
Engineering of Microbial Substrate Promiscuous CYP105A5 for Improving the Flavonoid Hydroxylation

Abstract
:
1. Introduction
2. Results
2.1. Bioinformatics Analysis
2.2. Cloning, Over-Expression, Purification, and Spectral Characterization of Proteins
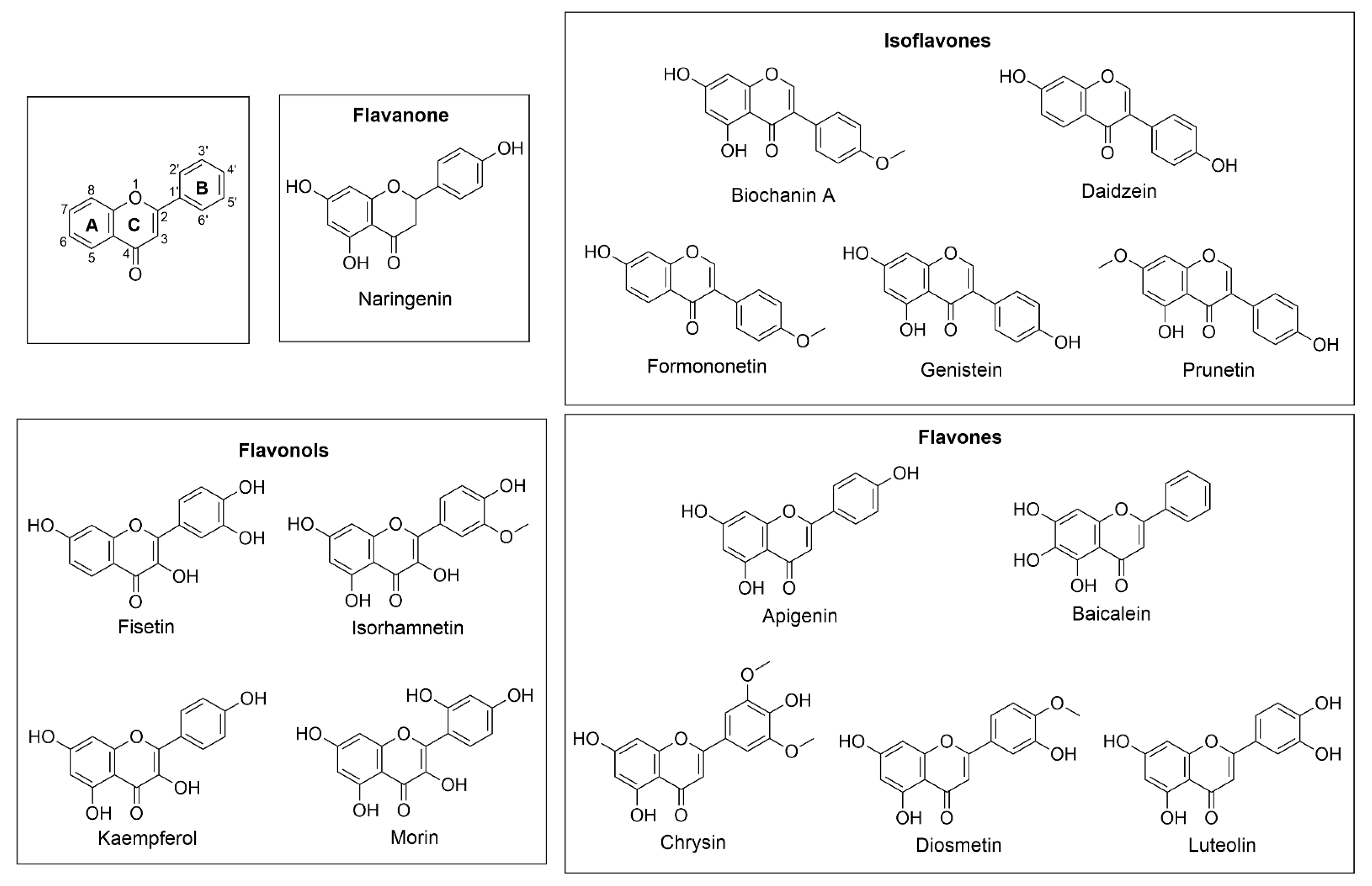
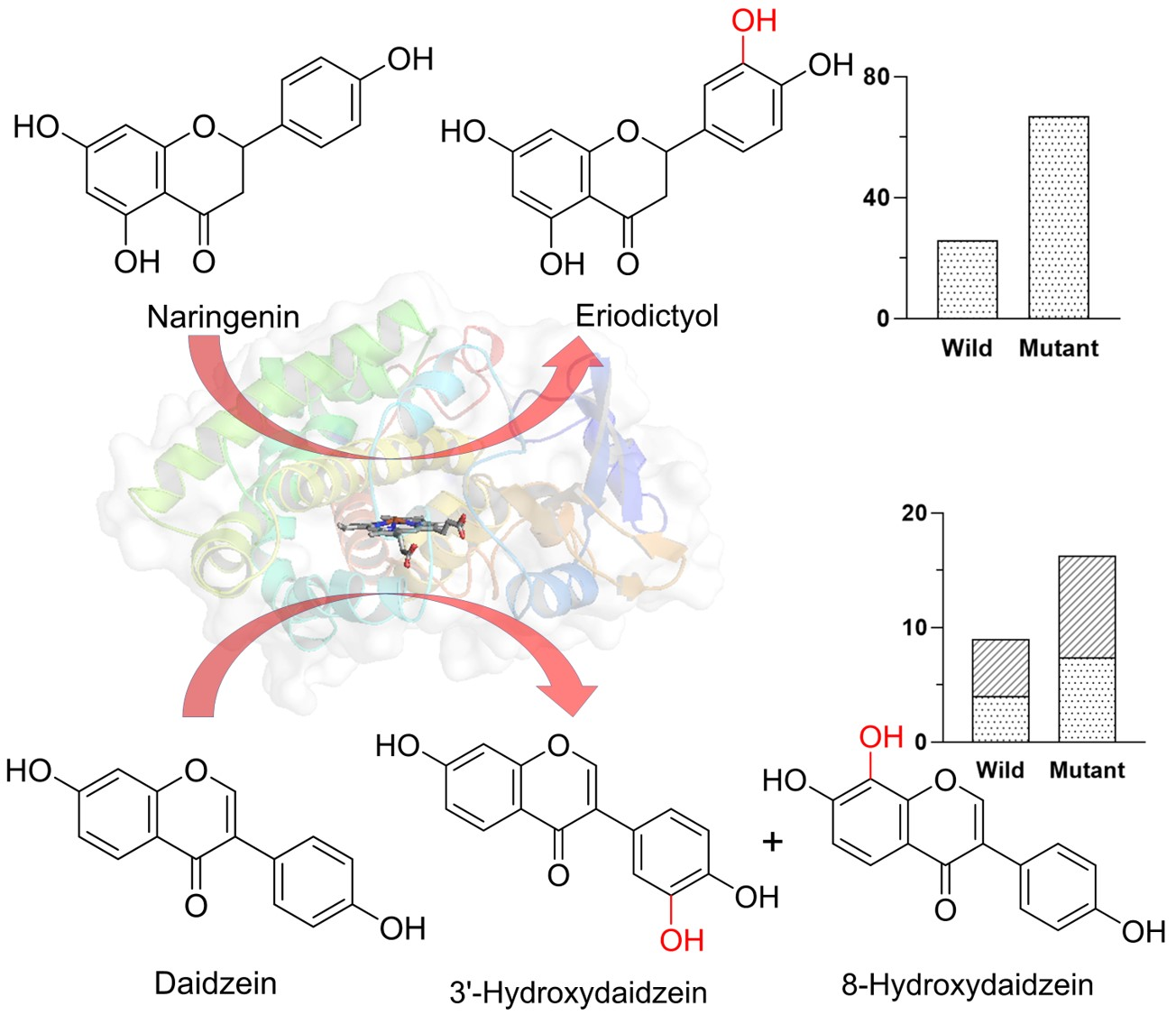
2.3. In Vitro Assay and Product Identification
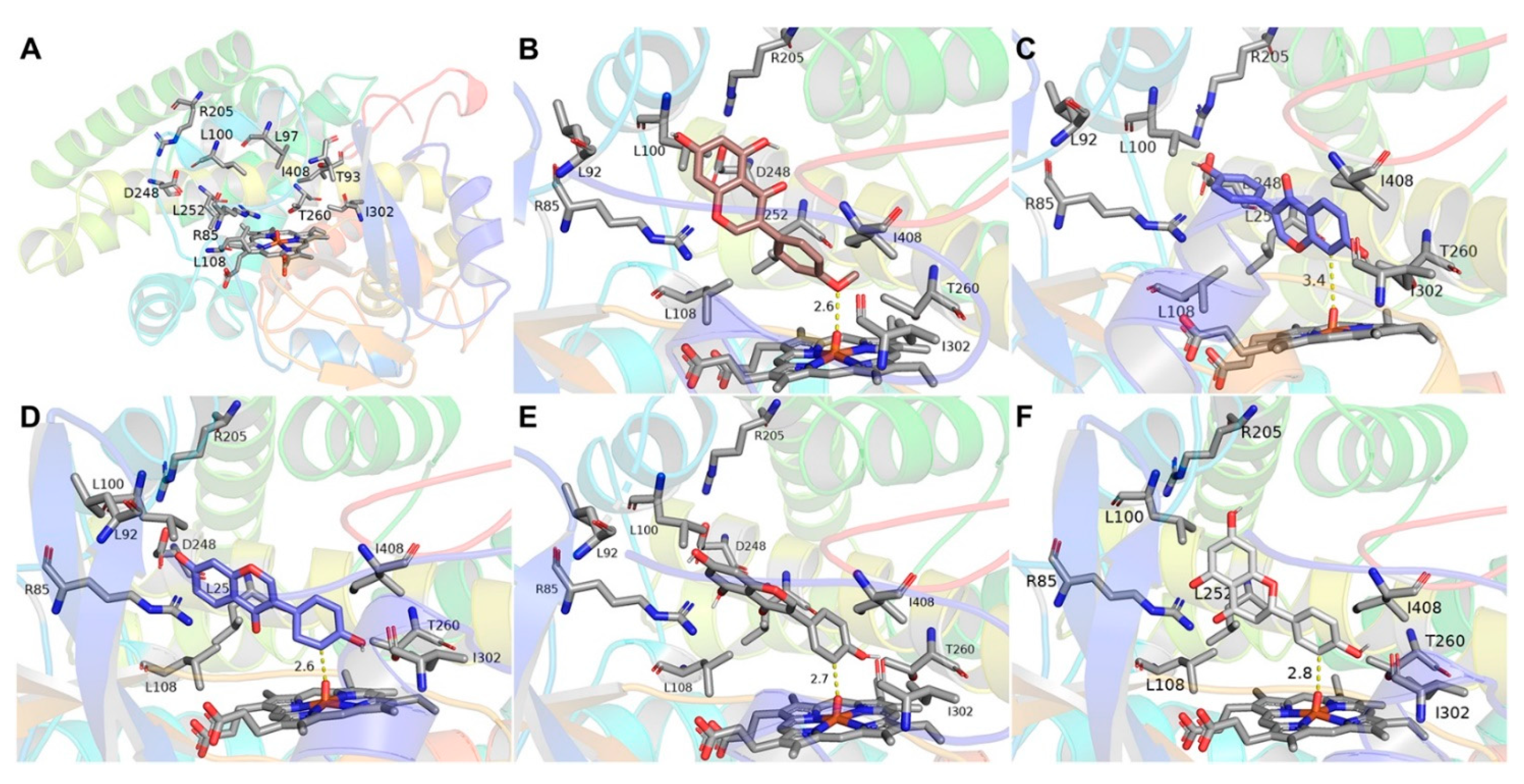
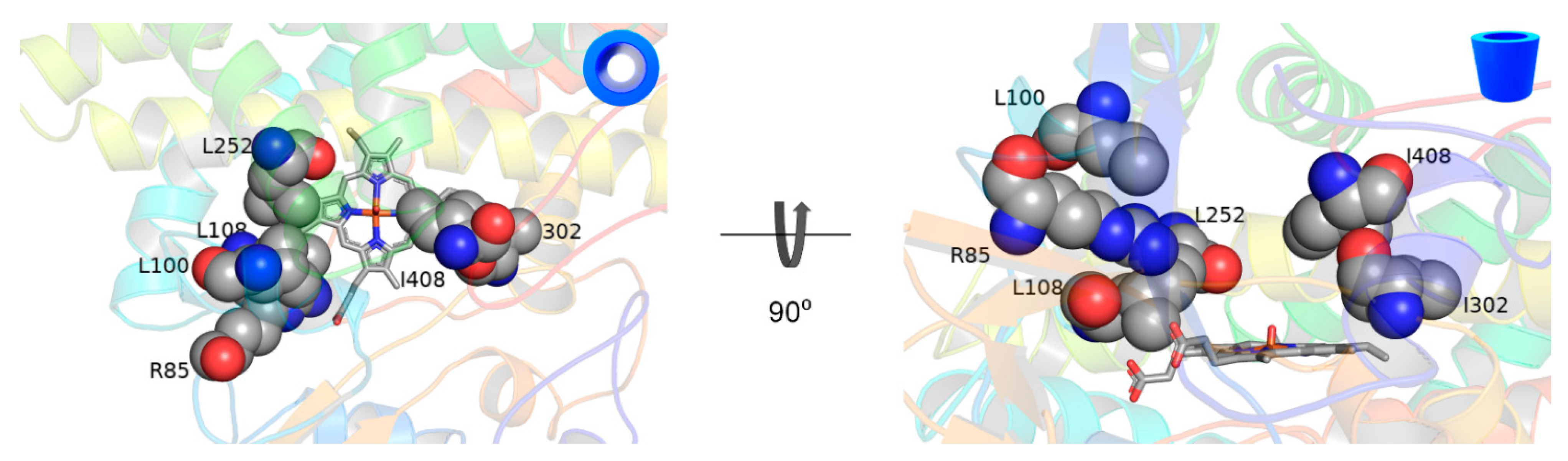
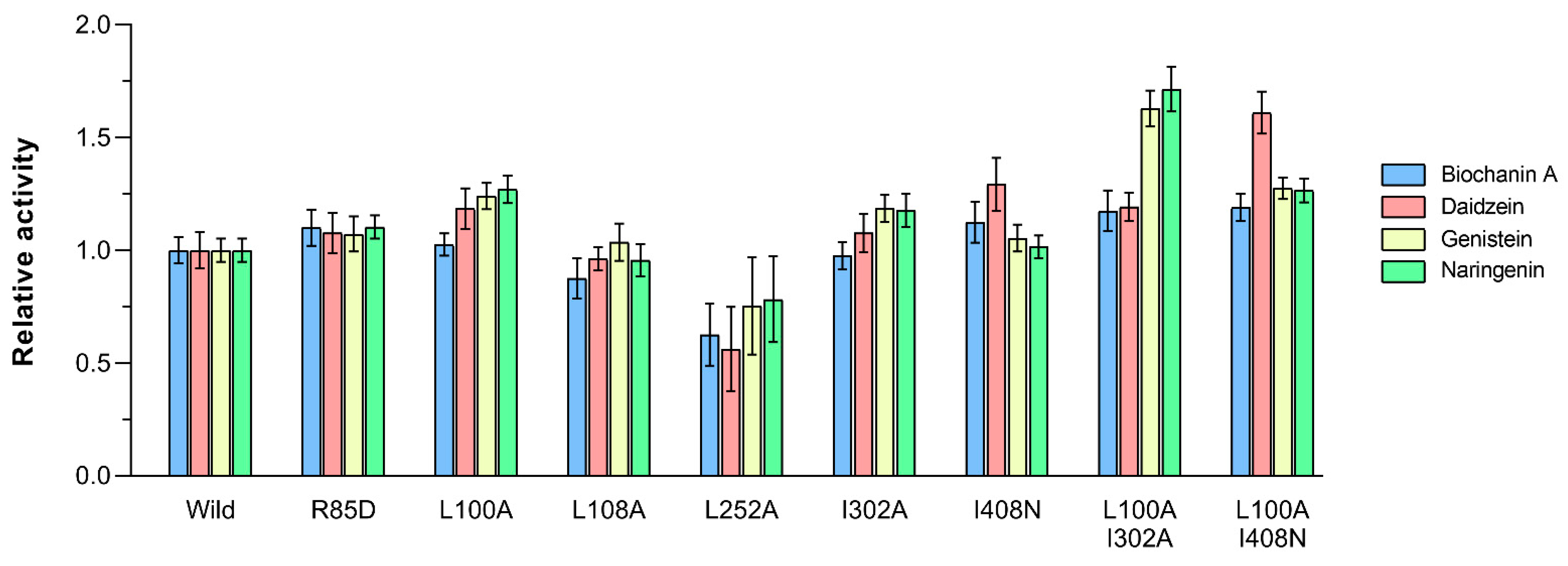
2.4. Docking and Mutagenesis Study
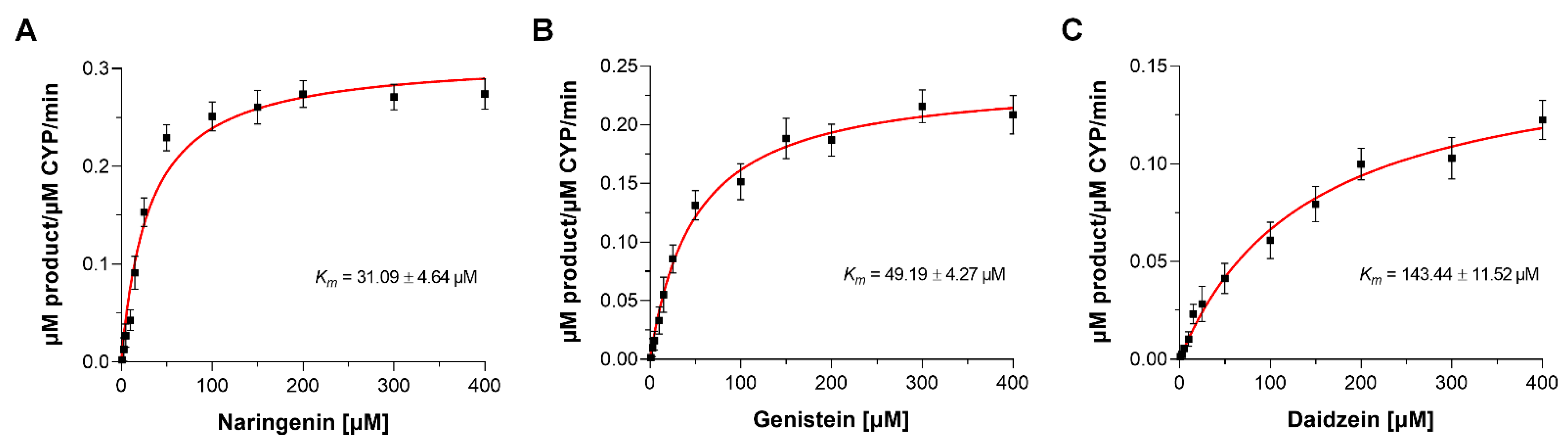
2.5. Enzyme Kinetic Studies
2.6. Whole-Cell Biotransformation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Bioinformatics Analysis
4.3. Molecular Cloning and Protein Over-Expression
4.4. Protein Purification and Determination of the Concentration
4.5. Enzyme Activity Assay
4.6. Site-Directed Mutagenesis
4.7. Determination of Kinetic Parameters
4.8. In silico Analysis: Homology Modeling and Docking
4.9. Whole-Cell Bioconversion
4.10. Analytical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guengerich, F.P. Cytochrome P450 enzymes in the generation of commercial products. Nat. Rev. Drug Discov. 2002, 1, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Isin, E.M.; Guengerich, F.P. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Dangi, B.; Oh, T.J. Bacterial CYP 154C8 catalyzes carbon-carbon bond cleavage in steroids. FEBS Lett. 2019, 593, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Urlacher, V.; Schmid, R.D. Biotransformations using prokaryotic P450 monooxygenases. Curr. Opin. Biotechnol. 2002, 13, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Volz, T.J.; Rock, D.A.; Jones, J.P. Evidence for two different active oxygen species in cytochrome P450 BM3 mediated sulfoxidation and N-dealkylation reactions. J. Am. Chem. Soc. 2002, 124, 9724–9725. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef] [PubMed]
