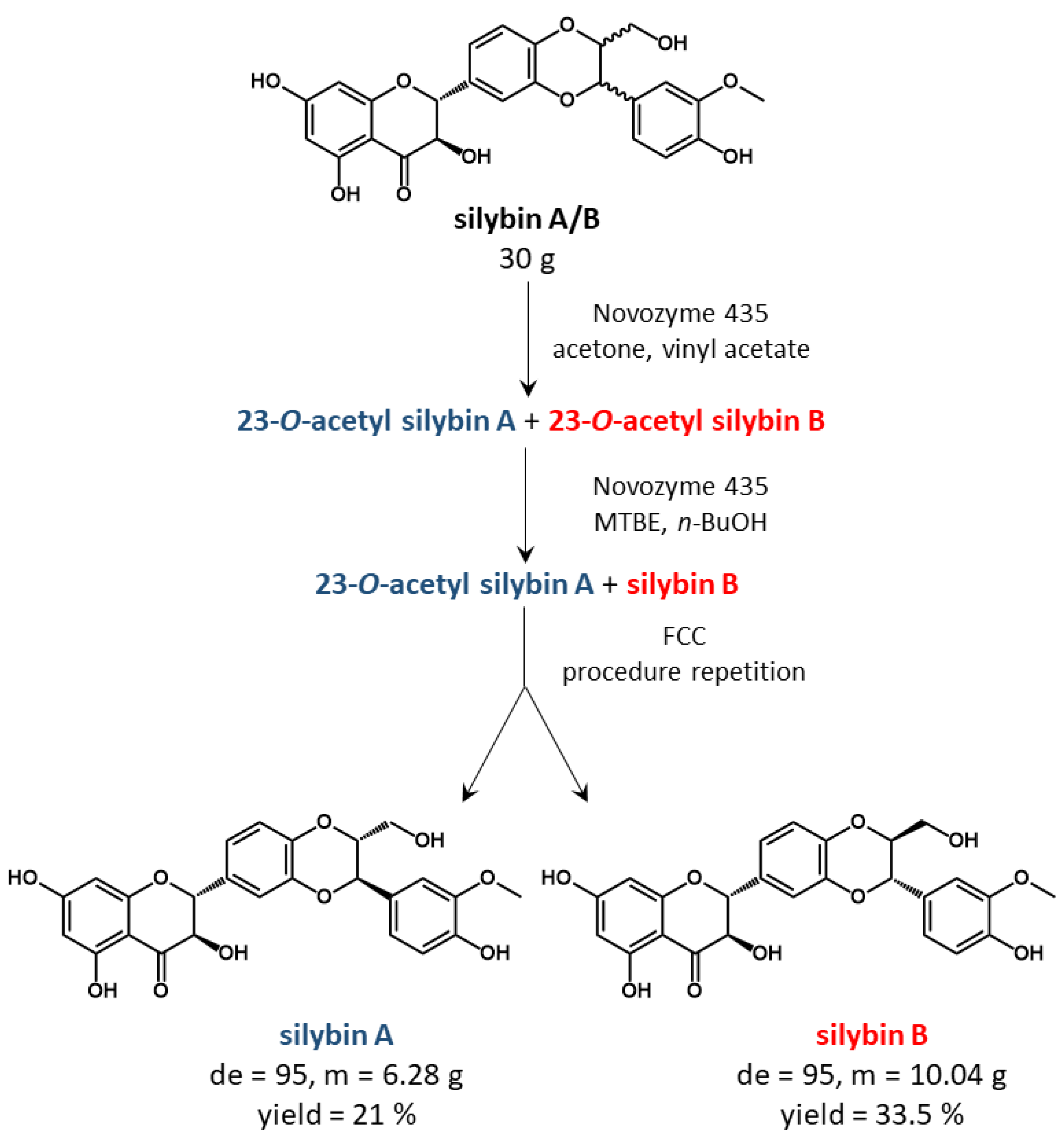
19] was pivotal as it allowed practical separation of gram quantities in good yield (about 50% total mass recovered, see
Scheme 1), although the separation of 30 g of silybin A/B took about 400 h (16 days). The simplified procedure shown in
Scheme 1 serves as a reference point; detailed information can be found in Ref. [
19].
This method was used only in batch mode. Due to the low diastereomeric ratio (D) value (around 6), repeated separation is required to increase the diastereomeric excess above 95. The low D value is probably due to the relative distance of the reaction center (C-23 OH) to the asymmetric centers (C-10 and C-11), which are separated by two to three bonds (
Figure 1).
Batch preparation methods capable of providing multigram quantities of pure silybin diastereomers for in vivo experiments have rather limited capacity and are excessively labor-intensive. In contrast, flow chemistry can be used to produce large quantities of optically pure material. The main advantage of flow chemistry is the easy catalyst reusability, rapid analysis, optimization and scale-up of chemical reactions. It also allows for enhanced control of reactions and provides access to a wider range of reaction variables resulting in better reproducibility of reactions due to better control. For a recent review on flow chemistry, see [
20].
