
Review of CO2 Reduction on Supported Metals (Alloys) and Single-Atom Catalysts (SACs) for the Use of Green Hydrogen in Power-to-Gas Concepts

Abstract
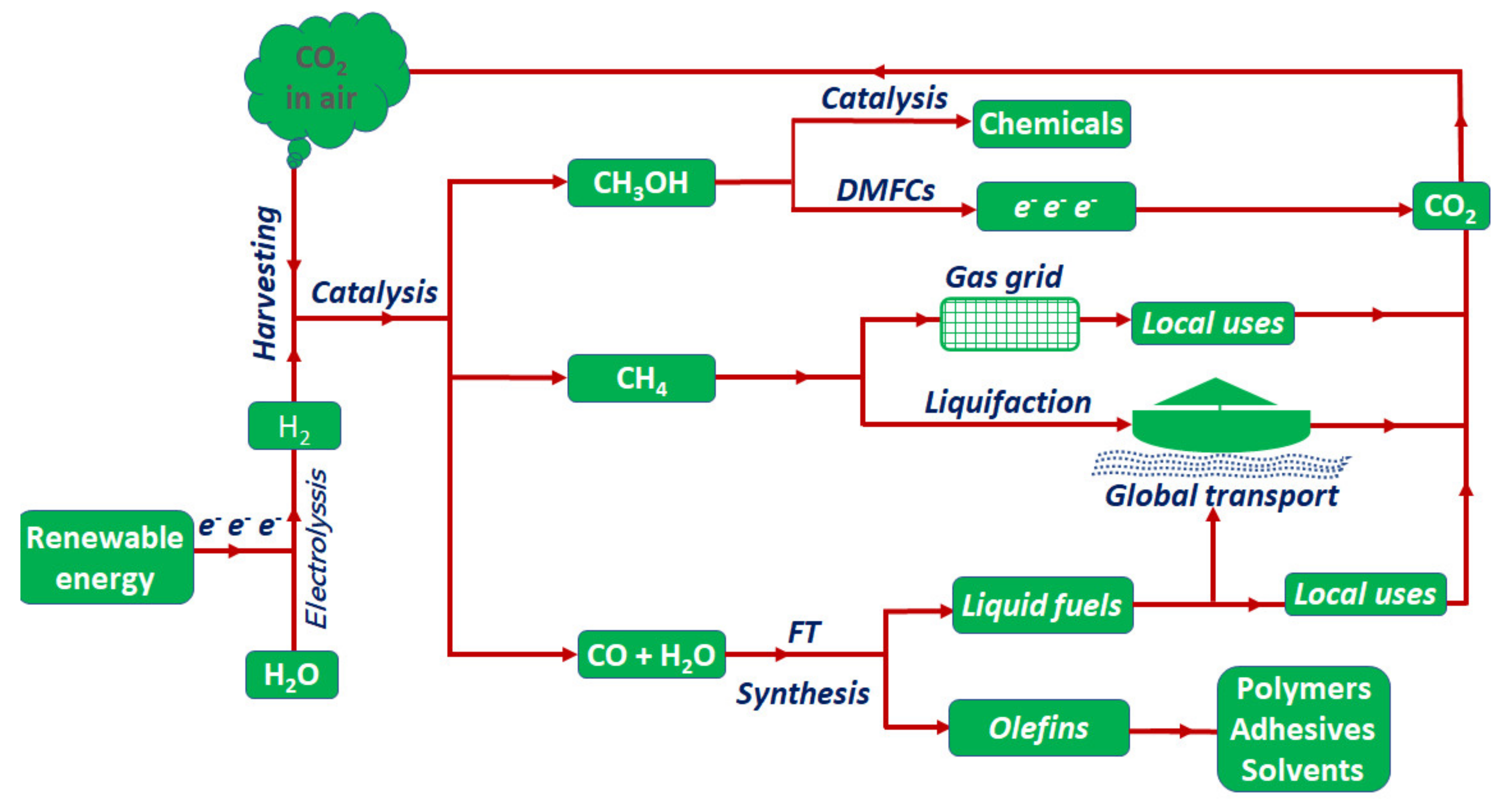
:1. Introduction—CO2 Reduction and Energy Perspectives
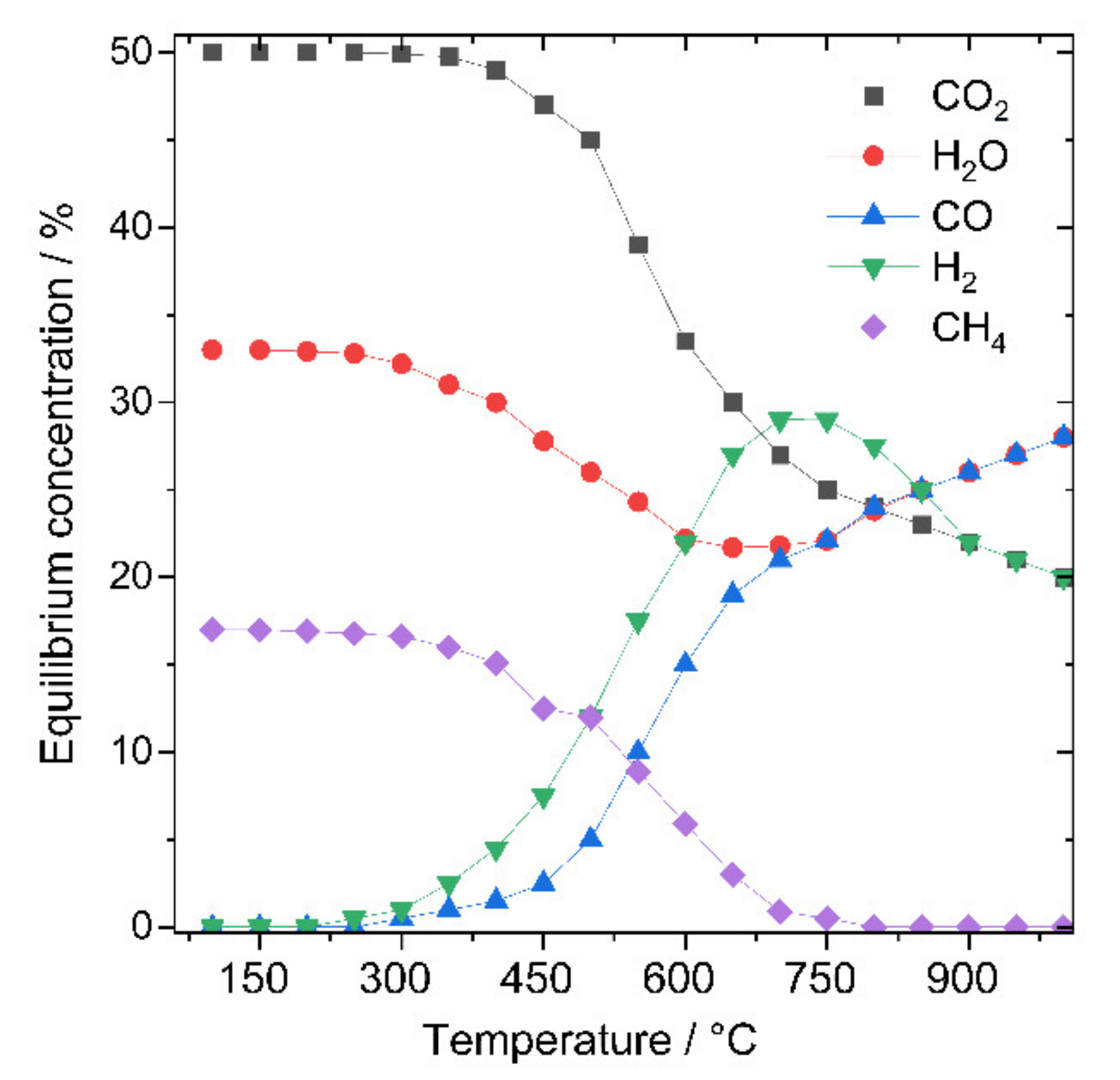
2. Thermodynamic and Kinetic Considerations
3. Catalysts for CO2 Reduction—Activity, Stability, and Selectivity of Different Supported Metals
3.1. Historical Background
3.2. Nickel-Based Catalysts
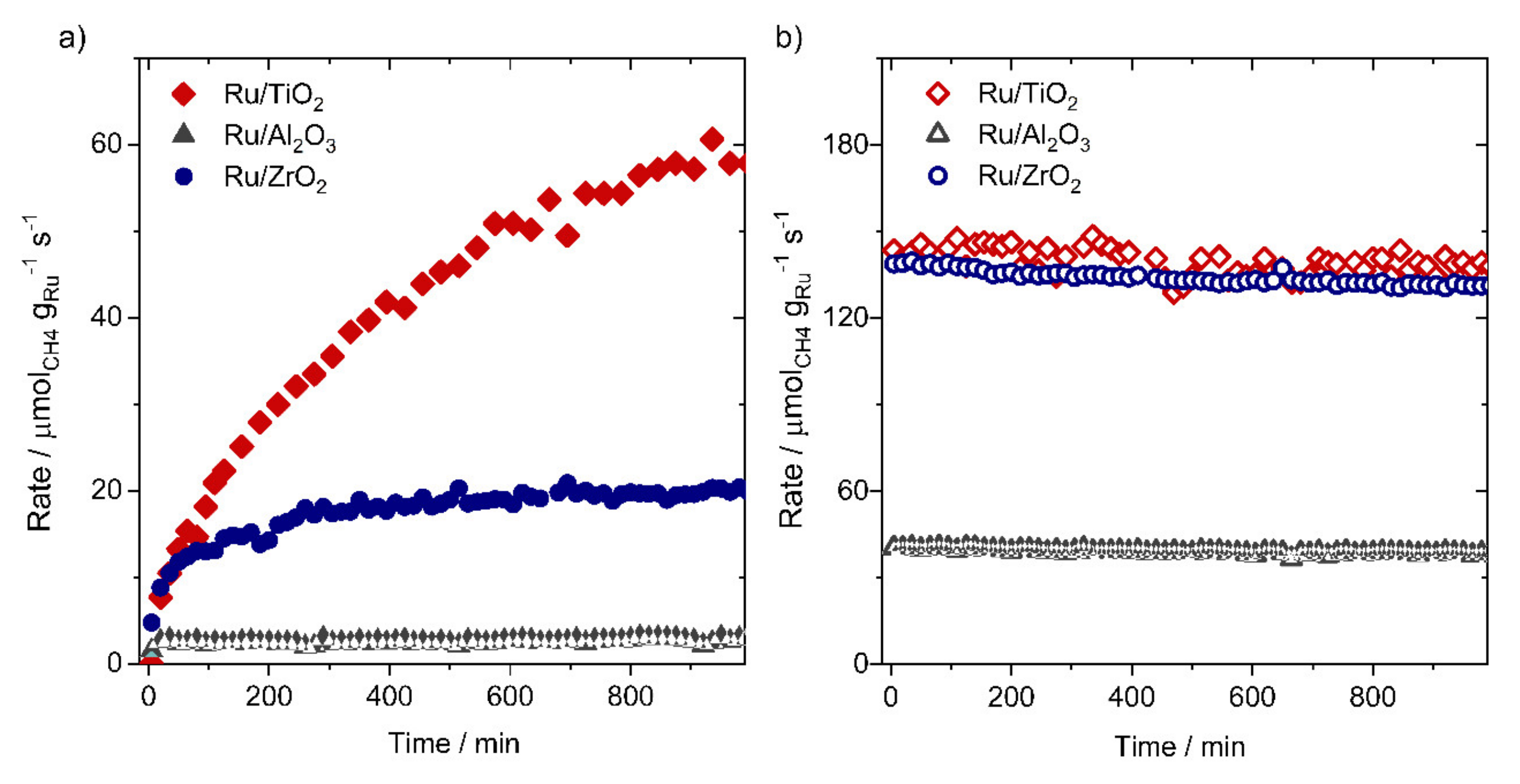
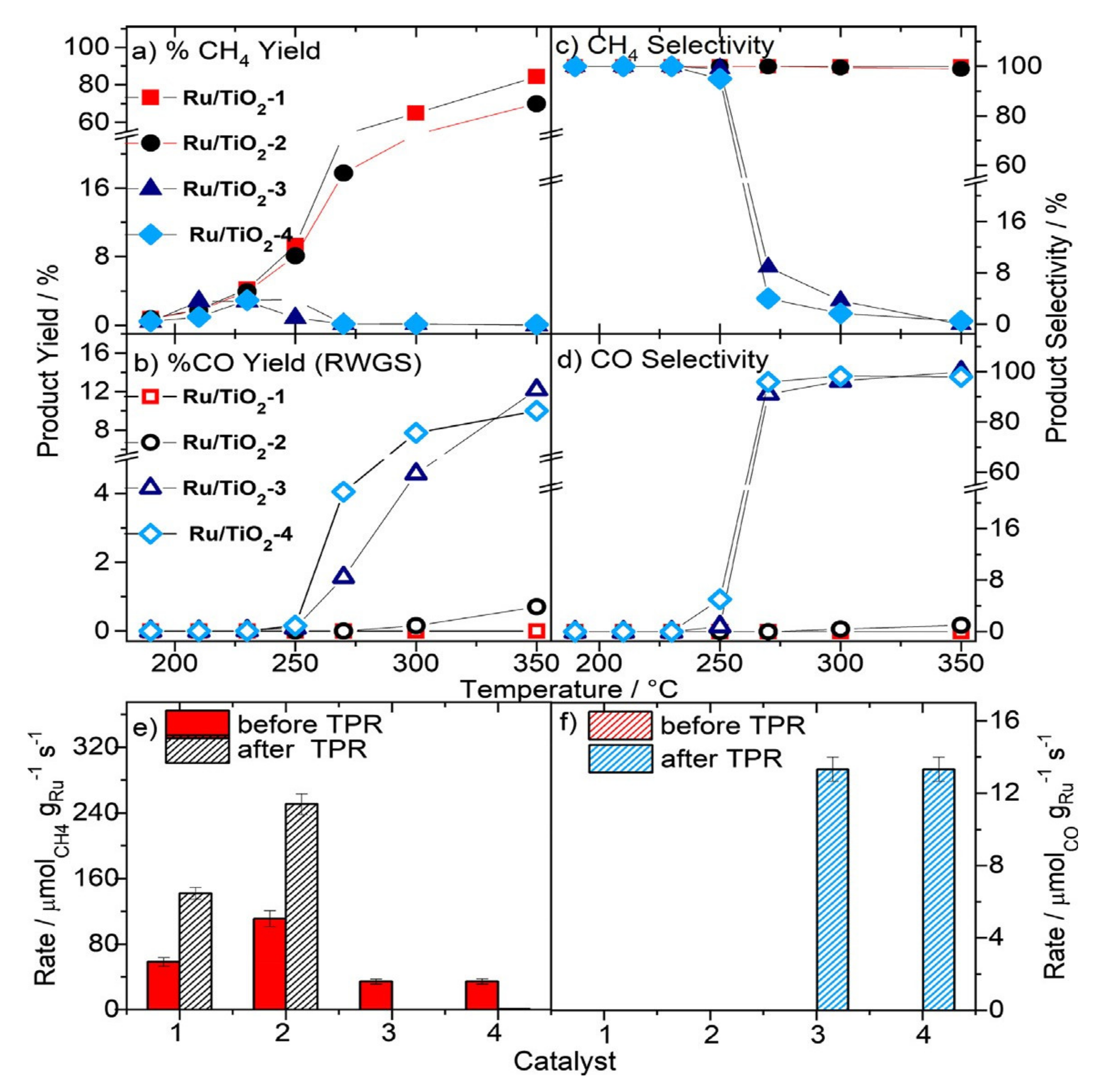
3.3. Ru-Based Catalysts
3.4. Rh-Based Catalysts
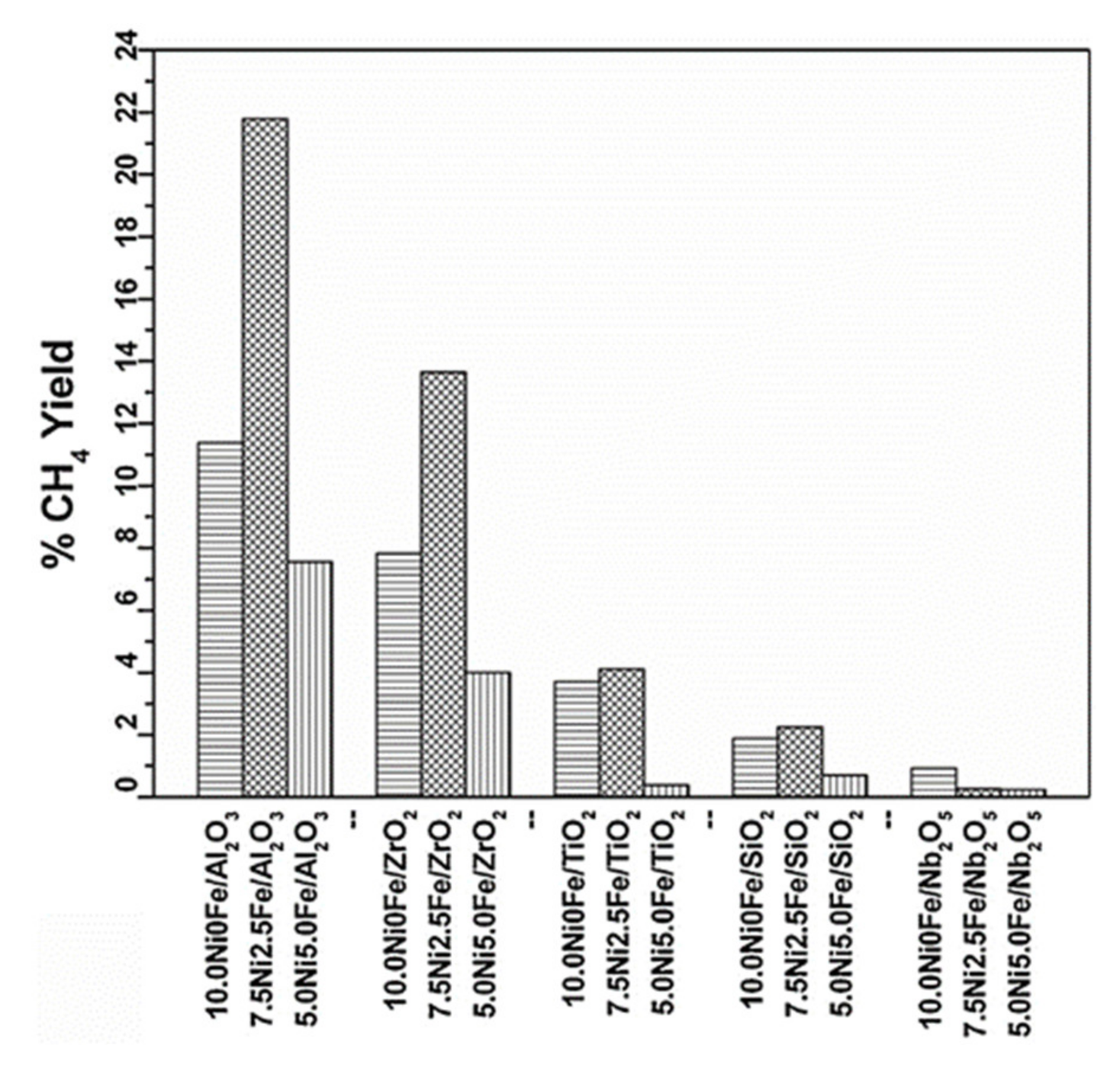
3.5. Bimetallic Alloy Catalysts
4. Support Effects on Different Metals
5. Nonconventional Approaches to Switch CO2 Reduction Selectivity
5.1. Heterogeneous Single Atom Catalysts (SACs) Supported on Metal Oxides for CO2 Reduction
5.1.1. Rh-Single-Atom Catalysts
5.1.2. Ru-Single-Atom Catalysts
5.1.3. Pt-Single-Atom Catalysts
5.2. Controlling the Metal-Support Interaction and Impact on CO2 Reduction Selectivity
6. Outlook, Challenges, and Requirements for Power-to-Gas Applications
- (i)
- reactor abrupt shutdowns due to loss of power, which leads to a rapid reduction in reactor temperature, and thus leads to the condensation of water vapor, existing as a byproduct of either syngas or methanation pathways.
- (ii)