- Klenk, J.M.; Dubiel, P.; Sharma, M.; Grogan, G.; Hauer, B. Characterization and structure-guided engineering of the novel versatile terpene monooxygenase CYP 109Q5 from Chondromyces apiculatus DSM 436. Microb. Biotechnol. 2019, 12, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Munro, A.W.; McLean, K.J.; Grant, J.L.; Makris, T.M. Structure and function of the cytochrome P450 peroxygenase enzymes. Biochem. Soc. Trans. 2018, 46, 183–196. [Google Scholar]
- Rude, M.A.; Baron, T.S.; Brubaker, S.; Alibhai, M.; Del Cardayre, S.B.; Schirmer, A. Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus species. Appl. Environ. Microbiol. 2011, 77, 1718–1727. [Google Scholar] [CrossRef] [Green Version]
- Moody, S.C.; Loveridge, E.J. CYP 105—Diverse structures, functions and roles in an intriguing family of enzymes in Streptomyces. J. Appl. Microbiol. 2014, 117, 1549–1563. [Google Scholar] [CrossRef] [Green Version]
- Sawada, N.; Sakaki, T.; Yoneda, S.; Kusudo, T.; Shinkyo, R.; Ohta, M.; Inouye, K. Conversion of vitamin D3 to 1α, 25-dihydroxyvitamin D3 by Streptomyces griseolus cytochrome P450SU-1. Biochem. Biophys. Res. Commun. 2004, 320, 156–164. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, D.P.; Romesser, J.A.; Leto, K.J. Identification of constitutive and herbicide inducible cytochromes P-450 in Streptomyces griseolus. Arch. Microbiol. 1988, 149, 406–412. [Google Scholar] [CrossRef]
- Omer, C.A.; Lenstra, R.; Litle, P.J.; Dean, C.; Tepperman, J.M.; Leto, K.J.; Romesser, J.A.; O’Keefe, D.P. Genes for two herbicide-inducible cytochromes P-450 from Streptomyces griseolus. J. Bacteriol. 1990, 172, 3335–3345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, M.; Yamashita, M.; Hashimoto, M.; Hino, M.; Fujie, A. Oxidative activities of heterologously expressed CYP107B1 and CYP105D1 in whole-cell biotransformation using Streptomyces lividans TK24. J. Biosci. Bioeng. 2005, 100, 567–572. [Google Scholar] [CrossRef]
