2.1. Structural and Surface Properties of Catalysts after Calcination
The properties of alumina-supported nickel catalysts promoted with cerium and tungsten are presented in
Table 1. The content of promoters varied from approximately 1 to 5 wt %. The effects of Ce and W introduction is compared for catalysts contained around 40 wt % Ni. Additionally, the influence of Ce is presented for catalysts contained smaller amounts of Ni (around 20 wt %).
The γ-Al
2O
3 used as support showed high specific surface area (251.7 m
2/g) (
Table 1). An introduction of 20 wt % Ni led to the decrease in specific surface area to 236.7 m
2/g. The course of nitrogen adsorption/desorption isotherms of the support and catalysts presented in
Figure 1 was similar, and can be considered as IV-type, in agreement with the IUPAC recommendations [
23]. The observed hysteresis loops, classified as H2(b), indicate the presence of mesopores. The decrease of mean pore diameter (D
BJH) and total pore volume (V
p) can be assigned to partial blockage of small pores of alumina support by nickel oxide or nickel aluminate species.
An introduction of larger amount of nickel (36.9 wt % Ni) increased the specific surface area, as well as further reducing the mean pore diameter and total pore volume (
Table 1). Such effects can be attributed to the formation of additional surface sites on the nickel oxide species, changes in the specific arrangement of small alumina particles around NiO nanoparticles and, at the same time, a decrease in the number of surface sites of the support due to the blockage of small pores by NiO nanoparticles. The introduction of promoters resulted in insignificant changes in the porosity of the catalysts or their specific surface area (
Figure 1,
Table 1). Small changes in the properties of the catalysts can be ascribed to variation in the size of nickel crystallites and/or formation of oxide phases of Ce and W.
The structural and surface properties of γ-Al
2O
3 support, as well as the applied preparation method, especially the addition of citric acid to impregnating solution, facilitated formation of relatively small NiO crystallites. X-ray diffraction curves of alumina support and nickel catalysts after calcination are presented in
Figure S1 in the Supplementary Materials. The presence of wide reflection peaks of γ-Al
2O
3 (PDF4 + 00-001-1303) point out small particle size of alumina support. The approximate size of Al
2O
3 domains was in the range of 4 nm. The reflection peaks of alumina partially overlapped NiO peaks (PDF4 + 04-016-6318), and hence it is difficult to determine the size of NiO crystallites with high accuracy. Their size was approximately in the range of 3–4 nm.
It is also difficult to unambiguously reject the possibility of the coexistence of dispersed nickel aluminate phases (NiAl
2O
4) due to similar position of reflection lines of such phases and width of X-ray diffraction peaks. Isolated XRD peaks of cerium and tungsten containing phases, such as CeO
2, WO
3, nickel aluminates, cerium aluminates, and aluminum or nickel tungstates, were not visible, indicating the presence of very small nanoparticles and their non-crystalline or amorphous nature. However, in comparing the narrow region of XRD curves for promoted Ni40-Al
2O
3 catalysts (
Figure 2), we were able to distinguish weaker shift of overlapping NiO and Al
2O
3 peaks towards lower angles for W-promoted catalysts (dashed lines) in comparison to unpromoted and Ce-promoted nickel catalysts (solid and dotted lines, respectively). This effect can be attributed to the presence of smaller NiO particles in W-promoted catalysts or participation of Ni in the dispersed nickel tungstate phases.
The crystalline structure of γ-Al
2O
3 is often presented as a cubic defect spinel. The oxygen atoms are in cubic close packing arrangement, and aluminum atoms occupy part of octahedral and tetrahedral sites [
24]. Nickel or cerium ions during initial stages of impregnation and thermal treatment can be in part located in the vacant sites of alumina spinel. Similar suggestions were recently proposed for more complex systems, including Ni/xCeO
2-Al
2O
3 [
25]. Moreover, such effects may result in the corresponding changes of the oxidation state of Ce. Damyanova et al. indicated the possibility of decrease in the Ce
3+/(Ce
3+ + Ce
4+) ratio as result of CeO
2 agglomeration with an increase of CeO
2 content [
25]. Treatment of tungsten precursor during calcination may lead to the formation of different oxide forms WO
x, 2 ≤ x ≤ 3. Bulk tungsten (VI) oxide WO
3 contains WO
6-octahedra connected by corners, while edge-shared octahedral are present in WO
2. Other intermediate oxide phases may show more complex structure with linear defects. Ostromecki et al. suggested that tungsten oxide surface species are deposited on the surface alumina at low loadings as tetrahedrally coordinated monomers and can form a mixture of tetrahedrally and octahedrally coordinated surface polymers at high loadings [
26]. They also indicated that the tungsten oxide species during impregnation may anchor to the alumina support through interactions with surface hydroxyls, whereas nickel oxides preferentially interact with the coordinatively unsaturated Al
3+ Lewis acid surface sites. This mutual spatial dispersion of ionic species in the initial stages of synthesis of catalysts may retard the growth rate of clusters of nickel precursors [
16]. Although the formation of nickel tungstates was reported in literature as a result of high temperature treatment (above 400 °C, i.e., the calcination temperature of catalysts used in this work), the presence of such surface or subsurface phases cannot be excluded, especially considering the high degree of dispersion and the presence of very small NiO crystallites formed under co-impregnation conditions [
27].