In this work, we report the use of a flow-chemical setup for the diastereomeric separation of silybins A and B. We optimized the conversion and diastereomeric ratio (D) by varying temperature (T, °C), flow rate (FR, mL/min), and starting material concentration (g/L). The setup also allowed for extensive enzyme recycling.
2. Results
2.1. Reaction Optimization
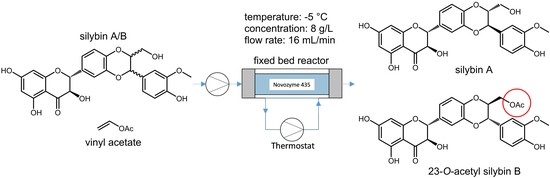
Only acetylation was used as the diastereomeric discrimination. Solvolysis as the discrimination step proved to be too slow at lower temperatures. The reaction was carried out in a fixed bed reactor. Novozym 435 was suspended in acetone for 12 h and then loaded into the reactor tube (approximately 170 cm3, 35 g dry weight). The length and diameter of the reactor tube were 1000 mm and 15 mm, respectively. The reactor tube was equipped with a jacket in which a thermostatic fluid was circulated to allow temperature control. The substrate was passed through the reactor bed dissolved in acetone with the reactant (vinyl acetate) as a co-solvent.
A partial factorial design with response surface methodology was used to maximize the diastereomeric ratio D. Since productivity (the rate at which a product can be obtained, a frequently optimized variable) was high in most cases, D and conversion were chosen as response variables. Optimization experiments were conducted with three variables and 3 to 5 sublevels, resulting in 23 experiments. The variables chosen were temperature (T, °C; from −10 to +20 °C), flow rate (FR, mL/min), and starting material concentration (g/L). The levels were chosen based on preliminary experiments.