- Goto, L.S.; Hokka, C.O.; Lima, J.F.; Prieto, T.; Araújo, A.P.U.; Nantes, I.L.; Nascimento, O.R. Structure and peroxidase activity of ferric Streptomyces clavuligerus orf10-encoded protein P450CLA: UV-visible, CD, MCD and EPR spectroscopic characterization. J. Braz. Chem. Soc. 2012, 23, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.H.; Fushinobu, S.; Takamatsu, S.; Wakagi, T.; Ikeda, H.; Shoun, H. Regio-and stereospecificity of filipin hydroxylation sites revealed by crystal structures of cytochrome P450 105P1 and 105D6 from Streptomyces avermitilis. J. Biol. Chem. 2010, 285, 16844–16853. [Google Scholar] [CrossRef] [Green Version]
- Fouces, R.; Mellado, E.; Díez, B.; Barredo, J.L. The tylosin biosynthetic cluster from Streptomyces fradiae: Genetic organization of the left region. Microbiology 1999, 145, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Wilson, D.; Zhao, L.; Liu, H.W.; Sherman, D.H. Hydroxylation of macrolactones YC-17 and narbomycin is mediated by the pikC-encoded cytochrome P450 in Streptomyces venezuelae. Chem. Biol. 1998, 5, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Ni, S.; Jia, C.; Wang, H.; Sun, G.; Wu, L.; Gan, M.; Shan, G.; He, W.; Lin, L.; et al. Identification of 4, 5-dihydro-4-hydroxygeldanamycins as shunt products of geldanamycin biosynthesis. J. Nat. Prod. 2012, 75, 1480–1484. [Google Scholar] [CrossRef]
- Palaniappan, N.; Kim, B.S.; Sekiyama, Y.; Osada, H.; Reynolds, K.A. Enhancement and selective production of phoslactomycin B, a protein phosphatase IIa inhibitor, through identification and engineering of the corresponding biosynthetic gene cluster. J. Biol. Chem. 2003, 278, 35552–35557. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Sugimoto, H.; Shinkyo, R.; Yamada, M.; Ikeda, S.; Ikushiro, S.; Kamakura, M.; Shiro, Y.; Sakaki, T. Structure-based design of a highly active vitamin D hydroxylase from Streptomyces griseolus CYP105A1. Biochemistry 2008, 47, 11964–11972. [Google Scholar] [CrossRef] [PubMed]