The obtained catalysts may likewise attain high nickel dispersion due to the large specific surface area of γ-Al
2O
3 and the presence of some steric hindrances, resulting from the micro-mesoporous structure and presence of promoter oxide species, which hinder agglomeration of nickel surface species during thermal treatment. Relatively high nickel dispersion, ascribed to citric acid presence in the impregnating solution, was recently presented in several articles [
20,
28,
29].
Citric acid has three carboxylic groups that can participate in the formation of bonds with nickel and cerium or tungsten. The equilibrium between citric acid and its deprotonated forms is related to the pH of the solution and nature of the ions [
13,
15,
16]. In the solutions of low pH, undissociated citric acid and nickel aqua complexes [Ni(H
2O)
6]
2+ may prevail. An increase in pH may facilitate the successive formation of additional nickel citrate complexes, such as [Ni(H
2Cit)(H
2O)
5]
+, [Ni(HCit)(H
2O)
4], [Ni(HCit)(Cit)(H
2O)
4]
3−, and [Ni(Cit)
2(H
2O)
4]
4−, which can be adsorbed on alumina support [
30]. The presence of citric acid may also influence viscosity during drying and calcination stages, hindering the migration of precursors, enhancing formation of “gel-like” rather than “crystalline-like” phases [
31].
Moreover, the impregnation at low pH or in the presence of some complexing agents may result in partial dissolution of the support (via etching) and redistribution of metal ions. Hence, during calcination of binary (Ni-Al) and ternary (Ni-Ce-Al or Ni-W-Al) systems, the formation of complex oxides, phase segregation, and mutual diffusion of nickel, cerium, tungsten, and aluminum ions can occur. The individual ions my diffuse through the interfacial region of oxide phases. The diffusion rate in the mixed oxides systems depends on temperature, the nature of oxide lattice, content of metals, the size of the ions, and the extent of the interfacial border. Therefore, the increase in NiO dispersion simultaneously leads to increase in the interface border and thus to the enhanced formation of nickel aluminate phases, as well as nickel tungstate phases (NiWO4) in the W-doped catalysts.
Nickel aluminate and tungstate species show low reducibility; hence, to restrict formation of such phases, the calcination of catalysts was performed at relatively low temperature (400 °C) [
20,
21,
32]. It seems, that the degree of the oxidation state of Ni
2+ in NiO crystallites essentially does not change during thermal treatment; however, it is worth noting that Ni
3+ sites can be formed under certain conditions [
33]. In turn, Ce
3+ ions introduced during impregnation of the alumina support can change the oxidation state to Ce
4+ in the drying and calcination stages. Formation of solid solution between NiO and CeO
x is relatively difficult, but potentially possible. The ionic radius of Ce ions is larger than Ni;
VIIIr(Ce
3+) = 1.14 Å,
VIIIr(Ce
4+) = 0.97 Å, and
VIr(Ni
2+) = 0.69 Å, respectively [
34]. An incorporation of Ni
2+ ions in the CeO
2 lattice would cause large distortion of the crystal lattice. The solubility of nickel in the particles with a micrometric size of CeO
2 is therefore relatively weak and falls below 1 atom % [
35]. However, several recent studies have reported the possibility of increasing the solubility limits of Ni
2+ in CeO
2 nanoparticles [
36,
37,
38]. In turn, formation other possible binary compounds, such as cerium aluminates (CeAlO
3), cannot be completely ruled out [
39,
40].