- hydrogen shortage during periods of low electricity production, which is intimately coupled with seasonal changes in temperature or speed of wind. This would lead to the decrease of the H2/CO2 ratios.
- (iii)
- considering that the anticipated supply of carbon dioxide would rely on the harvesting from air or from the outlet of industrial off-gases, any fluctuations in these two supplies would cause similar problems for the performance and stability of the catalyst. In this way, the reactor would operate instead at high H2/CO2 ratios.
- (iv)
- considering the collection of carbon dioxide from air the presence of some impurities of oxygen and water are highly possible in the CO2 feed, which may also affect the stability of these catalysts.
- (v)
- any loss of selectivity toward either syngas or methane would indispensably complicate the practical applications, especially for small scale uses, e.g., for methanation reactors applied for domestic uses (e.g., house heating or fueling farms), the formation of syngas as a byproduct would impost extra costs for elimination and may also result in safety risks.
6.1. Candidates for CO2 to Methanation
6.2. Candidates for CO2 to Syngas
7. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kerr, R.A. Global warming is changing the world. Science 2007, 316, 188–190. [Google Scholar] [CrossRef] [Green Version]
- Peters, G.P.; Andrew, R.M.; Boden, T.; Canadell, J.G.; Ciais, P.; Le Quéré, C.; Marland, G.; Raupach, M.R.; Wilson, C. The challenge to keep global warming below 2 C. Nat. Clim. Chang. 2013, 3, 4–6. [Google Scholar] [CrossRef]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Schlögl, R. Chemistry’s Role in Regenerative Energy. Angew. Chem. Int. Ed. 2011, 50, 6424–6426. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; Jones, J.-P.; Prakash, G.S.; Olah, G.A. Recycling of carbon dioxide to methanol and derived products—Closing the loop. Chem. Soc. Rev. 2014, 43, 7995–8048. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.B.; Denton, W.H.; Nicholls, C.M. Technology and Uses of Liquid Hydrogen; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Iulianelli, A.; Ribeirinha, P.; Mendes, A.; Basile, A. Methanol steam reforming for hydrogen generation via conventional and membrane reactors: A review. Renew. Sustain. Energy Rev. 2014, 29, 355–368. [Google Scholar] [CrossRef] [Green Version]
- McGrath, K.M.; Prakash, G.S.; Olah, G.A. Direct methanol fuel cells. J. Ind. Eng. Chem. 2004, 10, 1063–1080. [Google Scholar]
- Al-Saydeh, S.A.; Zaidi, S.J. Carbon Dioxide Conversion to Methanol: Opportunities and Fundamental Challenges; Carbon Dioxide Chemistry, Capture and Oil Recovery. 2018. Available online: https://www.semanticscholar.org/paper/Carbon-Dioxide-Conversion-to-Methanol%3A-and-Al-Saydeh-Zaidi/5f2e7b783c722bc21c338c7170ff1b9ca9b995c2 (accessed on 19 December 2021).