Enzymatic discrimination of enantiomers usually depends on the extent of conversion. Conversion represents the molar ratio of starting materials to starting materials and products. Diastereomeric excess (de) is not a suitable measure of enzymatic selectivity as it also depends on conversion. Therefore, the enantiomeric ratio (E) was introduced, which is independent of the conversion [
21]. For diastereomeric pairs of compounds, the analogous diastereomeric ratio (D) was used. Equation (1) was used to calculate the diastereomeric ratio (D), where c = conversion, d.e.p. = diastereomeric excess of the products.
The authors are aware that Equation (1) was recommended (by Kurt Faber) for conversion (c) values between 30 and 70% for best accuracy [
22]. However, for consistency, Equation (1) was used regardless of the conversion value, with conversion ranging from 28% to 97% (
Table 1). The high diastereoselectivity of the reaction was promoted by high starting material concentration and high flow rate. The highest diastereoselectivity (D = 11.3) was observed at −5 °C with a concentration of 8 g/L and a flow rate of 16 mL/min.
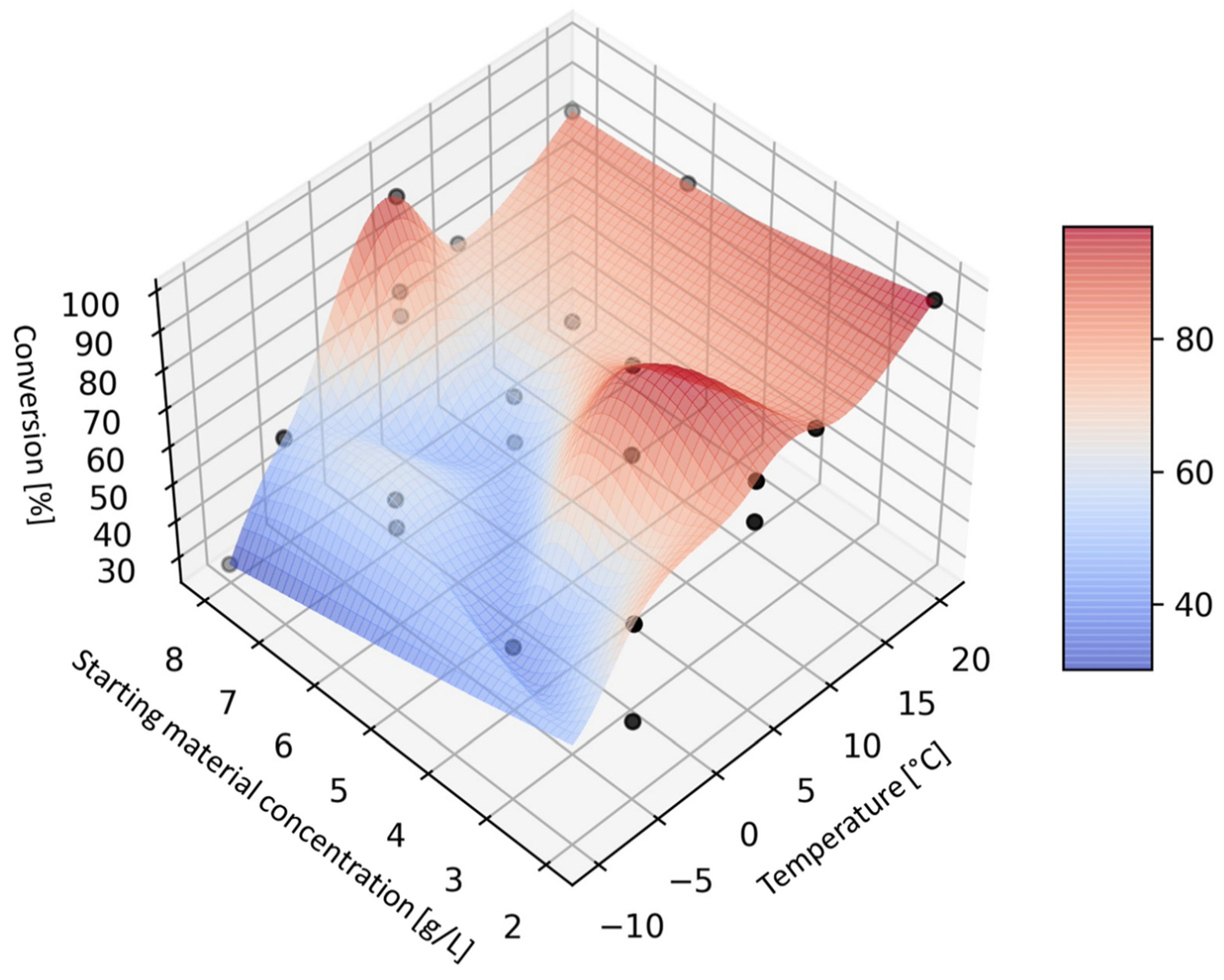
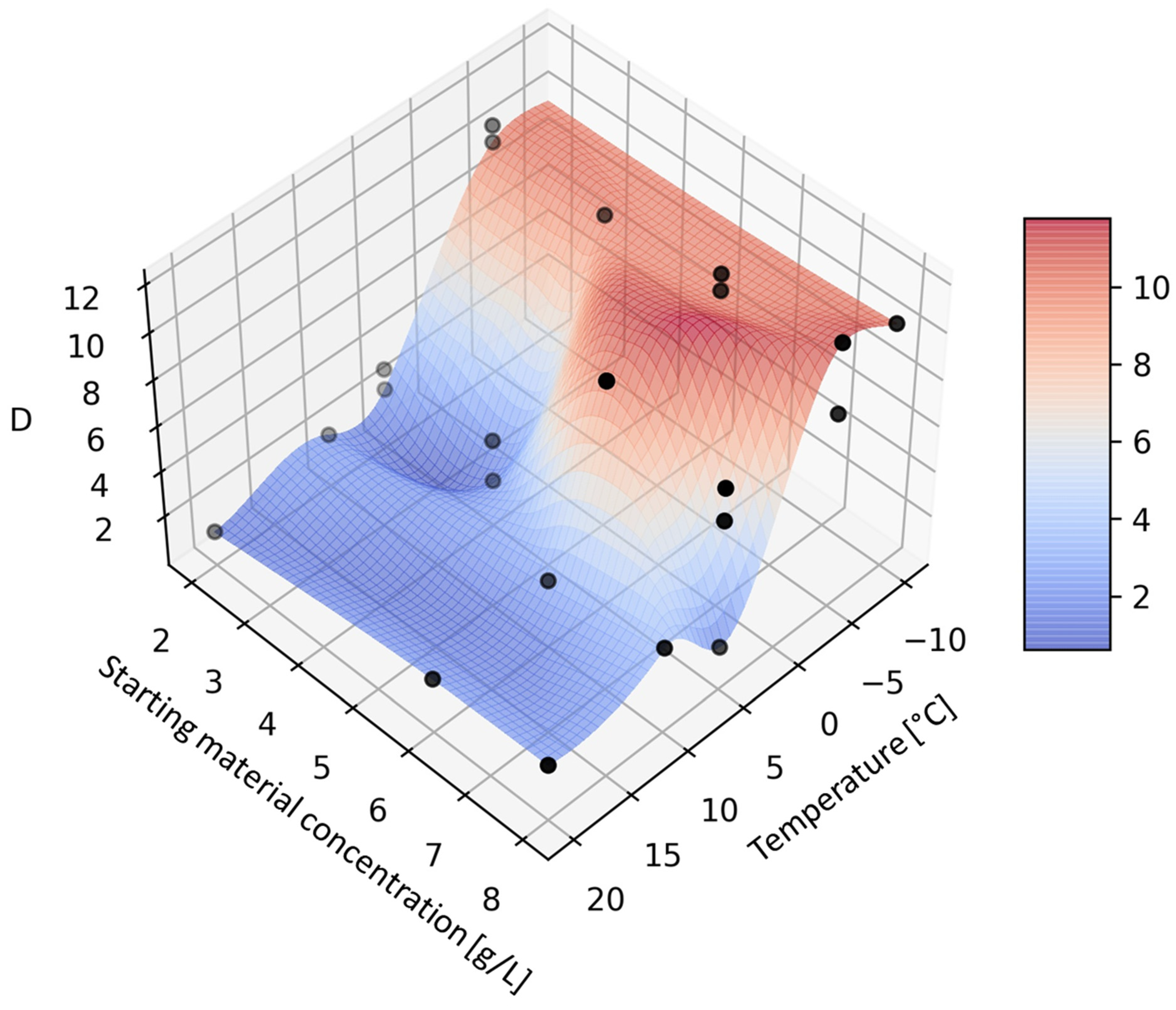
The resulting response surface graph (
Figure 2) shows very little dependence of conversion on concentration. The relative insensitivity of the conversion to the starting material concentration is probably due to the fact that enzyme saturation was not reached at the concentrations used (2 to 8 g/L). However, further increase of the starting material concentration was not possible due to the high viscosity of the reaction mixture, which resulted in increased backpressure in the reactor. The effect of temperature on the conversion was as expected—for every 10 °C an increase in conversion of about 20% was observed.