2.2. Active Phase Formation
The temperature-programed reduction curves of alumina-supported nickel catalysts are presented in the
Figure 3. The catalysts were reduced in a wide range of temperatures. One can observe small TPR peaks on the curve of Ni40-Al
2O
3 catalyst, located in the range from 250 to 350 °C. The predominant reduction peaks occurred between 350 and 800 °C, with large maximum at around 520 °C. The reduction of cerium-promoted catalysts depended on the content of Ni and Ce. The effects of the presence of cerium were more visible in the catalysts with low nickel loading and may have directly resulted from differences in the Ce/Ni ratio in the catalysts. The introduction of the small amounts of Ce to the catalysts with 20 wt % Ni led to the increase in reducibility. This effect was especially manifested in the increase in the intensity of low-temperature peaks and small shift of high-temperature reduction peaks to the lower temperatures. Similar effects were visible for the catalysts with high nickel loading, but the extent was much weaker. Low-temperature peaks were not visible in the W-promoted catalysts. The increase in the main peak intensity with an increase in W content was observed. In the literature, low-temperature reduction peaks on the TPR curves of nickel catalysts have been often attributed to the consumption of hydrogen in the reduction of “free NiO” crystallites, weakly interacting with alumina support, accordingly to the reaction NiO + H
2 → Ni + H
2O [
41,
42]. On the other hand, high-temperature peaks were attributed to the reduction of nickel oxide phases strongly interacting with alumina. High reduction temperature may also lead to the formation of oxygen vacancies and partial reduction of cerium (Ce
4+ → Ce
3+) in CeO
2 species [
43].
Low reducibility of alumina-supported catalysts has often been reported as the effect of the presence of surface and bulk nickel aluminate spinels [
44]. It is well established that the extent of interactions between NiO and Al
2O
3 may increase with an increase in NiO dispersion and increase in calcination temperature. The application of co-precipitation, sol–gel, or high-surface area support impregnation methods can lead to the formation of highly dispersed oxide systems. The presence of difficult-to-reduce phases allows formation of relatively small nickel crystallites, mainly due to retardation of diffusion processes during reduction stage.
It was discussed above that high calcination temperature may lead to the formation of spinel oxide species or solid solutions, which complete reduction can be achieved at very high temperatures, often above 800 °C [
44]. Cerium and nickel precursors used in the synthesis of Ni-Ce/Al
2O
3 catalysts by the co-impregnation method are simultaneously deposited on the alumina support in the presence of citric acid, and hence the formation of mixed Ni and Ce oxide phases can be expected. Thus, gradual increase in reducibility of catalysts with an increase in Ce content (in the low-temperature region) may result from the increase of the contribution of the direct interactions between NiO and CeO
2 phases [
25], while the shift of high-temperature reduction peaks to lower temperatures can be attributed to the decrease in the interactions between nickel oxide and alumina support or aluminate species. Such effects are more pronounced and observed for catalysts with high nickel loading, where the Ce/Ni ratio is much smaller.
Although in the obtained catalysts the molar ratio of W/Ni is relatively small, nickel and tungsten precursors and then corresponding oxide phases may interact during impregnation, drying, and calcination stages, leading to the formation of mixed nickel and tungsten oxides (NiO, WO
3) and nickel tungstate species (NiWO
4), together with suitable compounds of Ni and W with alumina, including NiAl
2O
4 and Al
2(WO
4)
3 [
45,
46]. Reduction of Ni-W-Al
2O
3 catalysts may occur through reduction of NiO species to metallic Ni
0 and successive transformation of WO
3 and NiWO
4 to WO
2, with tungsten on the lower oxidation state, accordingly to the reaction equation NiWO
4 + 2H
2 → Ni + WO
2 + 2H
2O, following WO
2 + 2H
2 → W + 2H
2O, and finally formation of Ni–W alloys. According to literature data, transformation of NiWO
4 and reduction of supported WO
3 and Al
2(WO
4)
3 species occurs at high temperatures, above 700 °C [
45,
46]. However, this does not exclude the possibility of partial reduction of tungsten phases at lower temperatures, facilitated by hydrogen atoms, activated on nickel crystallites, which can spillover to WO
x species (see further discussion on XPS results). This may explain the increase in the size of the TPR peaks in the temperature range 500–700 °C with increasing tungsten content. As a result, the catalysts after activation for 2 h at 600 °C in hydrogen may contain nickel crystallites, as well as partially reduced tungsten oxide phases or probably even metallic W and Ni–W alloys. Such species may interact with CO
2 or H
2O during methanation reaction and participate in the successive elementary surface reactions via redox-type mechanism, making W-promoted nickel catalysts similar to cerium promoted or ceria supported catalysts. Although the presence of WO
x or unreduced nickel aluminate species may prevent fast migration of nickel nuclei during reduction stage, leading to the formation of high-active catalysts, an increase in reduction temperature can simultaneously facilitate diffusion of surface atoms and increase the risk of catalysts sintering. An application of high reduction temperature (700–800 °C) may also bring some technical problems of the operation of methanation units.