- Fujiwara, N.; Tada, S.; Kikuchi, R. Power-to-gas systems utilizing methanation reaction in solid oxide electrolysis cell cathodes: A model-based study. Sustain. Energy Fuels 2020, 4, 2691–2706. [Google Scholar] [CrossRef]
- Sabatier, P.; Senderens, J. New methane synthesis. CR Acad. Sci. Paris 1902, 134, 514–516. [Google Scholar]
- Sabatier, P. The method of direct hydrogenation by catalysis. Nobel Lect. 1912, 11, 1901–1921. [Google Scholar]
- Mills, G.A.; Steffgen, F.W. Catalytic methanation. Catal. Rev. 1974, 8, 159–210. [Google Scholar] [CrossRef]
- Bailera, M.; Lisbona, P.; Romeo, L.M.; Espatolero, S. Power to Gas projects review: Lab, pilot and demo plants for storing renewable energy and CO2. Renew. Sustain. Energy Rev. 2017, 69, 292–312. [Google Scholar] [CrossRef]
- Schiebahn, S.; Grube, T.; Robinius, M.; Tietze, V.; Kumar, B.; Stolten, D. Power to gas: Technological overview, systems analysis and economic assessment for a case study in Germany. Int. J. Hydrogen Energy 2015, 40, 4285–4294. [Google Scholar] [CrossRef]
- Sterner, M.; Specht, M. Power-to-Gas and Power-to-X—The History and Results of Developing a New Storage Concept. Energies 2021, 14, 6594. [Google Scholar] [CrossRef]
- Snel, R. Olefins from syngas. Catal. Rev. Sci. Eng. 1987, 29, 361–445. [Google Scholar] [CrossRef]
- Lee, S. Methanol synthesis from syngas. In Handbook of Alternative Fuel Technologies; CRC Press: Boca Raton, FL, USA, 2007; pp. 313–338. [Google Scholar]
- Schulz, H. Short history and present trends of Fischer—Tropsch synthesis. Appl. Catal. A Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective methanation of CO over supported noble metal catalysts: Effects of the nature of the metallic phase on catalytic performance. Appl. Catal. A Gen. 2008, 344, 45–54. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Song, C.; Ji, P.; Wang, N.; Wang, W.; Cui, L. Recent advances in supported metal catalysts and oxide catalysts for the reverse water-gas shift reaction. Front. Chem. 2020, 8, 709. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, P. How I Have Been Led to the Direct Hydrogenation Method by Metallic Catalysts1. Ind. Eng. Chem. 1926, 18, 1005–1008. [Google Scholar] [CrossRef]
- Fischer, F.; Tropsch, H. Über die direkte Synthese von Erdöl-Kohlenwasserstoffen bei gewöhnlichem Druck.(Erste Mitteilung). Ber. Dtsch. Chem. Ges. 1926, 59, 830–831. [Google Scholar] [CrossRef]
- Ponec, V. Some aspects of the mechanism of methanation and Fischer-Tropsch synthesis. Catal. Rev. Sci. Eng. 1978, 18, 151–171. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrogen Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Kester, K.B.; Zagli, E.; Falconer, J.L. Methanation of carbon monoxide and carbon dioxide on Ni/Al2O3 catalysts: Effects of nickel loading. Appl. Catal. 1986, 22, 311–319. [Google Scholar] [CrossRef]
- Wu, H.-C.; Chen, T.-C.; Wu, J.-H.; Pao, C.-W.; Chen, C.-S. Influence of sodium-modified Ni/SiO2 catalysts on the tunable selectivity of CO2 hydrogenation: Effect of the CH4 selectivity, reaction pathway and mechanism on the catalytic reaction. J. Colloid Interface Sci. 2021, 586, 514–527. [Google Scholar] [CrossRef]
- Yan, Z.; Liu, Q.; Liang, L.; Ouyang, J. Surface hydroxyls mediated CO2 methanation at ambient pressure over attapulgite-loaded Ni-TiO2 composite catalysts with high activity and reuse ability. J. CO2 Util. 2021, 47, 101489. [Google Scholar] [CrossRef]
- Zhou, R.; Rui, N.; Fan, Z.; Liu, C.-j. Effect of the structure of Ni/TiO2 catalyst on CO2 methanation. Int. J. Hydrogen Energy 2016, 41, 22017–22025. [Google Scholar] [CrossRef]
- Zeng, L.; Wang, Y.; Li, Z.; Song, Y.; Zhang, J.; Wang, J.; He, X.; Wang, C.; Lin, W. Highly Dispersed Ni Catalyst on Metal–Organic Framework-Derived Porous Hydrous Zirconia for CO2 Methanation. ACS Appl. Mater. Interfaces 2020, 12, 17436–17442. [Google Scholar] [CrossRef] [PubMed]
- Ratchahat, S.; Sudoh, M.; Suzuki, Y.; Kawasaki, W.; Watanabe, R.; Fukuhara, C. Development of a powerful CO2 methanation process using a structured Ni/CeO2 catalyst. J. CO2 Util. 2018, 24, 210–219. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, Y.H.; Moon, D.H.; Ahn, J.Y.; Nguyen, D.D.; Chang, S.W.; Kim, S.S. Reaction mechanism and catalytic impact of Ni/CeO2–x catalyst for low-temperature CO2 methanation. Ind. Eng. Chem. Res. 2019, 58, 8656–8662. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrogen Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, H.; Cui, K.; Xie, H.; Jiao, Z.; Zhang, G.; Xiong, K.; Zheng, X. Methanation of carbon dioxide over Ni/CeO2 catalysts: Effects of support CeO2 structure. Int. J. Hydrogen Energy 2017, 42, 16108–16117. [Google Scholar] [CrossRef]
- Lin, J.; Ma, C.; Wang, Q.; Xu, Y.; Ma, G.; Wang, J.; Wang, H.; Dong, C.; Zhang, C.; Ding, M. Enhanced low-temperature performance of CO2 methanation over mesoporous Ni/Al2O3-ZrO2 catalysts. Appl. Catal. B Environ. 2019, 243, 262–272. [Google Scholar] [CrossRef]
- Patel, R.; Al-Fatesh, A.S.; Fakeeha, A.H.; Arafat, Y.; Kasim, S.O.; Ibrahim, A.A.; Al-Zahrani, S.A.; Abasaeed, A.E.; Srivastava, V.K.; Kumar, R. Impact of ceria over WO3–ZrO2 supported Ni catalyst towards hydrogen production through dry reforming of methane. Int. J. Hydrogen Energy 2021, 46, 25015–25028. [Google Scholar] [CrossRef]
- Chatla, A.; Abu-Rub, F.; Prakash, A.V.; Ibrahim, G.; Elbashir, N.O. Highly stable and coke-resistant Zn-modified Ni-Mg-Al hydrotalcite derived catalyst for dry reforming of methane: Synergistic effect of Ni and Zn. Fuel 2022, 308, 122042. [Google Scholar] [CrossRef]
- Daroughegi, R.; Meshkani, F.; Rezaei, M. Characterization and evaluation of mesoporous high surface area promoted Ni-Al2O3 catalysts in CO2 methanation. J. Energy Inst. 2020, 93, 482–495. [Google Scholar] [CrossRef]
- Ewald, S.; Kolbeck, M.; Kratky, T.; Wolf, M.; Hinrichsen, O. On the deactivation of Ni-Al catalysts in CO2 methanation. Appl. Catal. A Gen. 2019, 570, 376–386. [Google Scholar] [CrossRef]
- Munnik, P.; Velthoen, M.E.; De Jongh, P.E.; De Jong, K.P.; Gommes, C.J. Nanoparticle growth in supported nickel catalysts during methanation reaction—Larger is better. Angew. Chem. 2014, 126, 9647–9651. [Google Scholar] [CrossRef]
- Ranjbar, A.; Irankhah, A.; Aghamiri, S.F. Reverse water gas shift reaction and CO2 mitigation: Nanocrystalline MgO as a support for nickel based catalysts. J. Environ. Chem. Eng. 2018, 6, 4945–4952. [Google Scholar] [CrossRef]
- Ranjbara, A.; Aghamiri, F.; Irankhah, A. Effect of MgAl2O4 catalyst support synthesis method on the catalytic activity of nickel Nano catalyst in reverse water gas shift reaction. Iran. J. Chem. Eng. 2019, 16, 58–69. [Google Scholar]
- Yang, L.; Pastor-Pérez, L.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T. Highly efficient Ni/CeO2-Al2O3 catalysts for CO2 upgrading via reverse water-gas shift: Effect of selected transition metal promoters. Appl. Catal. B Environ. 2018, 232, 464–471. [Google Scholar] [CrossRef]
- Chen, X.; Su, X.; Su, H.-Y.; Liu, X.; Miao, S.; Zhao, Y.; Sun, K.; Huang, Y.; Zhang, T. Theoretical insights and the corresponding construction of supported metal catalysts for highly selective CO2 to CO conversion. ACS Catal. 2017, 7, 4613–4620. [Google Scholar] [CrossRef]