- Ba, L.; Li, P.; Zhang, H.; Duan, Y.; Lin, Z. Semi-rational engineering of cytochrome P450sca-2 in a hybrid system for enhanced catalytic activity: Insights into the important role of electron transfer. Biotechnol. Bioeng. 2013, 110, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wang, Q.; Ikeda, H.; Zhang, C.; Xu, L.H. Hydroxylation of steroids by a microbial substrate-promiscuous P450 cytochrome (CYP105D7): Key arginine residues for rational design. Appl. Environ. Microbiol. 2019, 85, 01530-19. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tungmunnithum, D.; Thongboonyou, A.; Pholboon, A.; Yangsabai, A. Flavonoids and other phenolic compounds from medicinal plants for pharmaceutical and medical aspects: An overview. Medicines 2018, 5, 93. [Google Scholar] [CrossRef]
- Pinto, C.; Cidade, H.; Pinto, M.; Tiritan, M.E. Chiral flavonoids as antitumor agents. Pharmaceuticals 2021, 14, 1267–1296. [Google Scholar] [CrossRef]
- Silva, C.F.; Pinto, D.C.; Silva, A.M. Chromones: A Promising Ring System for New Anti-inflammatory Drugs. ChemMedChem 2016, 1, 2252–2260. [Google Scholar] [CrossRef]
- Perez-Vizcaino, F.; Fraga, C.G. Research trends in flavonoids and health. Biochem. Biophys. 2018, 646, 107–112. [Google Scholar] [CrossRef]
- Hu, M. Commentary: Bioavailability of flavonoids and polyphenols: Call to arms. Mol. Pharm. 2007, 4, 803–806. [Google Scholar] [CrossRef] [Green Version]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Plaza, M.; Pozzo, T.; Liu, J.; Gulshan Ara, K.Z.; Turner, C.; Nordberg Karlsson, E. Substituent effects on in vitro antioxidizing properties, stability, and solubility in flavonoids. J. Agric. Food Chem. 2014, 62, 3321–3333. [Google Scholar] [CrossRef]
- Lo, Y.L.; Wang, W.; Ho, C.T. 7, 3′, 4′-Trihydroxyisoflavone modulates multidrug resistance transporters and induces apoptosis via production of reactive oxygen species. Toxicology 2012, 302, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Kwon, S.H.; Ma, S.X.; Seo, J.Y.; Lee, B.R.; Kim, K.; Kim, S.Y.; Lee, S.Y.; Jang, C.G. The memory-enhancing effects of 7, 8, 4′-trihydroxyisoflavone, a major metabolite of daidzein, are associated with activation of the cholinergic system and BDNF signaling pathway in mice. Brain Res. Bull. 2018, 142, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, B.G.; Ahn, J.H. Production of bioactive hydroxyflavones by using monooxygenase from Saccharothrix espanaensis. J. Biotechnol. 