The dependence of diastereoselectivity on the concentration of the starting silybin was also low, again probably due to the fact that enzyme saturation was not reached in the selected concentration range (
Figure 3). The dependence of diastereoselectivity on temperature was initially flat, with a fairly sharp change around +5 °C. At temperatures above +5 °C, diastereoselectivity D was low but increased rapidly when the temperature dropped below +5 °C, showing a plateau between −5 °C and −10 °C. The best diastereoselectivities were still relatively moderate (around 10), but they were the highest achieved so far with CALB.
2.2. Carrier Stability
The immobilized enzyme showed little to no degradation over several months. For stability testing, 128 g of silybin (natural mixture) was passed through the column for 30 days in 30 experiments (cycles). Between experiments, there were only small technical breaks. Using the same conditions (2 g silybin, 500 mL acetone, 500 mL vinyl acetate, −5 °C, 4 mL/min) at the beginning and end of the interval, no detectable deterioration in conversion (77.6% and 74.4%, respectively) or diastereomeric ratio D (3.28 and 3.26, respectively) was observed.
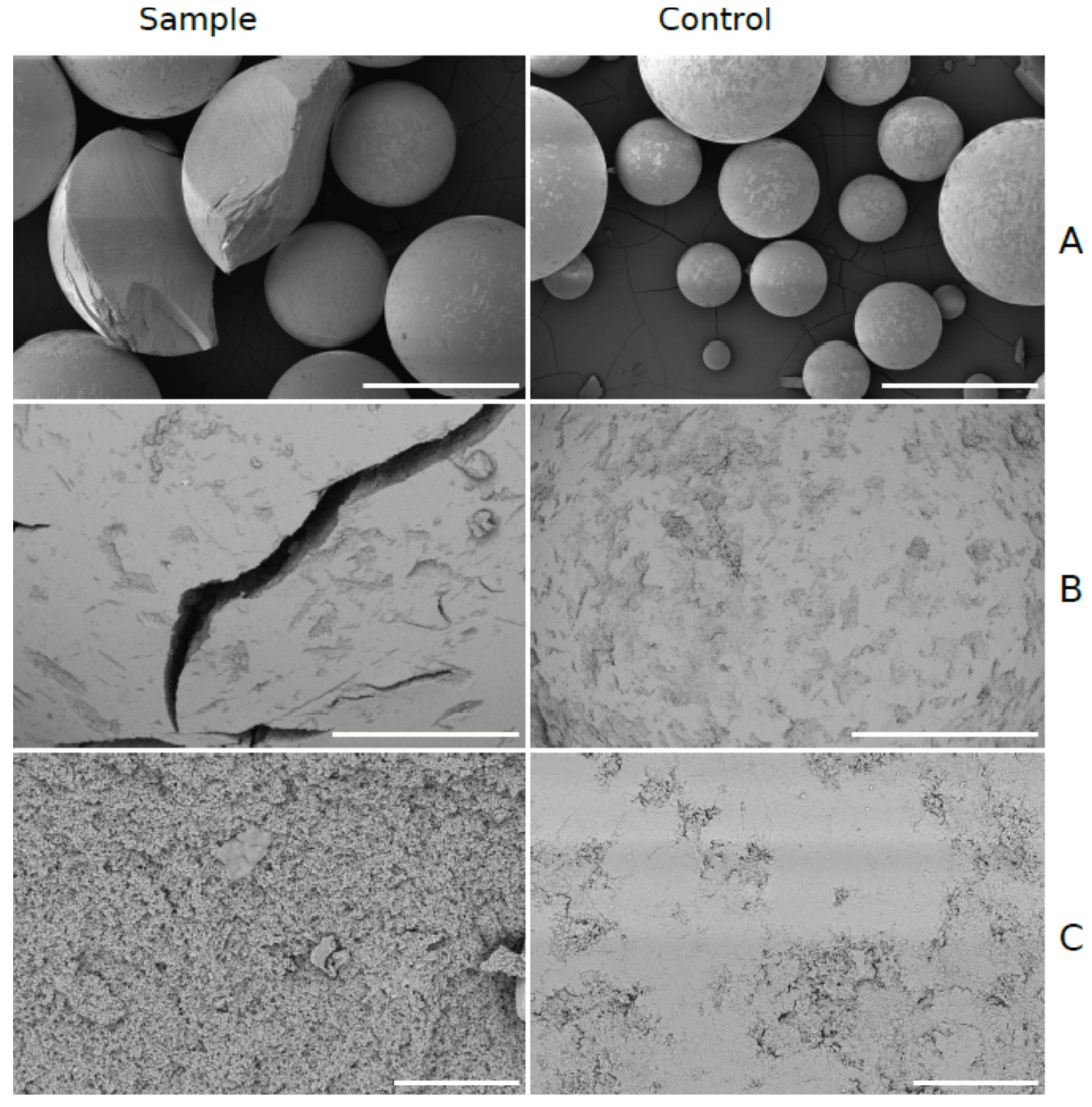
Optical microscopy also revealed no differences in the textural morphology and structural integrity of the immobilized enzyme (enzyme carrier, polyurethane beads, data not shown). On the other hand, some mechanical degradation of the enzyme carrier was observed by scanning electron microscopy, probably due to repeated manipulation (