- Chen, C.-S.; Budi, C.S.; Wu, H.-C.; Saikia, D.; Kao, H.-M. Size-tunable Ni nanoparticles supported on surface-modified, cage-type mesoporous silica as highly active catalysts for CO2 hydrogenation. ACS Catal. 2017, 7, 8367–8381. [Google Scholar] [CrossRef]
- Liu, M.-H.; Chen, H.-A.; Chen, C.-S.; Wu, J.-H.; Wu, H.-C.; Yang, C.-M. Tiny Ni particles dispersed in platelet SBA-15 materials induce high efficiency for CO2 methanation. Nanoscale 2019, 11, 20741–20753. [Google Scholar] [CrossRef]
- Wu, H.; Chang, Y.; Wu, J.; Lin, J.; Lin, I.; Chen, C. Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: The influence of particle size on selectivity and reaction pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdőhelyi, A. Hydrogenation of CO2 to CH4 over alumina-supported noble metals. J. Mol. Catal. 1980, 8, 471–474. [Google Scholar] [CrossRef] [Green Version]
- Rynkowski, J.M.; Paryjczak, T.; Lewicki, A.; Szynkowska, M.I.; Maniecki, T.P.; Jóźwiak, W.K. Characterization of Ru/CeO2-Al2O3 catalysts and their performance in CO2 methanation. React. Kinet. Catal. Lett. 2000, 71, 55–64. [Google Scholar] [CrossRef]
- Traa, Y.; Weitkamp, J. Kinetics of the methanation of carbon dioxide over ruthenium on titania. Chem. Eng. Technol. Ind. Chem.-Plant Equip.-Process. Eng.-Biotechnol. 1999, 22, 291–293. [Google Scholar] [CrossRef]
- Li, D.; Ichikuni, N.; Shimazu, S.; Uematsu, T. Hydrogenation of CO2 over sprayed Ru/TiO2 fine particles and strong metal–support interaction. Appl. Catal. A Gen. 1999, 180, 227–235. [Google Scholar] [CrossRef]
- Jiménez, V.; Sánchez, P.; Panagiotopoulou, P.; Valverde, J.L.; Romero, A. Methanation of CO, CO2 and selective methanation of CO, in mixtures of CO and CO2, over ruthenium carbon nanofibers catalysts. Appl. Catal. A Gen. 2010, 390, 35–44. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J.N. CO2 reduction on supported Ru/Al2O3 catalysts: Cluster size dependence of product selectivity. ACS Catal. 2013, 3, 2449–2455. [Google Scholar] [CrossRef]
- Wang, X.; Hong, Y.; Shi, H.; Szanyi, J. Kinetic modeling and transient DRIFTS–MS studies of CO2 methanation over Ru/Al2O3 catalysts. J. Catal. 2016, 343, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Sheng, W.; GAO, D.-N.; WANG, S.-D. Effect of support calcination temperature on the catalytic properties of Ru/Ce0.8Zr0.2O2 for methanation of carbon dioxide. J. Fuel Chem. Technol. 2014, 42, 1440–1446. [Google Scholar]
- Proaño, L.; Tello, E.; Arellano-Trevino, M.A.; Wang, S.; Farrauto, R.J.; Cobo, M. In-situ DRIFTS study of two-step CO2 capture and catalytic methanation over Ru,“Na2O”/Al2O3 Dual Functional Material. Appl. Surf. Sci. 2019, 479, 25–30. [Google Scholar] [CrossRef]
- Dongapure, P.; Bagchi, S.; Mayadevi, S.; Devi, R.N. Variations in activity of Ru/TiO2 and Ru/Al2O3 catalysts for CO2 hydrogenation: An investigation by in-situ infrared spectroscopy studies. Mol. Catal. 2020, 482, 110700. [Google Scholar] [CrossRef]
- Aitbekova, A.; Wu, L.; Wrasman, C.J.; Boubnov, A.; Hoffman, A.S.; Goodman, E.D.; Bare, S.R.; Cargnello, M. Low-temperature restructuring of CeO2-supported Ru nanoparticles determines selectivity in CO2 catalytic reduction. J. Am. Chem. Soc. 2018, 140, 13736–13745. [Google Scholar] [CrossRef]
- Aitbekova, A.; Goodman, E.D.; Wu, L.; Boubnov, A.; Hoffman, A.S.; Genc, A.; Cheng, H.; Casalena, L.; Bare, S.R.; Cargnello, M. Engineering of Ruthenium–Iron Oxide Colloidal Heterostructures: Improved Yields in CO2 Hydrogenation to Hydrocarbons. Angew. Chem. Int. Ed. 2019, 58, 17451–17457. [Google Scholar] [CrossRef]
- Wang, F.; He, S.; Chen, H.; Wang, B.; Zheng, L.; Wei, M.; Evans, D.G.; Duan, X. Active site dependent reaction mechanism over Ru/CeO2 catalyst toward CO2 methanation. J. Am. Chem. Soc. 2016, 138, 6298–6305. [Google Scholar] [CrossRef]
- Tada, S.; Ochieng, O.J.; Kikuchi, R.; Haneda, T.; Kameyama, H. Promotion of CO2 methanation activity and CH4 selectivity at low temperatures over Ru/CeO2/Al2O3 catalysts. Int. J. Hydrogen Energy 2014, 39, 10090–10100. [Google Scholar] [CrossRef]
- Nagase, H.; Naito, R.; Tada, S.; Kikuchi, R.; Fujiwara, K.; Nishijima, M.; Honma, T. Ru nanoparticles supported on amorphous ZrO2 for CO2 methanation. Catal. Sci. Technol. 2020, 10, 4522–4531. [Google Scholar] [CrossRef]
- Ruterana, P.; Buffat, P.; Thampi, K.; Graetzel, M. Selective Dispersion of the Ru-RuOx/TiO2 Catalyst for Methanation of CO2 at Room Temperature and Atmospheric Pressure. MRS Online Proc. Libr. 1989, 139, 327. [Google Scholar] [CrossRef]
- Abe, T.; Tanizawa, M.; Watanabe, K.; Taguchi, A. CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method. Energy Environ. Sci. 2009, 2, 315–321. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Wiese, K.; Parlinska-Wojtan, M.; Rabeah, J.; Brckner, A.; Behm, R., Jr. Encapsulation of Ru Nanoparticles: Modifying the Reactivity Toward CO and CO2 Methanation on Highly Active Ru/TiO2 Catalysts. Appl. Catal. B Environ 2020, 270, 118846. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Dyballa, M.; Parlinska-Wojtan, M.; Bansmann, J.; Pollastri, S.; Olivi, L.; Aquilanti, G.; Behm, R.J. Raising the COx Methanation Activity of a Ru/Al2O3 Catalyst by Activated Modification of Metal–Support Interactions. Angew. Chem. Int. Ed. 2020, 59, 22763–22770. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Abdel-Mageed, A.M.; Gauckler, C.; Olesen, S.E.; Chorkendorff, I.; Behm, R.J. Selective CO Methanation on Isostructural Ru Nanocatalysts: The Role of Support Effects. J. Catal. 2019, 373, 103–115. [Google Scholar] [CrossRef]
- Falbo, L.; Visconti, C.G.; Lietti, L.; Szanyi, J. The effect of CO on CO2 methanation over Ru/Al2O3 catalysts: A combined steady-state reactivity and transient DRIFT spectroscopy study. Appl. Catal. B Environ. 2019, 256, 117791. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Widmann, D.; Olesen, S.E.; Chorkendorff, I.; Biskupek, J.; Behm, R.J. Selective CO Methanation on Ru/TiO2 Catalysts: Role and Influence of Metal.Support Interactions. ACS Catal 2015, 5, 6753–6763. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Li, M.; Cisneros, S.; Bansmann, J.; Rabeah, J.; Brückner, A.; Groß, A.; Behm, R.J. Electronic metal-support interactions and their promotional effect on CO2 methanation on Ru/ZrO2 catalysts. J. Catal. 2021, 400, 407–420. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Widmann, D.; Olesen, S.E.; Chorkendorff, I.; Behm, R.J. Selective CO Methanation on Highly Active Ru/TiO2 Catalysts: Identifying the Physical Origin of the Observed Activation/Deactivation and Loss in Selectivity. ACS Catal. 2018, 8, 5399–5414. [Google Scholar] [CrossRef] [Green Version]
- Cisneros, S.; Chen, S.; Diemant, T.; Bansmann, J.; Abdel-Mageed, A.M.; Goepel, M.; Olesen, S.E.; Welter, E.S.; Parlinska-Wojtan, M.; Gläser, R. Effects of SiO2-doping on high-surface-area Ru/TiO2 catalysts for the selective CO methanation. Appl. Catal. B Environ. 2021, 282, 119483. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M. Operando investigations of particle size and support effects during the selective CO methanation over oxide supported Ru nanoparticles in idealized and realistic H2 feed gases, Universität Ulm. 2016. Available online: https://oparu.uni-ulm.de/xmlui/handle/123456789/4003 (accessed on 19 December 2021).