The XRD curves of the catalysts after reduction at 600 °C for 2 h are shown in the
Figure S2. Wide reflection peaks of γ-Al
2O
3 and metallic nickel are partially overlapped, indicating the presence of the small Ni crystallites. Reflection lines of cerium and tungsten phases are not visible. Consequently, it is difficult to unambiguously estimate the size of nickel crystallites, despite advanced Rietveld method. Mean size of Ni crystallites, determined from the XRD results in the catalysts with ca. 20 wt % Ni, was equal to 4.2 nm (
Table 1). An increase in Ni loading to ca. 40 wt % led to the increase in mean Ni crystallite size to 6.3 nm. XRD studies showed no strong changes in Ni crystallite size with increasing Ce or W content (
Figure 4,
Table 1).
Transmission electron microscopy images of selected catalysts after reduction at 600 °C are presented in
Figure 5. Ni40-Al
2O
3 catalyst contained round nickel crystallites deposited on highly dispersed alumina particles. More detailed inspection of catalysts’ morphology revealed the presence of some surface roughness and irregularities in the shape of nickel crystallites. Some crystallites appeared to be partially covered by the support. The catalyst promoted with cerium and tungsten (Ni40-Ce5-Al
2O
3 and Ni40-W5-Al
2O
3, respectively) showed similar morphology (
Figure 5). The inset in
Figure 5 reveals the presence of nickel particle in the Ni40-W5-Al
2O
3 catalyst partially covered by amorphous–like phases; however, it is difficult to state their chemical nature. STEM images confirmed strong mutual mixing of Ni, Al, Ce, and W elements in the catalysts.
The Ce and W promoters were in part located close to Ni, suggesting direct interaction between nickel particles and promoters. TEM images of promoted catalysts disclosed the presence of some irregular nickel crystallites/agglomerates of the size around 6–8 nm and very small, nearly atomically dispersed Ni crystallites. Electron microscopic studies indicated the decrease of the mean size of nickel crystallites from around 12.7 nm in Ni40-Al
2O
3 catalyst to 7.7 and 7.6 nm in Ni40-Ce5-Al
2O
3 and Ni40-W5-Al
2O
3 catalysts, respectively (
Figure 6).
The active surface area of catalysts (S
a) determined from hydrogen chemisorption studies gradually increased from 12.5 m
2/g for Ni20-Al
2O
3 catalyst with an increase in Ce content to 14.1 m
2/g for Ni20-Ce5-Al
2O
3 (
Table 1). Ni40-Al
2O
3 catalyst showed higher active surface area than Ni20-Al
2O
3 and similar increase in the active surface area of catalysts after introduction of cerium promoter was observed. The values of S
a varied from 18.1 m
2/g for N40-Al
2O
3 to 19.6 m
2/g for Ni40-Ce5-Al
2O
3 catalyst. The opposite direction was visible for tungsten-promoted catalysts. An increase in the content of tungsten from 1 to 5 wt % led to the decrease in the active surface area to 11.6 m
2/g. In the further part of the article, we pointed out the complex role of the tungsten promoter in catalysts, the possibility of the occurrence of metallic tungsten, tungsten alloys, and tungsten oxides, limiting the hydrogen chemisorption.
The curves of hydrogen temperature-programmed desorption are presented in
Figure 7. In the case of Ni20-Al
2O
3 catalyst, the overlapped peaks were observed between −50 and 700 °C. The peaks below 500 °C can be in general ascribed to the desorption of hydrogen from the sites of different strengths on the surface of nickel crystallites [
47,
48]. High-temperature peaks, above 500 °C, in the literature have been often ascribed to the re-oxidation of nickel by the contribution of hydroxyl groups from the support [
49]. The intensity of low temperature peak (−50–100 °C) and wide multiple peaks located between 100 and 500 °C increased with an increase in Ce content for Ni20-Ce-Al
2O
3 catalysts. Such effects indicate an increase in the number of active nickel surface sites, resulting from the decrease in nickel particle size. The intensity of desorption peaks increased with an increase in Ni loading (Ni40-Ce-Al
2O
3).
The shape of desorption peaks and position of maxima for unpromoted and Ce-promoted catalysts containing different amounts of nickel were similar, which may indicate that cerium promoter does not strongly influence the strength of nickel–hydrogen bonds and distribution of individual types of Ni sites [
50]. In turn, an introduction of the small amounts of tungsten led to the decrease in the intensity of the TPD peaks, indicating the decrease in the number of active surface sites, which is consistent with static hydrogen chemisorption studies presented above. Note that the intensity of high-temperature maxima increased with an increase with W content. This effect can be ascribed to the enhanced re-oxidation of nickel active phase or reduced tungsten species by the participation of surface hydroxyl groups or water desorbed at high temperatures from the support (Ni + H
2O → NiO + H
2, WO
x + yH
2O → WO
x+y + yH
2), suggesting changes in nickel dispersion, enhancement of interactions between alumina, and reduced tungsten oxide phases and nickel surface sites.