Figure 4). These morphological changes probably caused a slow but observable increase of the backpressure in the flow reactor. At the lowest magnification (250×), a fraction of the carrier beads were cracked or sheared off (
Figure 4A). This was probably caused by the mechanical stress during the manipulation with the carrier. At higher magnification (1500×), some of the carrier beads had visible cracks, possibly caused by repeated solvent changes in the flow reactor (
Figure 4B). Detailed microscopic images of the surface (magnification 10,000×) (
Figure 4C) showed the changes in surface quality and the opening of macropores. These changes probably did not affect the reaction but can be used to estimate the age of the enzyme-based on a very small amount of the carrier (single bead). No pH memory of the enzyme used under this setup was determined.
2.3. Optimal Reaction Conditions
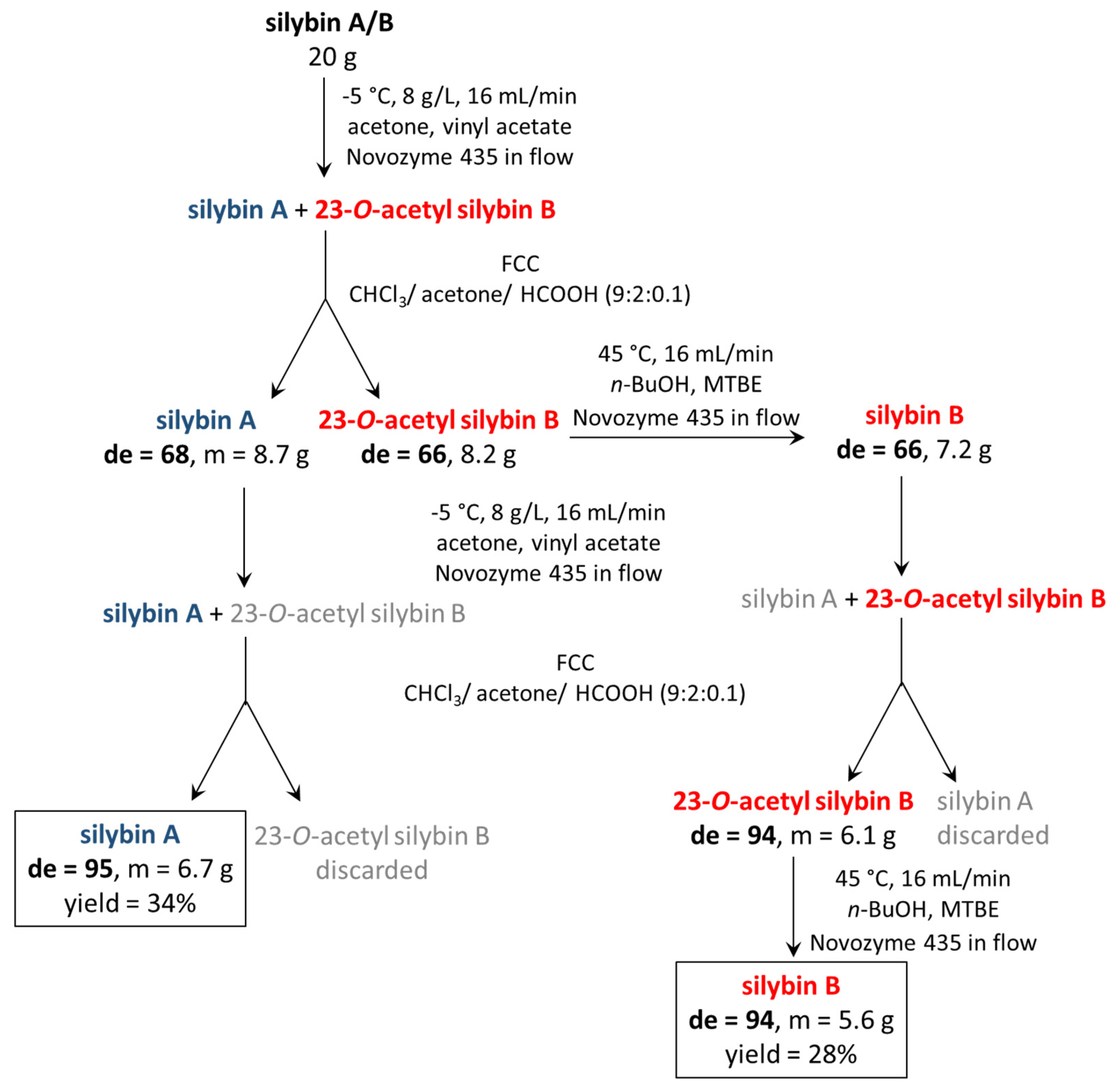
Based on the reaction optimization results, −5 °C, starting material concentration 8 g/L, and a flow rate of 16 mL/min were set as the optimal reaction conditions (
Scheme 2). With these conditions and a 1000 × 15 mm column, 20 g of the natural silybin diastereomeric mixture can be processed per day. After subsequent chromatography and hydrolysis of silybin B acetate (which is quantitative), we routinely obtain 6.7 g of silybin A (de = 