2014, 176, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Collins, S.M.; Vernacchio, V.R.; Lachance, D.M.; Koffas, M.A. Optimization of naringenin and p-coumaric acid hydroxylation using the native E. coli hydroxylase complex, HpaBC. Biotechnol. Prog. 2016, 32, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.L.; Pandey, R.P.; Jung, N.; Jung, H.J.; Kim, E.H.; Sohng, J.K. Hydroxylation of diverse flavonoids by CYP450 BM3 variants: Biosynthesis of eriodictyol from naringenin in whole cells and its biological activities. Microb. Cell Fact. 2016, 15, 135. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.L.; Kong, J.Q. Altering the Regioselectivity of Cytochrome P450 BM3 variant M13 toward genistein through protein engineering and variation of reaction conditions. ACS Omega 2020, 5, 32059–32066. [Google Scholar] [CrossRef]
- Liu, L.; Yao, Q.; Ma, Z.; Ikeda, H.; Fushinobu, S.; Xu, L.H. Hydroxylation of flavanones by cytochrome P450 105D7 from Streptomyces avermitilis. J. Mol. Catal. B Enzym. 2016, 132, 91–97. [Google Scholar] [CrossRef]
- Chiang, C.M.; Ding, H.Y.; Lu, J.Y.; Chang, T.S. Biotransformation of isoflavones daidzein and genistein by recombinant Pichia pastoris expressing membrane-anchoring and reductase fusion chimeric CYP105D7. J. Taiwan Inst. Chem. Eng. 2016, 60, 26–31. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Mak, P.J.; Denisov, I.G. Spectroscopic studies of the cytochrome P450 reaction mechanisms. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 178–204. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 3, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Shinkyo, R.; Hayashi, K.; Yoneda, S.; Yamada, M.; Kamakura, M.; Ikushiro, S.I.; Shiro, Y.; Sakaki, T. Crystal structure of CYP105A1 (P450SU-1) in complex with 1α, 25-dihydroxyvitamin D3. Biochemistry 2008, 47, 4017–4027. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Moss, D.S.; Thornton, J.M. Main-chain bond lengths and bond angles in protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Dangi, B.; Kim, K.H.; Kang, S.H.; Oh, T.J. Tracking down a new steroid-hydroxylating promiscuous cytochrome P450: CYP154C8 from Streptomyces sp. W2233-SM. ChemBioChem 2018, 19, 1066–1077. [Google Scholar] [CrossRef]
- Yasuda, K.; Sugimoto, H.; Hayashi, K.; Takita, T.; Yasukawa, K.; Ohta, M.; Kamakura, M.; Ikushiro, S.; Shiro, Y.; Sakaki, T. Protein engineering of CYP105s for their industrial uses. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 23–31. [Google Scholar] [CrossRef]
- Seitz, C.; Ameres, S.; Forkmann, G. Identification of the molecular basis for the functional difference between flavonoid 3′-hydroxylase and flavonoid 3′, 5′-hydroxylase. FEBS Lett. 2007, 581, 3429–3434. [Google Scholar] [CrossRef]
- Latunde-Dada, A.O.; Cabello-Hurtado, F.; Czittrich, N.; Didierjean, L.; Schopfer, C.; Hertkorn, N.; Werck-Reichhart, D.; Ebel, J. Flavonoid 6-hydroxylase from soybean (Glycine max L.), a novel plant p-450 monooxygenase. J. Biol. Chem. 2001, 276, 1688–1695. [Google Scholar] [CrossRef]
- Kagawa, H.; Takahashi, T.; Ohta, S.; Harigaya, Y. Oxidation and rearrangements of flavanones by mammalian cytochrome P450. Xenobiotica 2004, 34, 797–810. [Google Scholar] [CrossRef]
- Kasai, N.; Ikushiro, S.; Hirosue, S.; Arisawa, A.; Ichinose, H.; Wariishi, H.; Ohta, M.; Sakaki, T. Enzymatic properties of cytochrome P450 catalyzing 3′-hydroxylation of naringenin from the white-rot fungus Phanerochaete chrysosporium. Biochem Biophys Res. Commun. 2009, 387, 103–108. [Google Scholar] [CrossRef]
- Meunier, B.; De Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Zhang, X.; Peng, Y.; Zhao, J.; Li, Q.; Yu, X.; Acevedo-Rocha, C.G.; Li, A. Bacterial cytochrome P450-catalyzed regio-and stereoselective steroid hydroxylation enabled by directed evolution and rational design. Bioresour. Bioprocess. 2020, 7, 2. [Google Scholar] [CrossRef]