2.3. Catalytic Performance and the State of Catalysts
Conversion of carbon dioxide and selectivity towards methane determined at different temperatures in the CO
2 methanation reaction in the presence of Ce-promoted Ni20-Al
2O
3 catalysts are presented in
Figure 8a. Thermodynamic constrains are represented here by the dashed lines [
21]. The catalysts show high activity at low reaction temperatures. CO
2 conversion over Ni20-Al
2O
3 catalyst approached thermodynamic limit below 350 °C. The drop in CO
2 conversion and selectivity to methane was observed above 350 °C. Such effects are consistent with thermodynamic limits [
21]. Thermodynamic values of CO
2 conversion decreased from around 100% at 200 °C to around 65% at 600 °C (under 2 bar pressure). The catalysts showed excellent selectivity to methane at low reaction temperatures (close to 100%) [
21]. The selectivity to CH
4 decreased at higher temperatures, in line with the thermodynamics of the CO
2 methanation reaction. The only by-product of the reaction was carbon monoxide [
21]. An introduction of the small amount of Ce led to the increase in CO
2 conversion at low reaction temperatures. Recently, we have demonstrated high durability of unpromoted and Ce-promoted alumina-supported nickel catalysts in time-on-stream of CO
2 methanation reaction [
12,
21].
Figure 8b shows the changes in CO
2 conversion at selected temperatures (200, 240, 280, 320 °C) in the presence of catalysts containing different Ni and promoter amounts. An increase in Ce content up to 5 wt % leads to increase in CO
2 conversion at low reaction temperatures. An almost linear increase in CO
2 conversion with an increase in Ce content up to around 2 wt % was visible at low temperatures for Ni20-Ce-Al
2O
3 and Ni40-Ce-Al
2O
3 catalysts. An introduction of larger amounts of Ce led to the less pronounced changes, especially at higher temperatures, 280 and 320 °C, wherein the activity of Ni40-Ce-Al
2O
3 catalysts was very high. The effects of the presence of tungsten in the catalysts on CO
2 conversion were more complex. Although selectivity changes towards methane with an increase in reaction temperature were the same as in the case of unpromoted catalyst, CO
2 conversion reached maximum at certain tungsten loading (at low reaction temperatures) (
Figure S3 in
Supplementary Materials). A maximum of CO
2 conversion at different reaction temperatures was attained at 2 wt % of W.
It is worth referring at this point to the methanation reaction mechanisms presented in the literature [
51,
52,
53,
54,
55,
56]. The overall process has been often regarded as the set of surface reactions, starting from hydrogen and carbon dioxide dissociative adsorption on the surface of metals; subsequent formation of hydrogen (H
ad), carbonyl (CO
ad), and oxygen species (O
ad); and then, depending on the assumed models, dissociation of carbonyl groups to oxygen (O
ad) and carbon (C
ad) hydrogenated next to CH
x species (carbide mechanism) or hydrogenation of CO
(ad) (hydrogen-assisted mechanism) to corresponding intermediates, such as HCO and H
2CO species. Therefore, in accordance with this model, an increase in the activity of catalysts at low temperatures has been ascribed to the modification of the nature of metal surface sites or to an increase in their number. However, the role of peculiar surface sites and facets is still a matter of discussion [
57,
58]. Zhou et al. indicated that methanation reaction is sensitive to the presence of low-coordinated Ni atoms that enhances the C–O bond scission and correlated its rate on nickel planes in following order Ni(211) > Ni(100) > Ni(111) [
57]. On the other hand, Zhen et al. underlined the positive role of Ni(111) planes in the increase in activity of catalyst during methanation reaction with regards of low CO
2 dissociation barrier [
58]. Numerous studies of CO
2 methanation reaction were performed using supported metal catalysts, indicating the role of support in the activation and transformation of CO
2 to methane [
59,
60]. The associative reaction mechanism assumes that suitable carbonate, hydroxycarbonate, or carbonyl groups are formed on the support in the first step, which are then hydrogenated to methane with participation of hydrogen atoms adsorbed on the surface of metallic crystallites [
59,
60]. According to this model, the improvement in catalysts activity can be achieved not only by modification of support properties but also by increase in the dispersion of metal crystallites, which result in the increase in the total number of periphery atoms. In our opinion, similar effects may appear in the case of cerium-promoted alumina supported nickel catalysts, given that cerium oxide species can be found in close proximity to nickel crystallites.