- Abdel-Mageed, A.M.; Widmann, D.; Eckle, S.; Behm, R.J. Improved performance of Ru/α–Al2O3 catalysts in the selective methanation of CO in CO2-rich reformate gases upon transient exposure to water containing reaction gas. ChemSusChem 2015, 8, 3869–3881. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mageed, A.M.; Eckle, S.; Behm, R.J. Water assisted dispersion of Ru nanoparticles: The impact of water on the activity and selectivity of supported Ru catalysts during the selective methanation of CO in CO2-rich reformate. J. Catal. 2016, 335, 79–94. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Eckle, S.; Behm, R.J. High selectivity of supported Ru catalysts in the Selective CO Methanation—Water makes the difference. J. Am. Chem. Soc. 2015, 137, 8672–8675. [Google Scholar] [CrossRef]
- Stiegler, T.; Meltzer, K.; Tremel, A.; Baldauf, M.; Wasserscheid, P.; Albert, J. Development of a structured reactor system for CO2 methanation under dynamic operating conditions. Energy Technol. 2019, 7, 1900047. [Google Scholar] [CrossRef]
- Falbo, L.; Martinelli, M.; Visconti, C.G.; Lietti, L.; Bassano, C.; Deiana, P. Kinetics of CO2 methanation on a Ru-based catalyst at process conditions relevant for Power-to-Gas applications. Appl. Catal. B Environ. 2018, 225, 354–363. [Google Scholar] [CrossRef]
- Eckle, S.; Denkwitz, Y.; Behm, R.J. Activity, selectivity, and adsorbed reaction intermediates/reaction side products in the selective methanation of CO in reformate gases on supported Ru catalysts. J. Catal. 2010, 269, 255–268. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdöhelyi, A.; Kocsis, M. Surface interaction between H2 and CO2 on RhAl2O3, studied by adsorption and infrared spectroscopic measurements. J. Catal. 1980, 65, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Solymosi, F.; Knozinger, H. Infrared spectroscopic study of the adsorption and reactions of CO2 on K-modified Rh/SiO2. J. Catal. 1990, 122, 166–177. [Google Scholar] [CrossRef]
- Solymosi, F.; Tombácz, I.; Koszta, J. Effects of variation of electric properties of TiO2 support on hydrogenation of CO and CO2 over Rh catalysts. J. Catal. 1985, 95, 578–586. [Google Scholar] [CrossRef]
- Erdőhelyi, A. Hydrogenation of carbon dioxide on supported Rh catalysts. Catalysts 2020, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Solymosi, F.; Tombacz, I. Photocatalytic reaction of H2O+ CO2 over pure and doped Rh/TiO2. Catal. Lett. 1994, 27, 61–65. [Google Scholar] [CrossRef]
- Erdohelyi, A.; Cserényi, J.; Solymosi, F. Activation of CH4 and its reaction with CO2 over supported Rh catalysts. J. Catal. 1993, 141, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Au, C. Carbon dioxide reforming of methane to syngas over SiO2-supported rhodium catalysts. Appl. Catal. A Gen. 1997, 155, 239–252. [Google Scholar] [CrossRef]
- Tsipouriari, V.A.; Efstathiou, A.M.; Verykios, X.E. Transient Kinetic Study of the Oxidation and Hydrogenation of Carbon Species Formed during CH4/He, CO2/He, and CH4/CO2Reactions over Rh/Al2O3Catalyst. J. Catal. 1996, 161, 31–42. [Google Scholar] [CrossRef]
- Stevens, R.W.; Chuang, S.S. In situ IR study of transient CO2 reforming of CH4 over Rh/Al2O3. J. Phys. Chem. B 2004, 108, 696–703. [Google Scholar] [CrossRef]
- Matsubu, J.C.; Yang, V.N.; Christopher, P. Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity. J. Am. Chem. Soc. 2015, 137, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Matsubu, J.C.; Zhang, S.; DeRita, L.; Marinkovic, N.S.; Chen, J.G.; Graham, G.W.; Pan, X.; Christopher, P. Adsorbate-mediated strong metal–support interactions in oxide-supported Rh catalysts. Nat. Chem. 2017, 9, 120–127. [Google Scholar] [CrossRef]
- Andersson, M.P.; Bligaard, T.; Kustov, A.; Larsen, K.E.; Greeley, J.; Johannessen, T.; Christensen, C.H.; Nørskov, J.K. Toward computational screening in heterogeneous catalysis: Pareto-optimal methanation catalysts. J. Catal. 2006, 239, 501–506. [Google Scholar] [CrossRef]
- De Masi, D.; Asensio, J.M.; Fazzini, P.F.; Lacroix, L.M.; Chaudret, B. Engineering iron–nickel nanoparticles for magnetically induced CO2 methanation in continuous flow. Angew. Chem. Int. Ed. 2020, 59, 6187–6191. [Google Scholar] [CrossRef]
- Le Saché, E.; Pastor-Perez, L.; Haycock, B.J.; Villora-Picó, J.J.; Sepulveda-Escribano, A.; Reina, T.R. Switchable catalysts for chemical CO2 recycling: A step forward in the methanation and reverse water–Gas shift reactions. ACS Sustain. Chem. Eng. 2020, 8, 4614–4622. [Google Scholar] [CrossRef]
- Raseale, S.; Marquart, W.; Jeske, K.; Prieto, G.; Claeys, M.; Fischer, N. Supported FexNiy catalysts for the co-activation of CO2 and small alkanes. Faraday Discuss. 2021, 229, 208–231. [Google Scholar] [CrossRef]
- Meshkini Far, R.; Ischenko, O.V.; Dyachenko, A.G.; Bieda, O.; Gaidai, S.V.; Lisnyak, V.V. CO2 hydrogenation into CH4 over Ni–Fe catalysts. Funct. Mater. Lett. 2018, 11, 1850057. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Monforte, G.; Bonura, G.; Ferraro, M.; Dispenza, G.; Antonucci, V.; Aricò, A.; Antonucci, P. The role of Gadolinia Doped Ceria support on the promotion of CO2 methanation over Ni and NiFe catalysts. Int. J. Hydrogen Energy 2017, 42, 26828–26842. [Google Scholar] [CrossRef]
- Huynh, H.L.; Tucho, W.M.; Yu, X.; Yu, Z. Synthetic natural gas production from CO2 and renewable H2: Towards large-scale production of Ni–Fe alloy catalysts for commercialization. J. Clean. Prod. 2020, 264, 121720. [Google Scholar] [CrossRef]
- Tian, D.; Liu, Z.; Li, D.; Shi, H.; Pan, W.; Cheng, Y. Bimetallic Ni–Fe total-methanation catalyst for the production of substitute natural gas under high pressure. Fuel 2013, 104, 224–229. [Google Scholar] [CrossRef]
- Pandey, D.; Ray, K.; Bhardwaj, R.; Bojja, S.; Chary, K.; Deo, G. Promotion of unsupported nickel catalyst using iron for CO2 methanation. Int. J. Hydrogen Energy 2018, 43, 4987–5000. [Google Scholar] [CrossRef]
- Moghaddam, S.V.; Rezaei, M.; Meshkani, F.; Daroughegi, R. Carbon dioxide methanation over Ni-M/Al2O3 (M: Fe, CO, Zr, La and Cu) catalysts synthesized using the one-pot sol-gel synthesis method. Int. J. Hydrogen Energy 2018, 43, 16522–16533. [Google Scholar] [CrossRef]