- Gotoh, O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 1992, 267, 83–90. [Google Scholar] [CrossRef]
- Sirim, D.; Widmann, M.; Wagner, F.; Pleiss, J. Prediction and analysis of the modular structure of cytochrome P450 monooxygenases. J. BMC Struct. Biol. 2010, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Urban, P.; Lautier, T.; Pompon, D.; Truan, G. Ligand access channels in cytochrome P450 enzymes: A review. Int J. Mol. Sci. 2018, 19, 1617. [Google Scholar] [CrossRef] [Green Version]
- Bracco, P.; Wijma, H.J.; Nicolai, B.; Buitrago, J.A.R.; Klünemann, T.; Vila, A.; Schrepfer, P.; Blankenfeldt, W.; Janssen, D.B.; Schallmey, A. CYP154C5 Regioselectivity in Steroid Hydroxylation Explored by Substrate Modifications and Protein Engineering. ChemBioChem 2021, 22, 1099–1110. [Google Scholar] [CrossRef]
- Hayashi, T.; Harada, K.; Sakurai, K.; Shimada, H.; Hirota, S. A role of the heme-7-propionate side chain in cytochrome P450cam as a gate for regulating the access of water molecules to the substrate-binding site. J. Am. Chem. Soc. 2009, 131, 1398–1400. [Google Scholar] [CrossRef]
- Nicholas, K.B.; Nicholas, H.B.; Deerfield, D.; Gauch, H. Genedoc: A Tool for Editing and Annotating Multiple Sequence Alignments; Ieee Software: Piscataway, NJ, USA, 1997. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Academic Press: Cambridge, MA, USA, 1965; pp. 97–166. [Google Scholar]
- Bhattarai, S.; Liou, K.; Oh, T.J. Hydroxylation of long chain fatty acids by CYP147F1, a new cytochrome P450 subfamily protein from Streptomyces peucetius. Arch. Biochem. Biophys. 2013, 539, 63–69. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. The carbon-monoxide binding pigment of liver microsomes evidence for its haemoprotein nature. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Martin, M.V.; Sohl, C.D.; Cheng, Q. Measurement of cytochrome P450 and NADPH–cytochrome P450 reductase. Nat. Protoc. 2009, 4, 1245–1251. [Google Scholar] [CrossRef] [Green Version]