95, yield = 34%) and 5.6 g silybin B (de = 94, yield 28%). The yields are calculated from total silybin, the maximal possible yield is therefore near 50%. The same conditions were used for the negative control without the enzyme and the carrier, and no conversion was observed. Further scale-up depends mainly on the column used and should be feasible at least at the pilot plant scale.
3. Discussion
The separation of silybin diastereomers is a difficult problem that has not yet been fully solved. Here we present a contribution to the enzymatic resolution of these diastereomers that makes chromatography, rather than the enzymatic resolution itself, the rate-limiting step of the procedure.
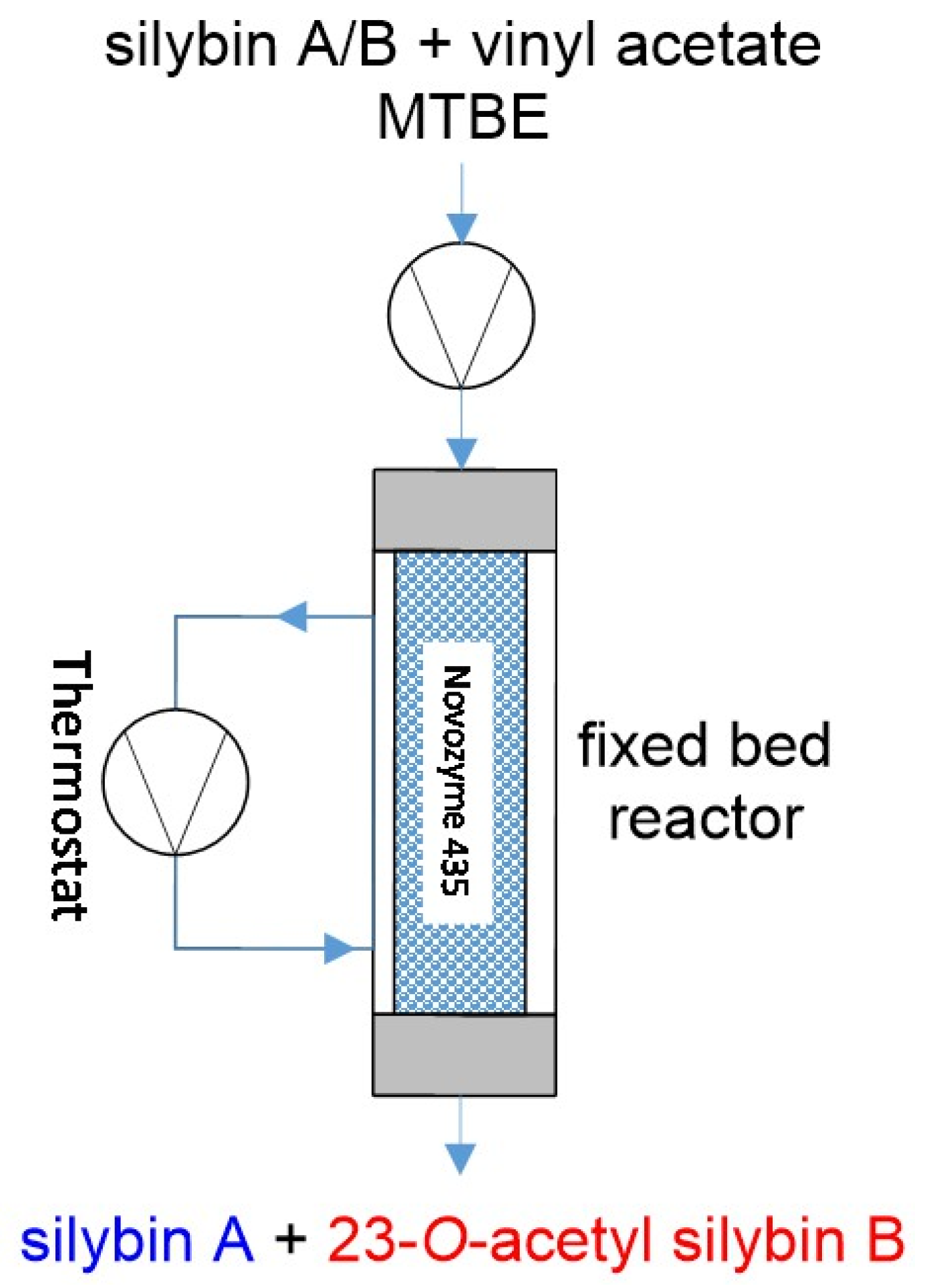
The continuous flow chemistry approach to the chemo-enzymatic resolution of silybin diastereomers represents (to our knowledge) the first report of a reaction in flow in the separation of silybin diastereomers (
Figure 5). Compared to previously used methods, the advantages of the flow-chemistry approach are clear. Key reaction parameters such as residence time (reaction time), temperature, and mixing are precisely controlled, resulting in better control of the reaction. The catalyst is separated in the fixed bed flow reactor and negligible degradation of the catalyst was observed over the course of 30 reactions. This simplifies the work-up to simple evaporation of the reaction mixture, and the number of flammable solvents is minimal.