- Huynh, H.L.; Zhu, J.; Zhang, G.; Shen, Y.; Tucho, W.M.; Ding, Y.; Yu, Z. Promoting effect of Fe on supported Ni catalysts in CO2 methanation by in situ DRIFTS and DFT study. J. Catal. 2020, 392, 266–277. [Google Scholar] [CrossRef]
- Serrer, M.-A.; Gaur, A.; Jelic, J.; Weber, S.; Fritsch, C.; Clark, A.H.; Saraçi, E.; Studt, F.; Grunwaldt, J.-D. Structural dynamics in Ni–Fe catalysts during CO2 methanation–role of iron oxide clusters. Catal. Sci. Technol. 2020, 10, 7542–7554. [Google Scholar] [CrossRef]
- Mutz, B.; Belimov, M.; Wang, W.; Sprenger, P.; Serrer, M.-A.; Wang, D.; Pfeifer, P.; Kleist, W.; Grunwaldt, J.-D. Potential of an alumina-supported Ni3Fe catalyst in the methanation of CO2: Impact of alloy formation on activity and stability. ACS Catal. 2017, 7, 6802–6814. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Zhao, G.; Yang, H.; Yuan, M.; An, X.; Zhou, H.; Qiao, Y.; Tian, Y. CO2 methanation over ordered mesoporous NiRu-doped CaO-Al2O3 nanocomposites with enhanced catalytic performance. Int. J. Hydrogen Energy 2018, 43, 239–250. [Google Scholar] [CrossRef]
- Lange, F.; Armbruster, U.; Martin, A. Heterogeneously-Catalyzed Hydrogenation of Carbon Dioxide to Methane using RuNi Bimetallic Catalysts. Energy Technol. 2015, 3, 55–62. [Google Scholar] [CrossRef]
- Tada, S.; Kikuchi, R.; Takagaki, A.; Sugawara, T.; Oyama, S.T.; Urasaki, K.; Satokawa, S. Study of RuNi/TiO2 catalysts for selective CO methanation. Appl. Catal. B Environ. 2013, 140, 258–264. [Google Scholar] [CrossRef]
- Chen, A.; Miyao, T.; Higashiyama, K.; Yamashita, H.; Watanabe, M. High catalytic performance of ruthenium-doped mesoporous nickel–aluminum oxides for selective CO methanation. Angew. Chem. 2010, 122, 10091–10094. [Google Scholar] [CrossRef]
- Qadir, M.I.; Weilhard, A.; Fernandes, J.A.; de Pedro, I.; Vieira, B.J.; Waerenborgh, J.C.; Dupont, J. Selective carbon dioxide hydrogenation driven by ferromagnetic RuFe nanoparticles in ionic liquids. ACS Catal. 2018, 8, 1621–1627. [Google Scholar] [CrossRef]
- Panaritis, C.; Edake, M.; Couillard, M.; Einakchi, R.; Baranova, E.A. Insight towards the role of ceria-based supports for reverse water gas shift reaction over RuFe nanoparticles. J. CO2 Util. 2018, 26, 350–358. [Google Scholar] [CrossRef]
- Schay, Z.; Guczi, L. Catalytic hydrogenation of carbon monoxide on RuFe/SiO2 catalysts. React. Kinet. Catal. Lett. 1980, 14, 207–212. [Google Scholar] [CrossRef]
- Proaño, L.; Arellano-Treviño, M.A.; Farrauto, R.J.; Figueredo, M.; Jeong-Potter, C.; Cobo, M. Mechanistic assessment of dual function materials, composed of Ru-Ni, Na2O/Al2O3 and Pt-Ni, Na2O/Al2O3, for CO2 capture and methanation by in-situ DRIFTS. Appl. Surf. Sci. 2020, 533, 147469. [Google Scholar] [CrossRef]
- Pawelec, B.; Damyanova, S.; Arishtirova, K.; Fierro, J.G.; Petrov, L. Structural and surface features of PtNi catalysts for reforming of methane with CO2. Appl. Catal. A Gen. 2007, 323, 188–201. [Google Scholar] [CrossRef]
- Kowalczyk, Z.; Stołecki, K.; Rarog-Pilecka, W.; Miśkiewicz, E.; Wilczkowska, E.; Karpiński, Z. Supported ruthenium catalysts for selective methanation of carbon oxides at very low COx/H2 ratios. Appl. Catal. A Gen. 2008, 342, 35–39. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective methanation of CO over supported Ru catalysts. Appl. Catal. B Environ. 2009, 88, 470–478. [Google Scholar] [CrossRef]
- Vance, C.K.; Bartholomew, C.H. Hydrogenation of carbon dioxide on group viii metals: III, Effects of support on activity/selectivity and adsorption properties of nickel. Appl. Catal. 1983, 7, 169–177. [Google Scholar] [CrossRef]
- Li, M.; Amari, H.; van Veen, A.C. Metal-oxide interaction enhanced CO2 activation in methanation over ceria supported nickel nanocrystallites. Appl. Catal. B Environ. 2018, 239, 27–35. [Google Scholar] [CrossRef]
- Mutz, B.; Carvalho, H.W.; Mangold, S.; Kleist, W.; Grunwaldt, J.-D. Methanation of CO2: Structural response of a Ni-based catalyst under fluctuating reaction conditions unraveled by operando spectroscopy. J. Catal. 2015, 327, 48–53. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdöhelyi, A.; Bánsági, T. Methanation of CO2 on supported rhodium catalyst. J. Catal. 1981, 68, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Büchel, R.; Baiker, A.; Pratsinis, S.E. Effect of Ba and K addition and controlled spatial deposition of Rh in Rh/Al2O3 catalysts for CO2 hydrogenation. Appl. Catal. A Gen. 2014, 477, 93–101. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Wiese, K.; Hauble, A.; Bansmann, J.; Rabeah, J.; Parlinska-Wojtan, M.; Brückner, A.; Behm, R.J. Steering the selectivity in CO2 reduction on highly active Ru/TiO2 catalysts: Support particle size effects. J. Catal. 2021, 401, 160–173. [Google Scholar] [CrossRef]
- Gac, W.; Zawadzki, W.; Rotko, M.; Greluk, M.; Słowik, G.; Kolb, G. Effects of support composition on the performance of nickel catalysts in CO2 methanation reaction. Catal. Today 2020, 357, 468–482. [Google Scholar] [CrossRef]
- Shen, L.; Xu, J.; Zhu, M.; Han, Y.-F. Essential role of the support for nickel-based CO2 methanation catalysts. ACS Catal. 2020, 10, 14581–14591. [Google Scholar] [CrossRef]
- Pandey, D.; Deo, G. Effect of support on the catalytic activity of supported Ni–Fe catalysts for the CO2 methanation reaction. J. Ind. Eng. Chem. 2016, 33, 99–107. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. [Google Scholar] [CrossRef]
- Cui, X.; Li, W.; Ryabchuk, P.; Junge, K.; Beller, M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 2018, 1, 385–397. [Google Scholar] [CrossRef]
- Wang, Y.; Arandiyan, H.; Scott, J.; Aguey-Zinsou, K.-F.; Amal, R. Single atom and nanoclustered Pt catalysts for selective CO2 reduction. ACS Appl. Energy Mater. 2018, 1, 6781–6789. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, H.; Yu, P.; Yuan, Y.; Shahbazian-Yassar, R.; Sheng, Y.; Wu, S.; Tu, W.; Liu, G.; Kraft, M. Isolated Ni single atoms in nitrogen doped ultrathin porous carbon templated from porous g-C3N4 for high-performance CO2 reduction. Nano Energy 2020, 77, 105158. [Google Scholar] [CrossRef]
- Xiong, X.; Mao, C.; Yang, Z.; Zhang, Q.; Waterhouse, G.I.; Gu, L.; Zhang, T. Photocatalytic CO2 Reduction to CO over Ni Single Atoms Supported on Defect-Rich Zirconia. Adv. Energy Mater. 2020, 10, 2002928. [Google Scholar] [CrossRef]