The obtained results of CO
2 methanation reaction performed in the presence of alumina-supported catalysts promoted with cerium indicate that the activity of catalysts is related to the number of active nickel surface sites. Indeed, a good correlation between the increase in active surface area of catalysts and increase in CO
2 conversion at selected reaction temperatures can be found for cerium-promoted catalysts (
Figure 8c). An increase in the active surface area of alumina supported catalysts (Ni20-Ce-Al
2O
3) from 12.5 to 14.1 m
2/g corresponds with an increase in CO
2 conversion from 4.5 to 10.2% at 200 °C (for Ni20-Al
2O
3 and Ni20-Ce5-Al
2O
3 catalysts, respectively). The increase in conversion is more evident at 240 °C (an increase from 18.2 to 37.9% is observed). The relative changes in CO
2 conversion become less evident with further increase in reaction temperature. Similar trends were observed for the second group of catalysts of higher Ni loading, promoted with cerium. An increase in the active surface area of Ni40-Al
2O
3 catalyst from 18.1 to 19.6 m
2/g observed in Ni40-Ce5-Al
2O
3 corresponded with an increase in CO
2 conversion from 6.6 to 14.4% at 200 °C, and from 27.2 to 53.0% at 240 °C, respectively. However, one can observe that some catalysts within Ni20-Ce-Al
2O
3 group, even with the lower active surface area, showed better catalytic performance than the catalysts containing 40 wt % Ni of high active surface area. For example, compare CO
2 conversion for Ni20-Ce5-Al
2O
3 catalyst and Ni40-Al
2O
3 at 240 °C (
Figure 8c).
The changes discussed above were not apparent for W-promoted catalysts. A decrease in the active surface area with increasing tungsten content was observed, but the CO
2 conversion decreased only after exceeding a certain W content (
Figure 8c). It was even possible to see an increase in CO
2 conversion at low W content for selected catalysts.
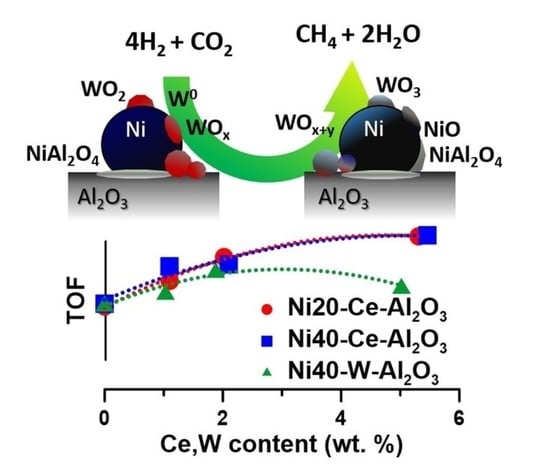
The turnover frequency values (TOF) determined at low conversion of CO
2 increased with increasing Ce content in Ni20-Ce(x)-Al
2O
3 catalysts (
Figure 8d). A similar trend was observed for Ni40-Ce(x)-Al
2O
3 catalysts. Such effects suggest that improved properties of cerium-promoted catalysts are not only related to the increased number of nickel surface sites, although an increase in the active surface area may increase the ability of the adsorption and dissociation of H
2 and CO
2 molecules, which are transformed to CO
ad or C
ad species, hydrogenated to formates, and finally transformed to CH
x species and desorbed from the surface of nickel crystallites [
61]. It seems probable that formation of oxygen vacancy sites or decrease in the oxidation state of Ce
4+ to Ce
3+ in the species located in the close proximity of nickel crystallites or on their surface may occur under reductive conditions of the methanation reaction. Such changes were proposed for ceria-supported nickel catalysts [
20,
21,
62,
63]. Hence, one can expect that the presence of vacancies or Ce
3+ sites in the dispersed cerium oxide species or their basic nature may facilitate activation of CO
2 molecules, dissociative adsorption, and conversion to carbonyl and formate species.
An increase in TOF was also observed for catalysts with low content of tungsten (
Figure 8d). In this case, the role of additional factors influencing activity of catalysts became even more evident. The catalysts (with low tungsten content) did not lose their activity, despite the decrease in active surface area. In our opinion a similar redox mechanism as in the Ce-promoted catalysts discussed above may occur in W-promoted catalysts. The oxidation state of tungsten may change under reaction conditions, leading to the facilitation of the activation and then conversion of CO
2. To confirm the presence of implied changes in the catalysts under different operation conditions, we applied quasi in situ XPS studies.
Survey XPS scans of selected catalysts (Ni40-Al
2O
3, Ni40-Ce5-Al
2O
3, and Ni40-W5-Al
2O
3) after calcination, activation in hydrogen at 600 °C, and after CO
2 methanation reaction performed at 350 °C are shown in
Figure S4 in the Supplementary Materials. The catalysts after reduction and methanation prior to the spectra recording were not exposed to air. The results of deconvolutions of corresponding spectra are presented in
Table S1.