The disadvantages are few. Compared to the batch setup [
19], the reaction requires different instrumentation. Instead of a thermostated shaker, the pump, a thermostat with external connections and a jacketed column (the fixed bed reactor) are required. This imposes additional input costs on the operator. The skillset is different and requires interdisciplinarity between chemistry and process engineering. In this setup, heating or cooling is not a problem due to the high thermal mass of the reactor, but the pressure should be closely monitored.
In comparison, separation using preparative HPLC is feasible, but the instrumentation is expensive and advanced. This method has been used in the past to obtain small amounts of pure diastereomers. Obtaining more than 100 mg (depending on column size) is possible with this method, but it is extremely impractical [
13,
14,
15,
16]. The batch chemo-enzymatic method is currently the method of choice for diastereomeric separation of silybin diastereomers. Developed by Monti et al. [
17], and Gažák et al. [
19], the method has been used to prepare several grams of each diastereomer. The method can be scaled up, but the authors use long reaction times (48 h). Alcoholysis, which requires higher temperatures, is used as the primary discrimination step. Using this method, a diastereoselectivity (D) of about 6 was achieved.
Using the flow method, a diastereoselectivity value above 10 can be achieved when the temperature is lowered below 0 °C, the optimal conditions allow for a reaction of 8 g of silybin per hour. Acetylation was used as the discrimination step, but since the separation must be repeated for complete diastereomeric separation, a practical rate of about 20 g per day can be achieved, limited by the chromatography step. We assume that the reaction productivity is linearly scalable by using larger fixed bed reactors and higher flow rates. In this work, we have only attempted to increase the diastereoselectivity as much as possible. The usual threshold for a “useful” resolution is a diastereoselectivity higher than 15 [
22], so the method still needs further optimization. To enable one-step resolution, a diastereoselectivity above 40 must be achieved, which would require a better lipase. Further research in this direction is therefore necessary.
The mechanical changes of the enzyme carrier observed on Scanning Electron Microscopy can be divided into two groups. At low magnification (250–1500×) some mechanical degradation of the enzyme carrier is observed (breakage and surface damage in the enzyme carrier), caused by general manipulation with the enzyme. This does not affect the reaction outcome but may slowly increase the backpressure of the fixed bed reactor. Published data [
23] suggest that although the carrier is stable enough for laboratory use, further stabilization (with silicone) is required for industrial use.
It is therefore advisable to keep manipulation with the enzyme to a minimum if recycling of the enzyme is desired. At higher magnification (10,000×), the surface changes become clear. The surface of the used enzyme carrier is less smooth and more textured compared to the unused enzyme carrier. Again, this has no observed effect on the reaction outcome, but allows determination of whether a particular enzyme carrier is new or has already been used.
4. Materials and Methods
4.1. Materials
Silybin A and silybin B were isolated as a natural mixture by the methanol extraction from silymarin Liaoning Senrong Pharmaceutical Co. (Panjin, People’s Republic of China, batch no.: 120501) [
24]. Briefly, crude silymarin was suspended in methanol and filtered over the celite layer. After washing with methanol, the filtrate was dried up. The filtrate contained silybin A and B as a roughly equimolar mixture with 95% purity (HPLC). Immobilized CALB lipase (Novozym 435 batch LC200256) was from Novozymes (Bagsvaerd, DK).
4.2. HPLC Analysis
The HPLC system consisted of a Shimadzu LC-20AB solvent delivery system, monolithic column RP-18 Chromolith Performance 100 mm × 3 mm, equipped with pre-column, the column was placed in the column thermostat Shimadzu CTO-20A, diode array detector Shimadzu SPD-M20A was used and signal at 285 nm was extracted. The chromatographic method used was isocratic, the mobile phase was formic acid, acetonitrile, methanol, water (0.1/2/37/61; v/v/v/v); the flow rate was 1.5 mL/min.