- Tang, Y.; Asokan, C.; Xu, M.; Graham, G.W.; Pan, X.; Christopher, P.; Li, J.; Sautet, P. Rh single atoms on TiO2 dynamically respond to reaction conditions by adapting their site. Nat. Commun. 2019, 10, 4488. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Fan, Y.-Z.; Li, X.; Wei, Z.; Xu, Y.-W.; Zhang, L.; Su, C.-Y. A porous rhodium (III)-porphyrin metal-organic framework as an efficient and selective photocatalyst for CO2 reduction. Appl. Catal. B Environ. 2018, 231, 173–181. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Li, D.; Bansmann, J.; Cisneros, S.; Biskupek, J.; Huang, W.; Behm, R.J. Morphology-Engineered Highly Active and Stable Ru/TiO2 Catalysts for Selective CO Methanation. Angew. Chem. Int. Ed. 2019, 58, 10732–10736. [Google Scholar] [CrossRef]
- Fan, M.; Jimenez, J.D.; Shirodkar, S.N.; Wu, J.; Chen, S.; Song, L.; Royko, M.M.; Zhang, J.; Guo, H.; Cui, J. Atomic Ru immobilized on porous h-BN through simple vacuum filtration for highly active and selective CO2 methanation. ACS Catal. 2019, 9, 10077–10086. [Google Scholar] [CrossRef]
- Wang, Q.; Santos, S.; Urbina-Blanco, C.A.; Hernández, W.Y.; Impéror-Clerc, M.; Vovk, E.I.; Marinova, M.; Ersen, O.; Baaziz, W.; Safonova, O.V. Solid micellar Ru single-atom catalysts for the water-free hydrogenation of CO2 to formic acid. Appl. Catal. B Environ. 2021, 290, 120036. [Google Scholar] [CrossRef]
- Chen, B.; Dong, M.; Liu, S.; Xie, Z.; Yang, J.; Li, S.; Wang, Y.; Du, J.; Liu, H.; Han, B. CO2 Hydrogenation to Formate Catalyzed by Ru Coordinated with a N, P-Containing Polymer. ACS Catal. 2020, 10, 8557–8566. [Google Scholar] [CrossRef]
- Huang, G.; Niu, Q.; Zhang, J.; Huang, H.; Chen, Q.; Bi, J.; Wu, L. Platinum single-atoms anchored covalent triazine framework for efficient photoreduction of CO2 to CH4. Chem. Eng. J. 2022, 427, 131018. [Google Scholar] [CrossRef]
- Tauster, S.; Fung, S.; Baker, R.; Horsley, J. Strong interactions in supported-metal catalysts. Science 1981, 211, 1121–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauster, S.; Fung, S.; Garten, R.L. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 1978, 100, 170–175. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Metal Loading (wt.%) | BET/m2 g−1 | Reaction Gas; Flow; GHSV | Metal Particle Size (nm)/Dispersion (%) | Selectivity (for CH4/for CO) ++ | Rate or TOF (Temperature) | Ref. |
|---|---|---|---|---|---|---|---|
| Ru/Carbon | 3 | 440 | 0.4% CO2/H2; 50 mL min−1 | 1.23/77 | 100/77 | 2.1 × 10−3 s−1 (240 °C) # | [114] |
| Ru/MgO | 5 | 94 | 0.4% CO2/H2; 50 mL min−1 | 1.28/74 | 100/0 | 7.9 × 10−3 s−1 (240 °C) # | [114] |
| Ru/MgAl2O4 | 5 | 96 | 0.4% CO2/H2; 50 mL min−1 | 0.98/93 | 100/0 | 8.55 × 10−3 s−1 (240 °C) # | [114] |
| Ru/Al2O3 | 10 | 225 | 0.4% CO2/H2; 50 mL min−1 | 0.98/93 | 100/0 | 10.5 × 10−3 s−1 (240 °C) # | [114] |
| Ru/Al2O3 | 5 | 83 | 1% CO, 15% CO2, 50% H2; balance He; 200 mL min−1; 48,000 h−1 | 2.2/43 | NR | 110.7 μmolCO2 g−1 s−1 (330 °C) § | [115] |
| Ru/Al2O3 | 0.1 | 200 | 5% CO2, 15% H2; balance He; 60 mL min−1 | NR/100 | 73/27 | 11 × 10−3 s−1 (300 °C) | [54] |
| Ru/Al2O3 | 2.2 | 63.3 | 15% CO2, 60% H2, 25% Ar 25%; 50 mL min−1 | NR/15.2 | 100/0 | 0.69 × 10−3 s −1 (250 °C) # | [61] |
| Ru/Al2O3 | 2.3 | 127 | 15.5% CO2, 80.9% H2, balance N2; 41.6 mL min−1; 18,000 h−1 | 1.7/71 | 100/0 | 6.8 × 10−3 s−1 (190 °C) # | [67] |
| Ru/TiO2 | 5 | 42 | 1% CO, 15% CO2, 50% H2; balance He; 200 mL min−1; 48,000 h−1 | 4.5/21 | NR | 64.8 μmolCO2 gRu−1 s−1 (330 °C) § | [115] |
| Ru/TiO2 | 2.2 | 64–46 | 15.5% CO2, 80.9% H2, balance N2; 41.6 mL min−1; 18,000 h−1 | 1.6/66 | 100/0 | 26.2 × 10−3 s−1 (190 °C) # | [66] |
| Ru/CeO2 | 5 | 3.3 | 1% CO, 15% CO2, 50% H2; balance He; 200 mL min−1; 48,000 h−1 | 5.1/19 | NR | 5.21 μmolCO2 g−1 s−1 (330 °C) § | [115] |
| Ru/CeO2 | 2.3 | 34.2 | 15% CO2, 60% H2, 25% Ar 25%; 40 mL min−1 | NR/35 | 100/0 | 0.71 × 10−3 s−1 (175 °C) # | [61] |
| Ru/ZrO2 | 2.1 | 114–64 | 15.5% CO2, 80.9% H2, balance N2; 41.6 mL min−1; 18,000 h−1 | 1.6/NR | 100/0 | 23.3 × 10−3 s−1 (190 °C) # | [71] |
| Ru/SiO2 | 5 | 144 | 1% CO, 15% CO2, 50% H2; balance He; 200 mL min−1; 48,000 h−1 | 5.5/17 | NR | 27 μmolCO2 g−1 s−1 (330 °C) § | [115] |
| Ni/Al2O3 | 3 | 150 | 1% CO2, 4% H2, & balance N2; 3000–70,000 h−1 | 9.8/9.9 | 88/12 | 22 × 10−3 s−1 (277 °C) # | [116] |
| Ni/SiO2 | 3 | 200 | 1% CO2, 4% H2, balance N2; 3000–70,000 h−1 | 2.5/39 | 60/40 | 13 × 10−3 s−1 (277 °C) # | [116] |
| Ni/SiO2 | 5 | 8 | 20% CO2/H2; 16,000–66,000 h−1 | 5.3/19.1 | 100/0// 85/15 | 1.1 × 10−3 s−1 (200 °C)//2.5 × 10−3 s−1 (250 °C) # | [117] |
| Ni/TiO2 | 3 | 50 | 1% CO2, 4% H2, balance N2; 3000–70,000 h−1 | 13/7.5 | 99/1 | 47.3 × 10−3 s−1 (277 °C) # | [116] |
| Ni/TiO2 | 5 | 53 | 20% CO2/H2 (1:4); 16,000–66,000 h−1 | 9.4/10.8 | 100/0// 100/0 | 6.1 × 10−3 s−1 (200 °C)//11.5 × 10−3 s−1 (250 °C) # | [117] |
| Ni/CaO-Al2O3 | 23 | 170 | 3.75% CO2, 15% H2, He balance; 40 mL min−1; 15,000 h−1 | 8/9 | 100/0 | 19.6 × 10−3 s−1 (250 °C) # | [118] |
| Ni/CeO2 | 5 | 11 | 20% CO2/H2; 16,000–66,000 h−1 | 8.7/11.6 | 100/0// 100/0 | 24.4 × 10−3 s−1 (200 °C)//76.4 × 10−3 s−1 (250 °C) | [117] |
| Rh/Al2O3 | 5 | 100 | 20% CO2/H2; 3000–6000 h−1 | NR/30.2 | 99/1 | 20.4 × 10−3 s−1 (200 °C) # | [119] |
| Rh/Al2O3 | 1 | 171 | 20% CO2/H2; 10 mL min−1; 6000 h−1 | NR/18 | 93/7 | 220 × 10−3 s−1 (400 °C) # | [120] |
| Rh/SiO2 | 5 | 240 | 20% CO2/H2; 3000–6000 h−1 | NR/22.8 | 99/1 | 4.4 × 10−3 s−1 (200 °C) # | [119] |
| Rh/TiO2 | 1 | 150 | 20% CO2/H2; 3000–6000 h−1 | NR/22.3 | 99/1 | 150 × 10−3 s−1 (200 °C) # | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Mageed, A.M.; Wohlrab, S. Review of CO2 Reduction on Supported Metals (Alloys) and Single-Atom Catalysts (SACs) for the Use of Green Hydrogen in Power-to-Gas Concepts. Catalysts 2022, 12, 16. https://doi.org/10.3390/catal12010016
Abdel-Mageed AM, Wohlrab S. Review of CO2 Reduction on Supported Metals (Alloys) and Single-Atom Catalysts (SACs) for the Use of Green Hydrogen in Power-to-Gas Concepts. Catalysts. 2022; 12(1):16. https://doi.org/10.3390/catal12010016
Chicago/Turabian StyleAbdel-Mageed, Ali M., and Sebastian Wohlrab. 2022. "Review of CO2 Reduction on Supported Metals (Alloys) and Single-Atom Catalysts (SACs) for the Use of Green Hydrogen in Power-to-Gas Concepts" Catalysts 12, no. 1: 16. https://doi.org/10.3390/catal12010016