- Roome, P.W., Jr.; Philley, J.C.; Peterson, J.A. Purification and properties of putidaredoxin reductase. J. Biol. Chem. 1983, 258, 2593–2598. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain/Mutant | Km (µM) | kcat (min−1) | kcat/Km (M−1 min−1) | Ratio kcat/Km | Coupling Efficiency [%] |
|---|---|---|---|---|---|
| Daidzein | |||||
| WT | 143.44 ± 11.52 | 0.09 ± 0.04 | 627.44 | 1.00 | 19.26 ± 3.07 |
| L100A | 124.73 ± 17.89 | 0.10 ± 0.05 | 801.73 | 1.28 | 24.94 ± 5.17 |
| I408N | 159.83 ± 15.37 | 0.12 ± 0.06 | 750.79 | 1.19 | 18.73 ± 6.30 |
| L100A/I408N | 131.57 ± 12.09 | 0.13 ± 0.04 | 988.06 | 1.57 | 26.38 ± 6.95 |
| Genistein | |||||
| WT | 49.19 ± 4.27 | 0.26 ± 0.07 | 5.296 × 103 | 1.00 | 53.83 ± 4.38 |
| L100A | 58.43 ± 6.74 | 0.34 ± 0.09 | 5.818 × 103 | 1.09 | 50.83 ± 6.72 |
| I302A | 67.01 ± 9.83 | 0.42 ± 0.10 | 6.267 × 103 | 1.18 | 44.30 ± 9.29 |
| L100A/I302A | 64.38 ± 8.56 | 0.49 ± 0.12 | 7.611 × 103 | 1.43 | 43.62 ± 8.16 |
| Naringenin | |||||
| WT | 31.09 ± 4.64 | 0.31 ± 0.09 | 9.971 × 103 | 1.00 | 61.27 ± 5.26 |
| L100A | 37.84 ± 8.02 | 0.43 ± 0.11 | 1.136 × 104 | 1.14 | 62.91 ± 9.51 |
| I302A | 32.96 ± 6.38 | 0.39 ± 0.07 | 1.183 × 104 | 1.18 | 50.36 ± 11.83 |
| L100A/I302A | 41.16 ± 7.16 | 0.69 ± 0.09 | 1.676 × 104 | 1.68 | 57.28 ± 10.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subedi, P.; Park, J.K.; Oh, T.-J. Engineering of Microbial Substrate Promiscuous CYP105A5 for Improving the Flavonoid Hydroxylation. Catalysts 2022, 12, 1157. https://doi.org/10.3390/catal12101157
Subedi P, Park JK, Oh T-J. Engineering of Microbial Substrate Promiscuous CYP105A5 for Improving the Flavonoid Hydroxylation. Catalysts. 2022; 12(10):1157. https://doi.org/10.3390/catal12101157
Chicago/Turabian StyleSubedi, Pradeep, Jong Kook Park, and Tae-Jin Oh. 2022. "Engineering of Microbial Substrate Promiscuous CYP105A5 for Improving the Flavonoid Hydroxylation" Catalysts 12, no. 10: 1157. https://doi.org/10.3390/catal12101157