High resolution Ni 2p XPS spectra are presented in
Figure 9. A good fit of the Ni 2p XPS peaks in the spectra of calcined Ni40-Al
2O
3 catalyst based on the peaks positions and energy separation was achieved, assuming the presence of surface NiAl
2O
4 spinel. Similar positions and shape of corresponding peaks can be found booth on the spectra of Ni40-Al
2O
3 and Ni40-Ce-5-Al
2O
3 calcined catalysts. The main component of Ni 2p
3/2 observed at 854.6 eV (FWHM equal 2.54 eV), with strong accompanied satellite peaks at 856.1 and 861.3 eV, was located in a slightly higher binding energy than corresponding peak of bulk NiO reported by Biesinger [
64,
65]. However, it should be stressed that chemical state of Ni
2+ in NiO phases on alumina support can be different from that in bulk NiO. As an example, Arranz et al. identified Ni 2p
3/2 peaks for NiO grown on polycrystalline aluminum at 854.0 and 856.2 eV (with FWHM value of 2.2 eV) [
66]. Simultaneously, they also observed the presence additional broader peak at around 856 eV (with FWHM value of 2.6 eV) attributed to Ni
int located at the aluminum oxide–nickel oxide interface. Hence, one can infer that nickel oxide particles in the obtained catalysts can be decorated by nickel aluminate species, which is in good agreement with the TPR results. The same fitting model was used for deconvolution of the spectra of calcined Ni40-W5-Al
2O
3 catalyst. In this case, the main and satellite lines were slightly shifted to higher energies (
Table S1). Moreover, the intensity of Ni 2p satellite peaks was much higher than on the spectra of unpromoted catalyst. Similar effects were reported by Solsona et al. for Ni–W–O mixed metal oxide catalysts and attributed to a different local environment of the Ni atoms resulting from the presence of tungsten in the structure of catalysts [
67]. However, in contrast to the mentioned work, we observed a very weak intensity of W 4f photoelectrons (
Figure 10), which could be attributed to the presence of diverse oxide species with tungsten in different chemical environment or the specific locations of tungsten species in the catalyst [
67,
68]. Similarly, very low intensities were observed for cerium, which prevented correct interpretation of the spectra (
Figure S4).
The spectra of reduced alumina-supported catalyst (denoted here as Ni40-Al
2O
3–H
2) showed distinct Ni 2p
3/2 peak located at 852.5 eV with corresponding smaller satellite peaks at 856.2 and 858.6 eV, which can be attributed to the presence of metallic nickel (Ni
0) [
64,
65]. In addition, it was possible to distinguish several peaks located at higher energies, ascribed to Ni
δ+, the nature of which is difficult to determine unambiguously (
Figure 9). The best fit was obtained through assuming the presence of NiOOH-like phases. The formation of such species could result from the reoxidation of surface nickel atoms lying in the immediate vicinity of the alumina support (or alumina species decorated the surface of crystallites) by the participation of hydroxyl groups from Al
2O
3. Similar phases were identified by the analysis of the XPS spectrum of Ni40-Ce5-Al
2O
3-H
2 catalyst sample. A good fit was achieved by additional assumption of the presence of small amounts of NiAl
2O
4 (
Table S1). Slightly smaller contribution of NiOOH phases could be a consequence of the presence of dispersed CeO
2 particles lying in the direct vicinity of surface nickel atoms (
Table S1).
XPS studies evidenced that activation of Ni40-Al2O3 and Ni40-Ce5-Al2O3 catalysts prior to the reaction resulted in the formation of metallic nickel crystallites and small amounts of nickel oxide or nickel aluminate species. More complex changes were identified in tungsten promoted catalyst. The first Ni 2p3/2 peak on the XPS spectrum of Ni40-W5-Al2O3-H2 catalyst was broader and its maximum was shifted to higher energies in comparison to the discussed above spectra. The best fit was achieved through assuming the presence of two types of metallic nickel atoms, which can be attributed to metallic nickel Ni0 identified by the presence of the peak at 852.6 eV, together with accompanying satellite peaks at similar position as in the unpromoted nickel catalyst, as well as metallic nickel atoms of different chemical environments, probably in contact with tungsten, denoted here as Ni0-W (where W is necessarily not a metallic tungsten). Additionally, small amounts of nickel aluminate spinel and oxide species were identified.