4.3. The Apparatus
The immobilized enzyme (Novozyme 435) was loaded into a jacketed column (i.d. 15 mm, length 1000 mm, enzyme load 35 g (dry enzyme). The column was equipped with a thermostat and solvents were delivered by LC-20 solvent pump (
Figure 5).
4.4. General Procedure for Silybin Acetylation
Natural silybin (2, 4, 6, or 8 g, 95% purity (HPLC), A/B 48:52) was completely dissolved in acetone (500 mL) and then an equal amount of vinyl acetate (500 mL) was added. The solvent mixture with silybin was then passed through the column reactor charged with the immobilized enzyme, the temperature set to −10, −5, 5, 10, or 20 °C, and the flow rate set at 4, 8, or 16 mL/min. After the reaction mixture was passed through, the column was washed with acetone (300 mL) at the same temperature and flow rate. The wash solvent and reaction mixture were evaporated to dryness in vacuo. The negative control was performed with a fixed bed reactor filled with 35 g of inert material. Two grams of silybin were passed through the reactor in 500 mL acetone, 500 mL vinyl acetate at −5 °C, and the flow rate was 4 mL/min. Conversion was 0% (HPLC).
4.5. Carrier Stability
The stability of the carrier was tested by comparing conversion and diastereomeric ratio before and after acetylation of 128 g of silybin in 30 days (30 experiments). Possible deterioration of the texture morphology and structural integrity of the carrier was investigated by scanning electron microscopy.
4.6. Scanning Electron Microscopy
Air-dried Novozyme samples mounted onto the standard SEM aluminum stubs with ultrasmooth carbon discs (SPI Supplies, Structure Probe, Inc., West Chester, PA, USA) were sputter-coated with 3 nm of platinum in a high-resolution Turbo-Pumped Sputter Coater Q150T (Quorum Technologies Ltd., Ringmer, UK). The samples were examined in an FEI Nova NanoSEM scanning electron microscope (FEI, Brno, Czech Republic) at 3 kV using SED, TLD, and CBS detectors. A beam deceleration mode [
25] with a stage bias of 335 V was used to minimize serious sample charging at higher magnifications.
4.7. Optimized Production of Silybin A and B Diastereomers
The starting material (silybin A/B, 20 g) was dissolved in acetone (1250 mL) and after being completely dissolved, vinyl acetate (1250 mL) was added and passed through the fixed bed reactor (temperature: −5 °C, flow rate: 16 mL/min, final concentration 8 g/L). After passing the solution, the fixed bed reactor was washed with another 300 mL of acetone (156 + 19 min). All solvents were evaporated in vacuo and the partially acetylated solid obtained was purified by flash column chromatography (250 g silica gel, chloroform/ acetone/ formic acid, 9:2:0.1) to afford silybin A (de = 68, m = 8.7 g) and 23-O-acetylsilybin B (de = 66, m = 8.2 g). Silybin A in high purity was prepared by repeating the procedure to obtain pure silybin A (de = 95, m = 6.7 g, total yield = 34%).
To obtain silybin B in high diastereomeric purity, 23-
O-acetylsilybin B (de = 66, m = 8.2 g) was first completely hydrolyzed. It was dissolved in methyl
tert-butyl ether (MTBE, 500 mL). Once the material was dissolved,
n-BuOH (500 mL) was added and the solution was passed through the fixed bed reactor (temperature: 45 °C, flow rate: 16 mL/min, final concentration 8 g/L). After passing the solution, the fixed bed reactor was washed with another 300 mL of acetone (61 + 19 min). The product (silybin B, de = 66, m = 7.2 g) was acetylated again. It was dissolved in acetone and vinyl acetate mixture (450 mL and 450 mL) and the solution was passed through the fixed bed reactor (temperature: −5 °C, flow rate: 16 mL/min, final concentration 8 g/L). After passing the solution, the fixed bed reactor was washed with another 300 mL of acetone (56 + 19 min). All solvents were evaporated under the reduced pressure and the partially acetylated solid obtained was chromatographed on silica gel (250 g) in the mobile phase chloroform:acetone:formic acid 9/2/0.1 to yield 23-
O-acetylsilybin B (de = 94, m = 6.1 g). This product was hydrolyzed completely when dissolved in MTBE (400 mL) and
n-BuOH (400 mL) and the solution was passed through the fixed bed reactor (temperature: 45 °C, flow rate: 16 mL/min, final concentration 8 g/L). After the solution passed, the fixed bed reactor was washed with another 300 mL of acetone (50 + 19 min). After evaporation, pure silybin B (de = 94, m = 5.6 g, total yield = 28%) was obtained (see also
Scheme 2).
Note: solvent change in the fixed bed reactor between acetone, MTBE or vice versa proceeds without pressure change.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}