The same samples after collecting the XPS spectra were transferred to the reaction chamber and exposed to the reaction mixture of H
2 and CO
2 at 350 °C for 1 h. The XPS spectra of catalysts, denoted here as Ni40-Al
2O
3-H
2-CO
2 and Ni40-Ce-5-Al
2O
3-H
2-CO
2, recorded after methanation reaction, revealed slight changes in the oxidation state of nickel, identified mainly by the increase in the contribution of NiOOH-type species (
Table S1). Such effects can be explained by partial reoxidation of surface nickel sites with surface hydroxyl groups or water formed during methanation reaction.
More visible changes were observed in the case of tungsten-promoted catalyst. In addition to the reoxidation of nickel phases discussed above, a decrease in the contribution of the Ni0-W peak was observed. This peak was also shifted towards lower energies. Such effect can be associated with a decrease in the contribution of W being in direct contact with nickel, probably due to reoxidation of tungsten and/or Ni0-W phase segregation.
Such changes correspond with perturbation of the W 4f XPS spectra (
Figure 10). The spectra of tungsten were fitted with the doublets of 4 f
7/2 and 4 f
5/2 peaks, together with corresponding W 5p
3/2 peaks of tungsten at different chemical environments, attributed to metallic tungsten (W
0), metallic tungsten interacting with surface oxygen atoms (W-O), tungsten dioxide (WO
2), and tungsten trioxide (WO
3) of different interactions with alumina support [
67,
68,
69,
70].
The XPS results indicate that reduction of tungsten-promoted catalysts may lead to the formation of oxides with the lower oxidation state of tungsten (WOx, x < 3) and also development of metallic tungsten. Such deep reduction can be ascribed to the interaction between dispersed tungsten oxide species and nickel crystallites, as well as enhanced breaking of W–O bonds by participation of hydrogen atoms activated on metallic nickel surface sites. Thus, metallic tungsten may interact with nickel (Ni0), forming corresponding alloys or composite oxide systems (Ni0-W). The presence of multiple forms of tungsten oxides may indicate that their contribution will depend on the presence of reducing and oxidizing agents in the reaction zone, including H2 and CH4, as well as CO, CO2, and H2O. The XPS spectra of catalysts recorded after the methanation reaction revealed the decrease in the contribution of metallic tungsten (W0) (which is line with discussed above Ni 2p spectra for Ni0-W forms), W–O-type species, and WO2, and simultaneously an increase in the contribution of WO3-like forms.
The increased activity of Ce-containing nickel catalysts has been often ascribed to the improved activation of CO
2 on the specific sites on CeO
2, regarded as redox and basic Lewis centers. In the case of W-promoted catalysts, the nature of oxides with different oxidation state of tungsten (and/or crystallographic structure of oxides) may vary, depending on the reaction conditions, including local and temporal changes in the concentration of the reducing and oxidizing agents over the catalysts surface (H
2, CH
4, H
2O, and CO
2). Such species may also participate in the elementary stages of the methanation reaction, affecting the rightward shift of the equilibrium state of surface reactions. It may be thought that under methanation reaction conditions, partially reduced forms of tungsten oxides (formed under activation steps of catalysts, WO
x + yH
2 → WO
x−y + yH
2O) or even metallic (W
0) or intermetallic Ni
0-W phases may undergo oxidation through binding surface oxygen atoms produced during dissociative adsorption of CO
2 or via the reaction with hydroxyl groups or water formed in the intermediate surface reactions (CO
2(ad) → CO
(ad) + O
(ad); WO
x−y + zO
(ad) → WO
x−y+z; WO
x−y + zH
2O → WO
x−y+z + zH
2) (schematically presented in
Figure 11) [
46,
67,
68,
69,
70,
71,
72].
XPS studies indicated that intermediate oxide species can be also formed on the surface of nickel crystallites. They can be reduced again with the participation of hydrogen atoms activated on the nickel surface sites in subsequent steps of the redox cycle (NiO(surf) + 2H(ad) → Ni0 + H2O; WOx + yH2 → WOx−y + yH2O). However, additional experimental and/or theoretical studies are necessary to verify such hypothesis.
Studies pointed out that positive effect of tungsten can be observed if suitable balance between corresponding surface oxidation-reduction reactions is achieved. They can participate in the redox cycles, enhancing activation and transformation of surface intermediate products. On the other hand, an increase in tungsten loading may lead to the partial coverage of nickel crystallites; formation of less-active W0 or WOx sites; and decrease in the number of Ni0 surface sites, capable for activation of H2 and CO2, thus participating in successive oxidation-reduction cycles of tungsten oxide species. It is likely that an increase in the concentration of oxidizing agents in the gas phase interacting with the catalyst surface of catalysts or the presence of catalysts additives hindering reversible redox changes may act in a similar direction.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}